
Abstract
Rechargeable room-temperature sodium–sulfur (Na–S) and sodium–selenium (Na–Se) batteries are gaining extensive attention for potential large-scale energy storage applications owing to their low cost and high theoretical energy density. Optimization of electrode materials and investigation of mechanisms are essential to achieve high energy density and long-term cycling stability of Na–S(Se) batteries. Herein, we provide a comprehensive review of the recent progress in Na–S(Se) batteries. We elucidate the Na storage mechanisms and improvement strategies for battery performance. In particular, we discuss the advances in the development of battery components, including high-performance sulfur cathodes, optimized electrolytes, advanced Na metal anodes and modified separators. Combined with current research achievements, this review outlines remaining challenges and clear research directions for the future development of practical high-performance Na–S(Se) batteries.
Graphic Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Fast global warming, heavy environmental pollution, vast natural resource wastage, uncontrolled urbanization and severe climate change have become increasingly urgent challenges that need to be resolved in the next few decades. Extensive use of renewable energy from natural resources (e.g., solar, wind and hydro) and reduced usage of fossil fuels constitute a sustainable approach to relieve the pressure from these challenges. However, these new types of renewable energy sources are season/weather dependent and intermittent [1]. To ensure affordable and continuous energy supply to customers, the development of stationary electrical energy storage (SEES) systems is essential to boost the utilization of renewable energies [1, 2]. SEES is a technique that can convert and store the power from a power network and convert the power back to the network when needed. Several SEES technologies have been developed to match the energy storage demand, including electrochemical storage (e.g., batteries, supercapacitors), thermal storage (e.g., molten salt, ice storage) or mechanical storage (e.g., pumped hydroelectric systems, flywheels) [2,3,4,5,6,7,8]. Among them, rechargeable batteries have attracted extensive interest, especially rechargeable lithium (Li)-ion batteries, which have dominated the market since their commercialization three decades ago, with applications ranging from small (e.g., smartphones, laptops) to large (e.g., electric vehicles, power walls) devices [9]. The fast-growing and higher demand energy storage market raises various concerns about (1) the limited raw material resources of lithium and cobalt (employed in cathode materials) or even nickel and copper and (2) the limited energy density of batteries based on graphite anodes and transition metal cathodes [9, 10]. Although employing Li metal anodes instead of graphite anodes could resolve the energy density limitation issue, the availability and cost of Li metal batteries remain problematic. Therefore, alternative battery technologies are being investigated, such as alkali metal (e.g., sodium (Na), potassium (K)) batteries and multivalent metal (e.g., zinc (Zn), magnesium (Mg), aluminum (Al), calcium (Ca)) batteries [11, 12].
Among all the alkali elements, Na has similar chemical characteristics to Li because it belongs to the same element group (group IA elements) and has attracted the most attention from researchers interested in studying beyond-lithium-ion batteries. The redox electrode potential of the Na metal anode is − 2.71 V (vs. the standard hydrogen electrode, SHE), which is only 0.35 V higher than that of the Li metal anode. In addition, Na metal anodes possess unique chemical and electrochemical properties, such as a high specific capacity of 1 165 mAh g−1, abundance in the Earth’s crust, and low cost (sodium carbonate—155 US$ t−1 vs. lithium carbonate—17 000 US$ t−1) [13,14,15,16,17,18,19,20]. Therefore, Na-based rechargeable batteries have recently attracted increasing attention.
To fulfill the low cost and high theoretical energy density requirements, room-temperature (RT) sodium–sulfur (selenium) (Na–S(Se)) batteries show the potential to be promising candidates for application in next-generation large-scale SEES systems. Sulfur (S) and selenium (Se) are promising cathode material candidates due to their high theoretical capacity (S—1 672 mAh g−1, Se—678 mAh g−1), abundant storage on the Earth, and low prices [18,19,20,21,22,23]. Sulfur is priced very competitively in the current market at only approximately 69 US$ t−1 [18], which makes the cathode based on sulfur practical for application in large-scale energy storage systems. Na–S batteries have already been commercialized in high-temperature (HT) rechargeable batteries with molten Na as the anode, Na-ion-conductive ceramics as the electrolyte, and molten sulfur as the cathode. The high operating temperature of such batteries (above 300 °C) impedes their facile and safe application in large-scale energy storage systems [24,25,26,27]. Therefore, a surge of interest in RT Na metal batteries has occurred in the past decade, in which Na metal is directly employed as the anode. Additionally, selenium is also very attractive as a new cathode material due to its relatively high ionic conductivity (1 × 10−3 S m−1) compared to sulfur and competitive theoretical specific energy (644 Wh kg−1), which has resulted in the flourishing development of Na–Se batteries in recent years [28,29,30].
However, these battery technologies still face various challenges for practical applications. On the anode side, Na metal suffers from vigorous side reactions and dendrite growth, which cause a short cycle life and severe safety issues. On the cathode side, sluggish reaction kinetics and complex reaction mechanisms lead to poor cycling performance. Extensive research is ongoing to overcome these challenges, including developing high-efficiency Na metal anodes, employing electrolyte engineering strategies and formulating optimized cathodes.
In this review, we comprehensively summarize the recent progress in achieving high-energy–density RT Na–S and Na–Se batteries. First, several high-performance cathode materials, including porous carbon-based composites, 2D material-based composites, polymeric sulfur composites, and other sulfur-integrated composites, are discussed with respect to associated structure optimization tactics and underpinning mechanisms. Then, the recent progress in electrolytes for sulfur cathodes and Na metal anodes is separately summarized and discussed. In particular, this review also highlights the advances in developing gel polymer electrolytes (quasi-solid-state electrolytes) and solid-state electrolytes by enhancing their conductivity, mechanical strength, solid‒solid interfacial contact with electrodes, and electrochemical and thermal stability, which can effectively improve the safety and electrochemical performance of Na–S(Se) batteries. Furthermore, we emphasize the development of advanced Na metal anodes from the points of view of nanostructure construction and advanced artificial protective layers that stabilize the solid electrolyte interphase (SEI). Additionally, several other strategies, such as separator modification and interlayer design that can simultaneously stabilize the cathode and anode, are reviewed, and the corresponding reaction mechanisms are simultaneously illustrated in each section. Finally, we provide future research directions to further improve the electrochemical performance of RT Na–S and Na–Se batteries.
2 History, Reaction Mechanisms and Challenges of Na Metal Batteries
2.1 Roadmap from Na-Ion Batteries to Na Metal Batteries
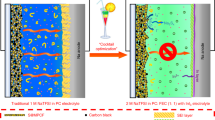
The development of Na-ion batteries started in the 1970s side-by-side with that of lithium-ion (Li-ion) batteries [31,32,33], as shown in Fig. 1a. However, based on the discoveries by John Goodenough et al. during the 1970s to 1980s, Li-ion batteries were commercialized in the 1990s [34, 35]. Afterwards, Li-ion batteries dominated the market, which led to the research on Na-ion batteries almost stopping during that period. Subsequently, the cost of the raw materials for Li-ion batteries continued to increase, which forced the interest in Na-ion batteries to be renewed in the early 2010s [24]. However, state-of-the-art prototype Na-ion batteries can only deliver a specific energy density of approximately 150 Wh kg–1, which is a small fraction of their theoretical value [24]. This made researchers shift their focus toward high-energy Na metal batteries, such as RT Na–S and Na–Se batteries. The advantages of RT Na–S and Na–Se batteries can be identified as two parts. The first part is the raw material abundance and the cost of the batteries. As shown in Fig. 1b, sodium has an approximately 1 180 times higher content than lithium in the Earth’s crust and a much lower price (approximately 110 times) than lithium, as does sulfur (approximately 18 times higher content and 246 times lower price than lithium). The second part is their electrochemical performance. RT Na–S batteries and Na–Se batteries have approximately 17 times and 8 times higher theoretical specific energy densities than Na-ion batteries, respectively. Moreover, Se has a higher conductivity than sulfur, and the theoretical volumetric capacity (3 253 mAh cm−3) is as high as that of sulfur (3 467 mAh cm−3) [36], which makes it promising for high-power and compact energy storage devices. These highlights of RT Na–S and Na–Se batteries give them the potential for further development in future stationary storage.

a Chronological review of the publications and the key points in the development of RT Na–S and Na–Se batteries. b Comparison of sodium, sulfur, and lithium in terms of the element abundance and current market prices of their raw materials, comparison of the ionic conductivity between sulfur and selenium, and comparison of the theoretical specific energy and specific capacity among RT Na–S, Na–Se and Na-ion batteries.
High-temperature sodium–sulfur (HT Na–S) batteries were first developed for electric vehicle (EV) applications due to their high theoretical volumetric energy density. In 1968, Kummer et al. from Ford Motor Company first released the details of the HT Na–S battery system using a β″-alumina solid electrolyte [37]. According to their report, HT Na–S batteries need to operate at a temperature of approximately 300 to 350 °C, in which sodium metal, sulfur and the resulting polysulfides are all in molten states with high chemical reactivity. This causes severe safety concerns and limits the application of HT Na–S batteries for mobile devices and residential use. Furthermore, the discharge of HT Na–S batteries stops at the formation of Na2S3, which is the ultimate liquid-phase catholyte at 300 °C. This leads to the practical capacity decreasing from 1 672 mAh g−1 (complete conversion from β-S8 to Na2S) to 557 mAh g−1 (conversion from β-S8 to Na2S3) [38]. In 2003, NGK Insulator Ltd. started the mass production of HT Na–S batteries [39]. After a few years of development, the production capacity of NGK Insulator Ltd. for HT Na–S batteries achieved 150 MW per year in 2010. However, a fire incident made them temporarily cease HT Na–S production in 2011 [40].
To decrease the operating temperature and improve the capacity of conventional HT Na–S batteries, Fielder et al. and Abraham et al. found that glymes could be suitable solvents for a catholyte that can dissolve both elemental sulfur and solid Na2Sx (x < 3) at 150–300 °C [41]. This enabled the discovery of intermediate-temperature sodium–sulfur (IMT Na–S) battery systems [42]. Subsequently, studies on catholyte additives, Na alloy anodes, improvement of the β″-Al2O3 purity, and solid inorganic electrolytes further enhanced the capacity and cycle life of IMT Na–S batteries [43]. However, IMT Na–S batteries still face similar safety problems owing to the employment of corrosive molten sodium, as well as a high maintenance cost.
RT Na–S batteries operating under ambient temperature (25 °C) were first proposed in 2006, suggesting the possibility of significantly reducing potential safety issues such as fire or explosion derived from highly reactive molten sodium and sulfur. Moreover, the maintenance cost of RT Na–S batteries is remarkably lower than that of HT Na–S and IMT Na–S batteries because they do not require stringent insulation cell packaging and extra heating device to maintain the high working temperature. More importantly, the theoretical energy density of RT Na–S batteries is up to 1 274 Wh kg−1 owing to the complete transition from β-S8 to Na2S, which is much higher than the theoretical energy density of HT Na–S batteries (760 Wh kg–1) [44]. All these advantages provide RT Na–S batteries with a promising future in the development of SEES systems.
As a chemical analogue of sulfur, Se has recently attracted growing research interest due to its much higher electronic conductivity (1 × 10−3 S m−1) than sulfur (5 × 10−30 S m−1) [45, 46]. Additionally, selenium also has an enhanced reaction activity with Na at RT. Meanwhile, Sodium–Selenium (Na–Se) batteries have a comparable high volumetric capacity, which makes them attractive for energy storage with limited space. However, Na–Se batteries have been less investigated due to the intrinsic low theoretical gravimetric capacity of Se (678 mAh g–1) compared to S. In the following sections, we will systematically introduce the mechanisms, challenges, and up-to-date research progress for both RT Na–S and Na–Se batteries.
2.2 Electrochemical Mechanisms and Challenges of RT Na–S and Na–Se Batteries
The RT Na–S battery commonly consists of a sulfur cathode, a sodium metal anode, and a separator soaked with a Na-ion-conducting electrolyte (Fig. 2). The working mechanism of the RT Na–S battery is similar to that of the Li–S battery system, which comprises a series of stepwise reactions starting from ring-opening of β-S8, followed by the formation of long-chain sodium polysulfides (Na2Sx, 4 \(\leqslant\) x \(\leqslant\) 8) and then short-chain sodium polysulfide species (Na2Sx, 1 \(\leqslant\) x \(\leqslant\) 4) [10, 26]. As shown in the inset of Fig. 2, a representative discharge curve of the RT Na–S battery can be identified as consisting of four stages.
-
1.
In stage I, the solid elemental sulfur β-S8 is reduced to a soluble long-chain Na polysulfide (Na2S8), corresponding to the plateau at ~ 2.2 V.
-
2.
Stage II corresponds to the sloping curve in the voltage region between 2.2 and 1.65 V, in which Na2S8 converts in solution to Na2S4.
-
3.
Stage III corresponds to the ~ 1.65 V plateau, where the liquid Na2S4 reduces to insoluble short-chain Na polysulfides, i.e., Na2S3, Na2S2, and Na2S.
-
4.
Stage IV corresponds to the sloping curve in the voltage region between 1.65 and 1.2 V, in which a solid-to-solid transition from Na2S2 to Na2S occurs [47].

Structure and challenges of RT Na–S batteries. Inset: reaction mechanisms of RT Na–S batteries
Although RT Na–S batteries possess a high theoretical energy density depending on the above reactions, several issues must be solved before their practical application. First, the extremely low electronic conductivity of sulfur leads to sluggish electrochemical reaction kinetics and low utilization of active sulfur during discharging. Second, the tremendous volumetric change of sulfur during sodiation/desodiation (~ 260% volume expansion upon full discharge) induces pulverization of the sulfur-based cathode, resulting in loss of active materials and fast capacity fading during cycling [26]. Third, the reaction kinetics of the transition from long-chain sodium polysulfide species to short-chain sodium polysulfides is slow, and vice versa, which results in low round-trip efficiency [48]. Notably, stage IV presents the highest polarization compared to the other stages, which can be ascribed to the slowest reaction kinetics caused by the poor conductivity of solid Na2S2 and Na2S. Fourth, the shuttle effect of the soluble sodium polysulfides (Na2S8, Na2S6 and Na2S4) in conventional liquid electrolytes induces low Coulombic efficiency, high self-discharge and poor cycling performance. These long-chain sodium polysulfide intermediates can easily move across the traditional separators (e.g., polypropylene microporous membrane, glass fiber) to react with the sodium metal anode and form insoluble Na2S2 and Na2S on the Na anode surface. The precipitates corrode the anode surface and cause the loss of both anode and cathode active materials [13]. Finally, sodium dendrite growth is a common and fundamental problem for all Na metal batteries [49]. These Na dendrites can cause continuous breaking and reconstruction of the SEI, which reduces the Coulombic efficiency of RT Na–S batteries. In addition, they can also penetrate the separator and lead to short-circuit of batteries and other associated safety issues [13].
The electrochemical reaction mechanism of Na–Se batteries is similar to that of RT Na–S batteries, with the formation of sodium polyselenides as intermediate products and sodium selenide (Na2Se) as the final discharge product. Likewise, the inherently low electronic conductivity and shuttling of high-order polyselenides (such as Na2Se4, Na2Se6 and Na2Se8) lead to poor electrochemical performance. The high similarity of RT Na–S and Na–Se batteries resulted in researchers employing similar strategies to overcome the shortcomings and improve the performance, which allows us to merge these two topics and summarize their recent progress together in the third section.
To date, tremendous efforts have been devoted to overcoming the shortcomings of RT Na–S and Na–Se batteries mentioned above. The massive successful experience in improving the electrochemical performance of Li–S batteries has shed light on the routes for enhancing the performance of RT Na–S and Na–Se batteries. However, the larger radius of Na+, higher solubility of long-chain sodium polysulfides and polyselenides, and lower reactivity of Na2S and Na2Se compared to the Li counterparts cause more challenges in realizing large-scale practical applications.
3 Cathode Design for RT Na–S and Na–Se Batteries
Although S and Se are the active cathode materials in the corresponding Na metal batteries, neat S or Se powders deliver poor electrochemical performance due to the limitations summarized in the previous section. Rational cathode material design has been demonstrated to be one of the most effective strategies to increase the conductivity and activity of sulfur-based cathodes, buffer their volume expansion, and alleviate the dissolution of polysulfides. Currently, the adopted conventional strategies include infusing S or Se into a porous carbon architecture, anchoring sulfur onto functional nanomaterials via physical and chemical adsorption, fabricating organosulfur composites, and integrating S or Se with other metallic (e.g., Mo) or nonmetallic (e.g., O) elements [44, 50]. In addition, developing catholytes consisting of soluble sodium polysulfides can increase the utilization of active sulfur and improve the electrochemical performance, which will be discussed in the electrolyte Sect. 4.1.3.
3.1 Porous Carbon-Based Composites
Porous carbon nanoarchitectures are the most popular sulfur and selenium hosts for RT Na–S and Na–Se batteries owing to their low cost and good conductivity. Beyond providing good electronic support for poorly conductive S and Se as well as the desirable final discharge products Na2S and Na2Se, the porous carbon structure can also offer space to accommodate the volume change and confine the intermediate polysulfides.
3.1.1 Structure Optimization of Porous Carbon
According to the diameter of the pores, porous materials can be divided into three types: macroporous (pore size > 50 nm), mesoporous (pore size of 2–50 nm) and microporous (pore size < 2 nm) structures. The various porous structures have different impacts on the electrochemical performance of RT Na–S and Na–Se batteries. Macroporous and mesoporous architectures can enable a high sulfur loading and promote electrolyte impregnation. However, these pores are too large to effectively suppress the dissolution of sodium polysulfides or polyselenides into the electrolyte. For example, Wang et al. fabricated hierarchical interconnected mesoporous hollow carbon nanospheres (iMCHSs) as cathode hosts for RT Na–S batteries. S was embedded in the mesopores and encapsulated in the inner hollow spaces of carbon spheres, achieving a high sulfur loading of approximately 60 wt% (wt% means the weight percentage) (Fig. 3a) [51]. Although the RT Na–S batteries using S@iMCHS cathodes displayed a significantly improved electrochemical performance (~ 292 mAh g−1 after 200 cycles at 100 mA g−1) compared to open carbon matrices, such as reduced graphene oxide or mesoporous carbon, self-discharge and capacity decay resulted from dissolution and shuttling of polysulfides, which has yet to be fully addressed.

Transmission electron microscopy (TEM) images and corresponding schematics of a iMCHSs and b MPC. iMCHS picture: reprinted with permission from Ref. [51]. Copyright © 2016, American Chemical Society. MPC picture: reprinted with permission from Ref. [53]. Copyright © 2017, American Chemical Society. c Electrochemical reactions between S/MPC and Na+ during discharge. Reprinted with permission from Ref. [38]. Copyright © 2014, John Wiley & Sons. d Atomic illustration of characteristic nitrogen groups, namely, pyridinic nitrogen (N-PD), pyrrolic nitrogen (N-PL), graphitic nitrogen (N-G), pyridine N-oxide (N-PDO), and pristine graphene (P-G), and optimized binding configurations of sodium sulfide with the corresponding N-doped carbon (where the large yellow ball represents Na, yellow ball represents S, brown ball represents C, blue ball represents N, red ball represents O, and pink ball represents H). e Binding energies of sodium sulfide, short-chain sodium polysulfides and sulfur allotropes to various N-doped carbons. Reprinted with permission from Ref. [55]. Copyright © 2021, American Chemical Society. f Atomic conformations and adsorption energies for Na2S, Na2S2, and Na2S4 species on S, N-doped carbon (right) and pristine carbon (left). Reprinted with permission from Ref. [56]. Copyright © 2020, John Wiley & Sons
In contrast, microporous carbon (MPC) materials are efficient in improving the cyclability of sulfur cathodes because the formation of soluble sodium polysulfides can be circumvented due to the micropore confinement. Theoretical calculations reveal that only small S2–4 molecules can stably exist in the micropores owing to space confinement (less than 0.7 nm) [52]. These micropores can also accommodate the formed final discharge product Na2S, which enables complete reduction of sulfur and leads to a high capacity. During charging, the in situ formed S2− species convert to S2–4 molecules rather than S8 due to the confinement inside the micropores, leading to a good cycling performance (approximately 684 mAh g−1 at 1 C after 2 000 cycles) (Fig. 3b). Xin et al. first proposed and demonstrated this concept by employing a carbon nanotube-supported sulfur-containing microporous carbon cathode (S/CNT@MPC) for RT Na–S batteries [38]. The electrochemical tests and theoretical simulation revealed a two-step reduction reaction from small sulfur molecules to Na2S2 and Na2S occurring at the composite cathode (Fig. 3c). Benefiting from the high conductivity of the CNT core and physical confinement of MPC, the S/CNT@MPC cathode presented a high reversible capacity of 1 148 mAh g−1 and an excellent rate and cycling performance of 580 mAh g−1 at 2 C after 200 cycles. However, the sulfur loading ratio in the MPC host is severely restricted by the micropore volume, and the maximum S loading amount in previous reports was approximately 50 wt% [53]. In addition, the average output voltage of the S/MPC cathode is low because of the absence of transformation of long-chain polysulfides at high voltage. Both issues remarkably decrease the energy density of RT Na–S batteries using the abovementioned materials. Thus, a hierarchical porous sulfur host that can combine the advantages of different pores and ameliorate their shortcomings will be desirable for practical application.
3.1.2 Defect Engineering of Porous Carbon
Although a rational porous carbon host architecture can alleviate dissolution of polysulfides, the weak physical interaction between nonpolar carbon and sulfur species is insufficient to address the shuttle effect. Doping heteroatoms (e.g., N, S) into the carbon material can increase its polarity, thereby suppressing the polysulfide shuttling owing to the enhanced chemical interaction between the polar host materials and sulfur species [54]. Seh et al. systematically investigated the interactions between various types of N-doped carbon and sulfur species and their influences on the performance of RT Na–S batteries [55]. As shown in Fig. 3d, density functional theory (DFT) calculations were performed to optimize the binding configurations and calculate the binding energies of N groups to different sulfur molecules (S4, S3 and S2) and sodium (poly)sulfides (Na2S4, Na2S3, Na2S2 and Na2S). The results in Fig. 3e show a stronger interaction between N-group-containing carbon and discharged sodium sulfide and polysulfides, while pristine carbon prefers to bind to sulfur in the charged state. The experimental results also demonstrate that the RT Na–S batteries with the highest proportion of pyridinic- and pyrrolic-type nitrogen groups deliver the best cycling performance.
Dual heteroatom doping or increasing the ratio of heteroatoms can further enhance the chemical adsorption capability of the carbon host. For instance, the adsorption energies of N, S-codoped carbon to sodium sulfide/polysulfides are much more negative than those of carbon, implying stronger entrapment of these discharge products [56]. Qiang et al. synthesized hierarchical porous carbon with a high doping concentration of 40 atom% (atom% means the atomic percentage) (N, S) for long-cycle-life RT Na–S batteries [57]. Due to the ultrahigh heteroatom concentration, the functional carbon could effectively immobilize sodium polysulfides and prevent side reactions between polysulfides and the carbonate electrolyte. Consequently, the RT Na–S batteries using the as-fabricated cathode maintained a capacity of 378 mAh g−1 at 230 mA g−1 after 350 cycles (98% capacity retention) [57]. Dual-doped (N, S, or O doping) porous carbon materials have also been employed as selenium hosts for high-performance Na–Se batteries [28, 58]. DFT calculations demonstrated the strong chemical affinity of dual-doped carbon to sodium polyselenides, which resulted in enhanced cycling performance. Note that extensive heteroatom doping of the carbon host would inevitably sacrifice the electronic conductivity required for fast reaction kinetics. Therefore, an optimum doping concentration is desired to achieve high-performance RT Na–S and Na–Se batteries.
3.1.3 Metal Catalyst Decoration of Porous Carbon
Decorating the carbon matrix with polar components, such as metal oxides, metal chalcogenides and metal nanoparticles (NPs) or single atoms (SAs), can also directly improve the chemical adsorption capability of the host for polysulfides owing to the strong polar–polar interactions. However, the interactions are restricted to the polar surface of the sulfur host and Na polysulfides, which cannot efficiently prevent the dissolution of Na polysulfides into the electrolyte, particularly at a high sulfur loading. Due to the poor conductivity of sulfur and sodium polysulfides, the conversion reactions from S species to short-chain sodium polysulfides are sluggish. This means that the intermediate polysulfides formed at the early stage cannot be reduced to short-chain polysulfides in a timely manner, and vice versa, resulting in the dissolution of an increasing amount of long-chain polysulfides into the electrolyte [13]. The use of carbon-supported transition-metal-based catalytic hosts can boost the conversion reaction of long-chain polysulfides to insoluble short-chain polysulfides and thereby fundamentally suppress polysulfide shuttling. For example, Liu et al. delicately designed and synthesized a core–shell nanoarchitecture with multisulfiphilic sites for RT Na–S batteries [59]. The inner cores consist of ZnS and CoS2 embedded in N-doped carbon, which not only provide good electronic conductivity to sulfur but also possess strong adsorption of polysulfides. Meanwhile, CoS2 and ZnS present good electrocatalytic capability, which speeds up sulfur conversion. Additionally, the outer MPC shell can further physically confine polysulfide dissolution and stabilize the cathode structure. Similarly, Xu’s group reported a hollow, polar and catalytic bipyramid prism consisting of carbon CoS2/carbon as a cathode host for high-performance RT Na–S batteries (Fig. 4a) [60]. Due to the unique porous structure that can trap sodium polysulfides and accelerate the reduction reaction, the sulfur/CoS2/C cathode achieves a high capacity of 675 mAh g−1 after 800 cycles at 0.5 C, with an ultralow capacity decay rate of 0.012 6%.

a Synthesis of bipyramid prism catalytic metal chalcogenide/C@S composites. Reprinted with permission from Ref. [60]. Copyright © 2020, Nature Publishing Group. b Schematic illustration of the construction of S@CNTs/Co@NC. c TEM image of CNTs/Co@NC. Reprinted with permission from Ref. [63]. Copyright © 2021, John Wiley & Sons. d Schematic illustration of the synthesis of S@Con-HS. e, f High-angle annular dark field (HAADF) images of S@Con-HS showing the Co SAs and clusters dispersed on carbon spheres. Reprinted with permission from Ref. [64]. Copyright © 2018, Nature Publishing Group
Beyond transition-metal chalcogenides, bare transition metal and precious metal NPs/SAs are also employed to modify the carbon host [48, 61, 62]. These metal NPs/SAs are expected to enhance the sulfur conductivity, catalyze the sulfur conversion, and capture polysulfides via chemical bonding. For example, Mou et al. reported metal organic framework (MOF)-derived hierarchical porous nanocubes with self-grown CNTs and Co NPs embedded in a N-doped carbon matrix to efficiently immobilize sulfur species (Fig. 4b) [63]. The Co NPs and N-doped carbon derived from bimetallic zeolitic imidazolate frameworks (ZIFs) have strong polarity and electrocatalytic activity, which significantly promote sulfur redox reactions and suppress polysulfide shuttling.
Zhang et al. fabricated a sulfur host with atomic cobalt-decorated hollow carbon sphere-encapsulated S (S@Con-HC) to enhance the sulfur activity and catalyze the reduction reaction from polysulfides to Na2S [64]. As shown in Fig. 4c, the shuttle effects were inhibited owing to the enhanced electrocatalysis of Co and the polar–polar interaction between Co clusters/SAs and polysulfides. As a result, the S@Con-HC cathode presented a significantly enhanced rate and cycling performance compared to the S@HC electrode. Lai et al. developed a general method to synthesize a range of N-doped carbon-supported SAs, such as Fe, Ni, Mn, Ge, Ru, and Pt, and evaluated their electrocatalytic behavior in RT Na–S batteries [65]. Among these candidates, the Fe SA-based sulfur cathode exhibited the best performance for polysulfide conversion and immobilization.
3.2 2D Material-Based Composites
Owing to the large active surface area, open channels for fast ion accessibility and good electronic conductivity, two-dimensional (2D) nanomaterials such as graphene, MXenes, and layered MOFs have attracted intensive attention as cathode hosts for RT Na–S and Na–Se batteries [66,67,68]. The 2D materials are generally assembled into porous structures to increase the electronic contact with sulfur and sodium sulfide while simultaneously providing physical and chemical confinement to Na polysulfides and polyselenides [69]. For instance, our group reported a wrinkled S-doped MXene (S-Ti3C2Tx) as the sulfur host and achieved high areal sulfur loading up to 4.5 mg cm−2 for RT Na–S batteries [70]. S-Ti3C2Tx improved the electronic conductivity of the sulfur species and effectively restricted the diffusion of sodium polysulfides due to its high polarity. The RT Na–S batteries using S-Ti3C2Tx as the cathode host offered an initial capacity of 822 mAh g−1, which remained at 577 mAh g−1 after 500 cycles at 2 C.
Ye et al. synthesized a 2D Ni-based MOF (Ni-MOF-2D) by exfoliating the bulk precursor and employed it as a sulfur host for RT Na–S batteries [71]. The 2D MOF exhibited strong adsorption and fast conversion kinetics for sodium polysulfides owing to their enhanced interaction. This can be ascribed to the dynamic electron states of Ni centers in the MOF, which cause electron transfer from the cathode to polysulfides during discharge and boost the sodiation process, and vice versa (Fig. 5a). The high redox capability of Ni centers is associated with the charge redistribution and semiconducting property of the 2D structure (Fig. 5b), which was not found in the bulk counterpart. The resultant RT Na–S batteries presented a stable cycling performance with a capacity of 347 mAh g−1 after 1 000 cycles (58% capacity retention). The electrocatalytic activity toward boosting the polysulfide redox reactions was also demonstrated in MoS2 nanosheets, which could be tuned by adjusting the sodiation depth [72].

a Schematic illustration of sodium polysulfide confinement on Ni-MOF-2D. b High-resolution TEM (HRTEM) image of Ni-MOF-2D and corresponding atomic schematic. Reprinted with permission from Ref. [71]. Copyright © 2020, John Wiley & Sons. c Probable chemical structure of copolymer CS90 obtained by reaction of the Ca monomer with sulfur. d TEM image of the CS90-rGO composite. Reprinted with permission from Ref. [75]. Copyright © 2017, American Chemical Society. e Illustration of the synthetic procedures for the composite cathode of Na2S nanospheres embedded in a spongy carbon matrix. f TEM (top) and scanning electron microscopy (SEM) (bottom) images of the composite. Reprinted with permission from Ref. [79]. Copyright © 2019, John Wiley & Sons
3.3 Polymeric Sulfur Composites
Polymeric sulfur composites are advantageous in suppressing polysulfide shuttling via the formation of covalent bonds between sulfur and the polymer. Wang et al. reported the first polymeric sulfur composite, amorphous sulfur NPs embedded in sulfurized polyacrylonitrile (S-PAN), as the cathode for RT Na–S batteries [73]. The composite was prepared by mixing sublimed S with PAN at 300 °C. During this heat treatment, PAN cyclizes into a heterocyclic structure and is dehydrogenated by S, resulting in the formation of S-PAN. The RT Na–S with S-PAN cathode delivered an initial discharge capacity of ~ 655 mAh g−1, which remained at 500 mAh g−1 after 18 cycles at a current density of 0.1 mA cm−2 (based on the mass of the composite). Hwang et al. increased the synthetic temperature to 450 °C to obtain a carbonized-polyacrylonitrile and sulfur composite (S@c-PAN), which improved the electrochemical reaction kinetics due to the good conductivity of carbon and unique 1D porous architecture [74]. Consequently, the RT Na–S with S@c-PAN cathode retained 70% of the capacity after 500 cycles at 1 C. However, the cathode reported by Wang et al. only offered a sulfur ratio of approximately 45 wt%, and that reported by Hwang et al. provided an even lower sulfur loading of approximately 31 wt%, which is a concern in that it limits the energy density of polymeric sulfur composites.
Incorporating sulfur chains into the polymer backbones can prominently improve the sulfur content in polymeric sulfur composites and thus provide a higher capacity in RT Na–S battery systems. Ghosh et al. synthesized a sulfur-rich copolymer with reduced graphene oxide (CS90-rGO) as a cathode, and this type of polymeric material-based cathode offered a high sulfur loading of approximately 90 wt% [75]. CS90 was prepared through thermal ring-opening polymerization of cardanol benzoxazine, which in situ reacted with sulfur (Fig. 5c). The CS90 copolymer enhanced the reversibility of low-order polysulfides due to the plasticizing effects of small organosulfur units formed during deep discharge (the organosulfur units evenly dispersed in the insoluble polysulfides and converted them into a soluble phase). After coupling with high-conductivity rGO (Fig. 5d), the CS90-rGO cathode delivered an initial discharge capacity of 542 mAh g−1, which remained at 335 mAh g−1 after 50 cycles at 200 mA g−1 [75]. Furthermore, small organosulfur molecules can also be applied as cathode materials to enhance the cycling stability of RT Na–S batteries. Wu et al. used the sulfur-carbon complex (SC-BDSA) prepared by using benzenedisulfonic acid (BDSA) and K2SO4 as the raw materials to form a cathode that effectively suppressed the shuttle effect by confining the sulfur in the bridging bonds (C–Sx–C) [76]. The RT Na–S battery using the SC-BDSA cathode provided a high initial discharge capacity of 1 050 mAh g−1, which was maintained at 750 mAh g−1 after 200 cycles at 250 mA g−1.
3.4 Other Sulfur-Based Composites
In addition to using traditional sulfur as an active cathode material, sodium sulfide (Na2S) can also be employed as the starting cathode material for RT Na–S batteries. Na2S in the fully sodiated state shrinks when it is initially desodiated, generating space for subsequent volume expansion during cycling [77]. Therefore, the cathode structural stability is significantly improved compared to using elemental S. However, the insulating property of Na2S still requires the aforementioned functional materials as a conductive support and a polysulfide immobilizer. Fan et al. reported an all-solid-state RT Na–S battery by using cast-annealed Na2S–Na3PS4–CMK-3 mesoporous carbon as the cathode, which reduced the interfacial resistance and improved the electrochemical performance [78]. Furthermore, Wang et al. applied hollow Na2S nanospheres embedded in a polyvinyl pyrrolidone (PVP)-derived carbon matrix to substitute the sulfur cathode (Fig. 5e, f) [79]. The hollow nanoarchitecture of Na2S and its intimate contact with the spongy carbon matrix ensure fast Na+ and electron transport, which improves the reaction kinetics and utilization of active materials. By pairing this material with a tin-based anode material, a Na metal-free RT Na–S battery was achieved with superior rate capability and cyclability.
Additionally, attention should also be paid to several emerging materials that have superior polysulfide capture ability. For instance, Sun’s group first introduced a ferroelectric material (BaTiO3) with spontaneous polarization as a cathode additive to trap polysulfides [21]. Mitra et al. deposited a thin sheath of aluminum oxyhydroxide (AlOOH) onto the surface of a S/carbon composite, and the AlOOH layer efficiently inhibited dissolution of polysulfides due to the structural encapsulation and chemical immobilization via Lewis acid–base interactions (AlOOH is a Lewis acid, and sodium polysulfides are considered to be Lewis bases) [80].
Overall, a great range of sulfur host materials has been reported thus far to tackle the challenges in developing RT Na–S batteries. A desirable cathode material should have the following features: (1) a hierarchical porous architecture to accommodate the volume change of S during discharge/charge and physically confine polysulfide dissolution; (2) good conductivity to enable electron percolation to the active S and insoluble low-order sodium sulfides; (3) high polarity or chemical adsorption capability to immobilize polysulfides; and (4) excellent electrocatalytic behavior to boost the conversion reactions from long-chain polysulfides to Na2S, as well as the desodiation process from Na2S to S molecules. Note that thoroughly addressing all the concerns with one type of material is difficult, and one promising solution is to synergize the merits of different components in the composite cathode and the efforts on electrolytes and metal anodes [81]. The cathode design for Na–Se batteries follows very similar strategies, which will not be separately summarized due to the small number of publications in the field.
4 Electrolyte Engineering for RT Na–S and Na–Se Batteries
Although the research on electrolytes for RT Na–S and Na–Se batteries is still at its beginning, understanding the contribution of electrolytes to the electrochemical performance of batteries is essential. As an electron-insulator buffer between the anode and the cathode, the electrolyte is in contact with all the battery components. Ideally, electrolytes should be electrochemically inert to the electrodes, current collectors, and separators, serving only as a medium for ionic mobility. Practically, during the first cycle, the electrolyte decomposes at the surface of the electrodes and forms a passivation layer called the SEI, which prohibits continuous electrolyte decomposition during cycling. The electrolyte should be thermally and electrochemically stable to ensure battery operation over the wide temperature and voltage windows, dictated by the electrode couples. Finally, the cost and environmental impact should be minimized by selecting inexpensive, abundant and eco-friendly chemicals [82]. RT Na–S and Na–Se batteries, as mentioned earlier, still face many challenges, such as polysulfide/polyselenide dissolution and Na dendrite growth. Electrolyte optimization can contribute to overcoming these challenges. Many approaches have been explored to understand and improve the electrolyte composition, such as tuning the composition of liquid electrolytes (e.g., cosolvent, additives, ionic liquids), gelling electrolytes to form quasi-solid-state electrolytes, and developing solid-state electrolytes.
4.1 Liquid Electrolyte Optimization for Sulfur Cathodes
Generally, batteries employ liquid electrolytes owing to multiple advantages, such as high ionic conductivity, ease of synthesis, and availability of chemicals. RT Na–S and Na–Se liquid electrolytes consist of one or several Na salts dissolved in a solvent. Additives and cosolvents are frequently added to the electrolyte to enhance the performance of batteries. In the following section, we categorize the latest advancements in liquid electrolytes applied in RT Na–S and Na–Se batteries according to the nature of the solvent, such as carbonate-based electrolytes, ether-based electrolytes, catholytes, ionic liquids and inorganic liquid electrolytes.
4.1.1 Carbonate-Based Electrolytes
Carbonate solvents, including linear carbonates (e.g., diethyl carbonate (DEC), dimethyl carbonate (DMC), ethyl methyl carbonate (EMC)) and cyclic carbonates (e.g., ethylene carbonate (EC), propylene carbonate (PC)), have been primarily employed in Na-based batteries owing to their high electrochemical stability and good solvation properties toward Na salts. Investigation of the interactions between Na+ cations and carbonate solvents demonstrated that the sodium ion pair dissociation energy is smaller than that of the Li counterpart with the same solvent due to the weaker Lewis acidity of Na+ [83]. Therefore, the knowledge acquired on Li SEI formation is not directly transferable to Na-based batteries [84]. Furthermore, a Na+ ion possesses a larger ionic radius (< 30%) and a larger coordination shell than a Li+ ion. Thus, the electrolyte properties, such as viscosity and ionic conductivity, are also different between Li-based carbonate electrolytes and Na-based carbonate electrolytes [85]. Ponrouch et al. explored the properties of variable electrolyte composition with different Na salts (e.g., NaClO4, NaPF6 and sodium bis(trifluoromethylsulfonyl)imide (NaTFSI)) dissolved in several common solvents (e.g., PC, EC, DMC, dimethoxymethane (DME), DEC, tetrahydrofuran (THF) and triglyme) or bisolvent mixtures (e.g., EC:DMC, EC:DME, EC:PC and EC:triglyme) [86]. They discovered that in terms of the electrolyte performance, i.e., ionic conductivity, viscosity and thermal stability, the choice of Na salt had a small influence in comparison with the solvent. Based on a 1 M (1 M = 1 mol L−1) NaClO4 sodium salt concentration, the ionic conductivity of the as-prepared electrolytes followed the order of DMC < triglyme < EC:DEC < PC < EC:triglyme < EC:PC < EC:DMC < EC:DME, while the viscosity order was DMC < EC:DME < EC:DMC < EC:DEC < EC:triglyme < EC:PC < triglyme < PC. Furthermore, the EC:PC and EC:DMC mixture demonstrated an electrochemical stability window of ~ 5 V, comparable to that of the single DEC or PC solvent. This result illustrates the synergistic effect of solvents in enhancing the electrolyte properties.
Wang et al. prepared a “cocktail optimized” electrolyte for RT Na–S batteries by introducing an InI3 additive into a concentrated 2 M NaTFSI in PC:fluoroethylene carbonate (FEC) electrolyte [44]. As schematically illustrated in Fig. 6a, on the cathode side, the dissolution of polysulfides is inhibited by the synergistic effects of the high-concentration Na salt and FEC-rich solvent. In addition, during the charging process, I− from the InI3 additive is oxidized to I3− and subsequently reacts with the irreversible Na2S end-product to form Na polysulfides, thereby avoiding Na2S deposition on the cathode. On the anode side, increasing salt concentration contributes to decreasing free solvent molecules in the electrolyte. Therefore, the decomposition of the electrolyte at the surface of the electrodes is diminished. Figure 6b visually reveals the absence of polysulfides in the “optimized cocktail” electrolyte (#5) through the conservation of the yellow color from InI3 after aging for 36 h. However, FEC has also been observed to have a positive effect on inhibiting polysulfide dissolution.

a Schematic illustration of the operation of RT Na–S batteries with (left) a 1 M (1 M = 1 mol L−1) NaTFSI in a PC electrolyte and (right) a 2 M NaTFSI in PC:FEC (with a volumetric ratio of 1:1) with a 10 mM InI3 additive electrolyte. b Visual observation of Na polysulfide formation in different electrolytes (1#—1 M NaTFSI in PC; 2#—1 M NaTFSI in PC:FEC (with a volumetric ratio of 1:1); 3#—2 M NaTFSI in PC; 4#—2 M NaTFSI in PC:FEC (with a volumetric ratio of 1:1); 5#—2 M NaTFSI in PC:FEC (with a volumetric ratio of 1:1) with 10 mM InI3) with the aging time at 60 °C. Reprinted with permission from Ref. [44]. Copyright © 2018, Nature Publishing Group. c Schematic illustration of the in situ electrochemical treatment (in situ ET) method. The inserted TEM image shows the in situ ET cathode composite with an FEC-containing electrolyte. Reprinted with permission from Ref. [88]. Copyright © 2018, John Wiley & Sons. d Combustion tests of 2 M NaTFSI in EC:DEC (with a volumetric ratio of 1:1) and 2 M NaTFSI in TMP:FEC (with a volumetric ratio of 7:3) electrolytes. Reprinted with permission from Ref. [90]. Copyright © 2019, Elsevier
The two electrolyte samples (#2 and #4) containing FEC remained mostly transparent. This observation has also been proven by Zhao et al., who employed a 1 M NaClO4 in EC:PC with a 5 wt% FEC electrolyte [87]. They discovered that the FEC additive had a positive impact on the cycling performance of the full Na–S battery by creating a strong SEI and allowing an easier transformation of S into Na2S. The analogous RT Na–Se batteries demonstrated a similar mechanism. By introducing 3 wt% FEC additive into the electrolyte, the anodic corrosion due to polyselenides was diminished [22]. FEC not only protects the Na anode by creating a strong SEI layer but also prevents polysulfide/polyselenide dissolution in the electrolyte. Lee et al. protected the cathode electrode by exploring an “in situ electrochemical treatment” (in situ ET) method to form an SEI layer on the sulfur-carbon cathode [88]. The in situ ET consisted of a galvanostatic and potentiostatic discharge followed by a galvanostatic and potentiostatic charge of the RT Na–S batteries. Figure 6c shows the mechanism of the in situ formation of a protective layer on the cathode electrode. When 8 wt% FEC is introduced into a 1.5 M NaClO4 in PC electrolyte, the pretreated cathode displays an SEI layer of approximately 20 nm (SEM images in Fig. 6c). During the in situ ET discharge, the SEI is formed on the cathode by reduction of the FEC-based electrolyte and is maintained after the in situ ET charge process. The in situ ET method and FEC additive benefit the full cell, with a capacity increase of about 120% (from 270 to 590 mAh g−1 at the 50th cycle) and a high Coulombic efficiency of 99.5% over 200 cycles at 2 C, confirming the efficiency of this new in situ ET method.
Carbonate-based electrolytes, although widely employed in batteries, are highly flammable. Therefore, nonflammable or fire-retardant additives have been introduced into carbonate-based electrolytes to reduce the ignition risk. Phosphate-based species demonstrate good fire-retardant properties owing to their hydrogen radical scavenging ability [11]. Trimethyl phosphate (TMP) was introduced into a 1.0 M NaClO4 in the EC/PC electrolyte to address the safety concerns of carbonate-based electrolytes in RT Na–S batteries [89]. When 15 wt% TMP was introduced into the electrolyte, the latter exhibited nonflammability, good thermal stability and electrochemical compatibility, allowing RT Na–S batteries to achieve stable capacities of 441 and 177 mAh g−1 for 200 cycles at rates of 1 C and 5 C, respectively. However, with 25 wt% TMP added to the electrolyte, the specific capacity of the batteries significantly declined due to an unstable SEI. An excessive amount of TMP leads to poorer cycling performance due to the lack of compatibility of phosphate compounds with electrodes [89]. Wu et al. prepared a nonflammable electrolyte based on 2 M NaTFSI dissolved in a mixture of TMP/EC (with a volumetric ratio of 7:3) solvents [90]. As shown in Fig. 6d, the electrolyte containing TMP can extinguish the fire, whereas a common carbonate-based electrolyte, such as 2 M NaTFSI in EC/DEC, burns until all the combustible is consumed. The RT Na–S battery with the electrolyte of 2 M NaTFSI in TMP/FEC delivered a reversible capacity of 788 mAh g−1 after 300 cycles at 1 C, with a negligible capacity decay below 0.04% per cycle.
4.1.2 Ether-Based Electrolytes
Ether-based electrolytes were overshadowed by the more successful carbonate-based electrolytes in Li-ion batteries for a long time due to their lack of electrochemical stability at high voltage. In the past decade, ether solvents, particularly the glyme family (e.g., monoglyme, diglyme, triglyme, tetraglyme), have been crucial in the development of electrolytes for Li–S batteries owing to their better compatibility with metal anodes (i.e., dendrite growth prevention) and sulfur composite cathodes [91, 92]. Moreover, long-chain ether solvents such as tetraethylene glycol dimethyl ether (TEGDME) are less flammable than carbonate solvents, reducing safety concerns. For instance, an ether-based electrolyte composed of 1 M NaTFSI salt dissolved in TEGDME resisted direct flame exposure for 30 s. The nonflammable electrolyte possesses a high ionic conductivity of 3 × 10−3 S cm−1 at RT, a high Na-ion transference number (t+ = 0.72), and an electrochemical stability window suitable for RT Na–S battery applications [93].
Inspired by the discoveries generated in Li–S batteries, researchers developed ether-based electrolytes for RT Na–S batteries, where ether compounds also contributed to enhancing the stability of sulfur composite cathodes and Na metal anodes [94, 95]. Ether species contain C–O–C groups, while carbonates possess O–C(=O)–O groups; thus, the electrolyte solvation properties will differ, as will the Na–S chemistry mechanism. Furthermore, the sulfur content in the composite cathode should be over 70% to reach a satisfactory energy density [96]. At such high sulfur loading, sulfur molecules inevitably exist on the surface of the composite cathode; thus, side reactions between sulfur molecules and the electrolyte occur to a greater extent, which results in a low specific capacity and capacity decay. When a common composite cathode is used (CNT/S, ~ 70 wt% S), an irreversible solution-phase reaction has been demonstrated to occur in carbonate solvents. However, in ether-based electrolytes, sulfur reduction involves several conversion reaction processes with the formation of polysulfide intermediates, which allows higher electrochemical reversibility [97, 98]. Liu et al. also observed that a high-sulfur-loading cathode performed better in ether-based than carbonate-based electrolytes (Fig. 7a) [99]. In the TEGDME electrolyte, when elemental sulfur is deposited on the surface of the composite cathode (i.e., the cathode with high sulfur loading), a “solid‒liquid” reaction occurs in which elemental sulfur reacts with Na to form liquid polysulfides (Na2Sx). In comparison, in carbonate-based electrolytes, side reactions will occur between the surficial sulfur and the electrolytes, leading to low specific capacity and Coulombic efficiency. When elemental sulfur is confined inside the pores of the composite cathode (i.e., the cathode with low sulfur loading), a “solid‒solid” reaction, in which elemental sulfur is directly transformed into Na2S, occurs in both ether-based and carbonate-based electrolytes (Fig. 7b). However, the soluble liquid polysulfides formed in ether electrolytes contribute to the shuttle effect, which leads to side reactions with the Na metal anode. To address this issue, additives can be introduced into the electrolytes to prevent the dissolution or shuttling of the liquid polysulfides.

a Cycling performance of the cathode with a high sulfur loading at 0.1 A g−1 based on the mass of sulfur. b Schematic illustrations of the mechanisms in ether and carbonate electrolytes for the cathode with a high sulfur loading (sulfur on the surface) and the cathode with a low sulfur loading (sulfur in the pores). c Cycling performance of the cathode with a low sulfur loading in a 1.0 M NaClO4 and 0.2 M NaNO3 in a TEGDME electrolyte with or without a 5 wt% FEC additive at 1.0 A g−1. d Cycling performance of the cathode with a high sulfur loading in TEGDME with or without a NaNO3 additive based on the mass of sulfur. Reprinted with permission from Ref. [99]. Copyright © 2021, Springer. e Photo of undissolved Na2S and P2S5 as well as the Na2S–P2S5 complex. f Raman analysis of prepassivated Na (surface), the Na2S–P2S5 complex, Na2S powder, P2S5 powder, and the blank solvent. Reprinted with permission from Ref. [79]. Copyright © 2018, John Wiley & Sons. g Proposed impact of activated carbon cloth (ACC) on the reaction mechanism during discharge of Na2S6. h Reaction mechanism describing the catalytic effect of ACC in the Na2S6@ACC cathode. i Second cycle discharge profiles of the Na2S6 catholyte-impregnated ACC cathode (blue) and Na2S6 catholyte-impregnated pristine carbon cloth cathode (red) at 0.2 C. j Long-term cycling of the Na2S6 catholyte-impregnated ACC cathode at 0.5 C. Reprinted with permission from Ref. [107]. Copyright © 2020, American Chemical Society
Similar to additives in carbonate electrolytes, FEC and NaNO3 additives demonstrate a positive effect in TEGDME-based electrolytes (Fig. 7c, d). FEC effectively protects the Na anode by creating a F-rich SEI layer on the anode, while the NaNO3 additive limits polysulfide dissolution and contributes to the formation of a Na–O-containing SEI on the Na anode, which further inhibits Na2S deposition on the anode [99]. Another approach consists of introducing soluble polysulfidophosphate complexes (Na2S/P2S5, with a molar ratio of 1:1) into ether-based electrolytes, which enables higher Coulombic efficiency owing to stronger phosphate-containing anode protection [100]. P2S5 species have been introduced as an additive into Li–S and RT Na–S batteries owing to their ability to facilitate the dissolution of the insoluble lithium Li2S or sodium Na2S by forming Li2S/P2S5 or Na2S/P2S5 complexes, respectively [101]. Although Na2S and P2S5 are separately insoluble in diglyme, the complex becomes soluble when they are mixed in a specific ratio, as shown in Fig. 7e. Raman spectroscopy was employed to determine the effect of the Na2S–P2S5 complex on the surface of the Na anode (Fig. 7f). When the Na anode was submerged overnight in a solution of Na2S–P2S5, a chemical passivation layer was observed on the surface of the anode, which further protected the anode. According to Wang et al., the reaction between the Na–P–S complex and Na forms an amorphous Na–P–S protective layer, which may consist of Na3PS4, Na2PS4, and Na4PS6 species [79].
4.1.3 Catholytes
Due to the inevitable polysulfide dissolution in ether-based electrolytes and the low utilization of sulfur in the cathode material, several research groups have focused on developing catholytes for RT Na–S batteries. Catholytes consist of an electrolyte containing the active material, namely, a “liquid-phase cathode”, which enables a homogeneous distribution of sulfur in the form of liquid polysulfides in the positive electrode (e.g., a carbon cloth current collector), thus improving the sulfur utilization. Yu et al. investigated the origin of capacity fading in RT Na–S batteries [102]. By employing a Na2S6 in TEGDME catholyte, the first discharge reaction step corresponding to the transformation of elemental sulfur into long-chain sodium polysulfides is suppressed. As a result, the electrochemical reaction mechanism in the lower-voltage-plateau region corresponding to the transformation of long-chain polysulfides to short-chain polysulfides can be more easily examined. The transformation between elemental sulfur and long-chain polysulfides (Na2Sn, 4 \(\leqslant\) n \(\leqslant\) 8) has been demonstrated to be highly reversible, whereas the transformation from Na2S4 into short-chain polysulfides (Na2Sn, 1 \(\leqslant\) n \(\leqslant\) 4) remains mostly irreversible mainly due to the deposition of the end-product on the anode, resulting in loss of active material [103].
To avoid side reactions between polysulfides and anodes, catholytes are generally employed with ion-selective membranes such as Na-β”-aluminate or Nafion-like polymers as separators [104, 105]. However, such ion-selective membranes are expensive. Kumar et al. employed a Na2S6 catholyte with a carbon cloth current collector decorated with MnO2 nanoarrays [106]. This method enabled the reduction of shuttling of polysulfides owing to polysulfide adsorption on MnO2. The Na|Na2S6 catholyte|MnO2-carbon cloth battery displayed an initial energy density of 946 Wh kg–1 and an energy density after 500 cycles of 728 Wh kg–1. More recently, the same group employed an activated carbon cloth current collector combined with a Na2S6 in TEGDME catholyte and a Na metal anode [107]. They claimed that the activated carbon cloth serves as a catalyst to stabilize free-radical species by enhancing the kinetics of conversion of long-chain polysulfides into small-chain polysulfides (Na2S2) through a two-step mechanism (Fig. 7g, h). This was further confirmed by the appearance of three plateaus on the discharge curve profile of the Na|Na2S6 catholyte|activated carbon cloth battery, whereas the discharge curve profile of the Na|Na2S6 catholyte|pristine carbon cloth battery displayed the usual two plateaus corresponding to the two reaction steps of the regular Na–S chemistry (Fig. 7i). The catholyte combined with the effective catalysis of the activated carbon cloth permitted achievement of long cycling stability with low specific capacity fading (Fig. 7j).
Ether compounds are preferentially used as solvents to synthesize catholytes owing to the facile solubility of long-chain polysulfides. However, Medenbach et al. innovated this approach by employing tetramethylurea as a solvent with a mixture of 0.5 M NaOTf and 1 mM Na2S5 sodium salts and a P2S5 additive [104]. This novel catholyte could achieve a specific capacity of approximately 800 mAh gS−1 over 30 cycles at 0.1 C, without any notable fading. Only a limited number of research articles have been published about catholyte electrolytes for Na–S or Na–Se batteries. Additional investigations on the catholyte composition, such as studying other solvents, cosolvents and additives, could significantly improve the battery cycling performance by achieving better protection of the Na anode to suppress side reactions between short-chain polysulfide species and the Na anode.
4.1.4 Ionic Liquids
Ionic liquids (ILs) are molten salts with a melting point below 100 °C. Recently, they have engendered enormous research owing to their advantageous solvent properties, such as ultralow volatility, nontoxicity, thermal and electrochemical stability, and intrinsic high ionic conductivity. In addition, ILs that are composed of only cationic and anionic species (i.e., no solvent) can be tailored to meet specific requirements [108]. Among the many ILs that have been discovered, only a restricted number can be applied as electrolytes for battery applications [109]. Typically, ILs employed as electrolytes in batteries are made of bulky ions such as imidazolium, pyrrolidinium or phosphonium cations and fluorinated anions such as TFSI−, PF6−, and FSI− [110]. ILs have been used as solvents, cosolvents or additives in Na-ion and Li-based batteries; however, fewer than a handful of studies have been reported for Na–S/Se batteries. An IL based on NaTFSI dissolved in N,N-diethyl-N-methyl-N-(2-methoxyethyl) ammonium bis(trifluoromethanesulfonyl)amide (DEME TFSA) was employed in RT Na–S batteries, which achieved an initial capacity of 700 mAh g−1 with Coulombic efficiency > 95% [111]. Although the as-prepared IL demonstrated low polysulfide dissolution, capacity fading was observed during cycling, which has been attributed to the degradation of the cathode material resulting from the repetitive volume change arising from elemental sulfur conversion into Na2S.
More recently, Wei et al. introduced a 1-methyl-3-propylimidazolium-chlorate IL tethered to SiO2 NPs into a 1 M NaClO4 in an EC/PC liquid carbonate electrolyte [112]. The combination of the IL and SiO2 NPs stabilized the Na electrodeposition on the Na anode, thus allowing smooth Na plating/stripping processes. Furthermore, the solubility of polysulfides was extremely reduced. The introduction of an IL/SiO2 additive was clearly beneficial in terms of the specific capacity and Coulombic efficiency. Interestingly, when the percentage of IL/SiO2 additive was increased from 5 wt% to 10 wt%, the battery delivered a smaller specific capacity due to the increase in viscosity. Very recently, Wang et al. introduced an IL into a β″-alumina solid electrolyte [113]. The IL provided high ionic conductivity, thermal and chemical stability and a wide electrochemical stability window. The dual IL‒solid electrolyte enabled Na–S battery operation at an IMT of 150 °C.
4.1.5 Liquid Inorganic Electrolytes
Inorganic electrolytes generally consist of liquid inorganic solvents (e.g., ammonia, SO2) in which Na salts are dissolved at high concentrations. This family of electrolytes is close to ILs. An ammonia solvate benefits high Na salt concentration (< 7 M), high ionic conductivity (~ 100 × 10−3 S cm−1) and safety (low flammability) [114]. Ruiz-Martínez et al. reported three ammoniate-based electrolytes for Na metal batteries, including NaI·3.3NH3, NaBH4·1.5NH3 and NaBF4·2.5NH3 [115]. The ionic conductivities of these electrolytes followed the trend in the sodium concentrations (i.e., NaBH4·1.5NH3 > NaBF4·2.5NH3 > NaI·3.3NH3). Moreover, NaBH4 and NaBF4 are less expensive than NaI salts. Regarding the cycling performance, the Na||Na symmetric cells are promising, especially that with NaBF4·2.5NH3, as the voltage hysteresis remains lower than that of the two other electrolytes. However, to further attest to the cycling performance of these electrolytes, the full cell configuration has yet to be tested. Another liquid inorganic electrolyte, more precisely a catholyte, based on SO2-solvated chloroaluminate (NaAlCl4·2SO2) was recently proposed and applied in RT Na–SO2 batteries [116].
4.2 Liquid Electrolyte Engineering for Na Metal Anodes
The quest to find a suitable liquid electrolyte is essential for the proper operation of Na metal anodes since the electrolyte is in direct contact with the anodes and regulates the reactions occurring at the anode/electrolyte interface. Extensive efforts in the research community have been devoted to optimizing the composition of liquid electrolytes. The nature of the solvents (e.g., carbonate, ether, IL), salts (e.g., organic, inorganic) and additives (e.g., contributing to SEI layer formation, dendrite suppression, fire extinguishment) employed in electrolytes is acknowledged to have significant impacts on the performance of Na metal batteries.
4.2.1 Solvent Selection
Based on the knowledge acquired on electrolytes applied in Li metal anodes, carbonate electrolytes have been investigated in Na metal anodes. However, several researchers have proven that Na anodes generally present poor electrochemical performance in carbonate-based electrolytes [117, 118]. This observation could result from the unstable SEI layer formed on Na metal anodes; however, notably, the stability of Na metal anodes in carbonate electrolytes can be improved by introducing electrolyte additives, which will be discussed in the next section. Recent studies demonstrated that vigorous decomposition of the electrolyte at the electrode surface could produce gases, which disturbed the formation of a uniform SEI. Chen et al. investigated the impact of ion–solvent complexes on the gas evolution in carbonate electrolytes [119]. As observed with in situ optical microscopy (Fig. 8a, b), the gas evolution is approximately tenfold higher in a 1.0 M NaClO4 in a PC electrolyte (denoted as the Na+-PC solution, Fig. 8b) than that in pure PC solvent (Fig. 8a), attesting to more violent decomposition of the electrolyte, which can only be ascribed to the presence of Na+-PC complexes. This phenomenon was explained with frontier molecular orbital theory analysis by comparing the lowest unoccupied molecular orbital (LUMO) energy levels of PC as a single molecule, PC as a solvent (PC + Sol), the Na atom-PC complex ([NaPC]) and the Na+-PC complex ([NaPC]+).

a, b In situ optical microscopy images of the gas evolution on Na in pure PC solvent and the Na+-PC solution using frontier molecular orbital theory analysis. c Frontier molecular orbital levels of PC (PC as a single molecule), PC + Sol (PC as a solvent), [NaPC] (Na atom-PC complex) and [NaPC]+ (Na+-PC complex). The red and green regions represent the positive and negative parts of the LUMO and highest occupied molecular orbital (HOMO) wave functions, respectively (isovalue: 0.02). The hydrogen, lithium, carbon, and oxygen atoms are marked with white, purple, gray, and red, respectively. d and e Schematics of the orbital hybridization between PC and a Na+ ion or a Na atom. Reprinted with permission from Ref. [119]. Copyright © 2017, John Wiley & Sons. f Coulombic efficiencies of Na plating and stripping using 1 M NaPF6 in various electrolyte solvents (the upper panel) and 1 M of various Na salts in diglyme (the lower panel). Reprinted with permission from Ref. [123]. Copyright © 2015, American Chemical Society. h Total gas amount generated in different electrolytes during cycling in Na|Na cells. The gas amount was calculated from the first three plating/stripping cycles and is normalized with respect to the surface area of the copper mesh on which Na was plated. Reprinted with permission from Ref. [124]. Copyright © 2019, American Chemical Society
As seen in Fig. 8c, when a PC molecule forms a complex with a Na atom, the LUMO energy level rises; however, when a PC molecule complexes with a Na+ ion, the LUMO energy level sharply decreases. This explains the poorer stability of ion complexes against Na metal than that of the pure solvent. The difference in energy levels was further explained with schematics of the orbital hybridization within Na+-PC complexes (Fig. 8d) and Na atom-PC complexes (Fig. 8e). However, the impact of the anions was not taken into consideration. Other publications believe that the stability of the Na metal anode also depends on the anions present in the electrolyte since metal corrosion is generally caused by the anions [120].
Similar to Li metal anodes, ether-based electrolytes seem to be preferential for Na metal stabilization. Na+ ion solvation was studied in common carbonate solvents (e.g., PC, DEC, DMC and EMC) and ether solvents (e.g., 1,2-dioxolane (DOL), THF and DME) by DFT, which established that Na+ cations are more easily solvated by carbonate solvents than by ether solvents [121]. Although ether solvents are usually less polar and have a lower dielectric constant, when combined with Na salts, the ionic conductivity and viscosity of such electrolytes remain satisfactory for battery applications [122]. Wei Seh et al. investigated the reversibility of Na plating and stripping processes in both carbonate- and ether-based electrolytes [123]. Two carbonate-based electrolytes (1 M NaPF6 in EC:DEC (with a volumetric ratio of 1:1) and 1 M NaPF6 in EC:DMC (with a volumetric ratio of 1:1)) and three ether-based electrolytes (1 M NaPF6 in diglyme, 1 M NaPF6 in monoglyme and 1 M NaPF6 in tetraglyme) were synthesized. The average Coulombic efficiency of Na metal plating and stripping after 300 cycles was approximately 99.9% in the ether-based electrolytes, whereas the cells with carbonate-based electrolytes could not surpass 20% Coulombic efficiency for less than 25 cycles. This confirms the excellent stability of ether-based electrolytes when in contact with Na metal anodes (Fig. 8f).
Furthermore, these authors also studied the influence of the nature of Na salts on the Na metal anode in ether solvents. According to their report, the Na salt stability against Na anodes can be ordered as NaPF6 > NaSO3CF3 (NaOTF) > NaN(SO2F)2 (NaFSI) > NaClO4 > NaN(SO2CF3)2 (NaTFSI) (Fig. 8g). The high stability of the Na anode in the NaPF6 in glyme electrolytes arose from the formation of a stable SEI layer consisting of Na2O and NaF species. Later, Goktas et al. confirmed that NaPF6 and NaOTf (NaSO3CF3) present better compatibility with Na metal anodes in diglyme electrolytes by studying the gas evolution during cycling in Na||Na symmetric cells (Fig. 8h) [124]. As previously mentioned, the lower the gas release is, the higher the stability of the electrolyte during Na plating and stripping processes. Apart from NaPF6 and NaOTf salts, NaBF4 in an ether electrolyte also allowed reversible Na plating and stripping cycles owing to the formation of a dense and amorphous SEI layer on the Na metal anode [125]. Wang et al. further explored the effect of NaBF4 in different glyme electrolytes (e.g., diglyme, triglyme and tetraglyme) [126]. When diglyme was employed as the solvent, the electrolyte displayed superior properties, such as higher ionic conductivity, lower interfacial resistance, and thinner SEI containing more B-O and NaF species, to the longer-chain glyme electrolytes (e.g., triglyme or tetraglyme).
To better understand the impact of Na salts and solvents on the plating and stripping processes of Na metal anodes, Zhou et al. extensively investigated the Na+ solvation structure, with a focus on the nature and location of the anions [120]. Similar to previous results, they confirmed that both the solvents and Na salts of the electrolytes dictated the reversibility of Na plating and stripping. Furthermore, they found that the location of the anion in the solvation shell played a particular role due to the difference in the binding energy of different Na+-solvent interactions, as schematically illustrated in Fig. 9. Two aspects are essential to the bonding interaction: the first is the degree of freedom of the anion (i.e., low steric hindrance), and the second is the strength of the Na+-solvent and Na+-anion interactions. For instance, ClO4− with a high degree of freedom and CF3SO3− with a strong interaction with Na+ ions can easily migrate close to the Na metal anode regardless of the nature of the solvent, thereby causing side reactions (Fig. 9a–c). In contrast, PF6− stays away from the Na metal anode when DME is employed as the solvent owing to the strong bonding interaction between DME molecules and Na+ ions (Fig. 9d). However, the bonding interactions between EC, DEC and PC solvents and Na+ ions are not strong enough to prevent PF6− from contacting the Na metal anode, resulting in low Coulombic efficiency (Fig. 9e, f).

Anionic interfacial model describing the interface between electrolytes and sodium metal anodes. Model of a DME-based electrolyte using different metal salts, including a 1.0 M NaClO4, c 1.0 M NaCF3SO3, and d 1.0 M NaPF6. b Model of a 1.0 M NaClO4 in a PC electrolyte. e Model of a 1.0 M NaPF6 in an EC/DEC electrolyte. f Model of a 1.0 M NaPF6 in a PC electrolyte. Reprinted with permission from Ref. [120]. Copyright © 2020, American Chemical Society
4.2.2 Salt Concentration Optimization
As discussed above, the solvation structure is one of the main driving factors for high-efficiency Na plating and stripping. Increasing the salt concentration in the electrolyte dramatically changes the solvation shell of the cation owing to fewer free solvent molecules in the solution, which results in many specific electrochemical and physical properties. Several studies have investigated the solvation structure of NaFSI dissolved in several glymes (e.g., monoglyme, diglyme and triglyme) at different concentrations [127, 128]. As shown in Fig. 10a, in the dilute electrolyte, NaFSI is totally dissociated into the solvent, forming solvent-separated ion pairs (SSIPs). As the concentration of NaFSI increases, contact ion pairs (CIPs) and aggregates (AGGs) are formed due to the lack of free solvent molecules in the solution. Lee et al. further applied a highly concentrated ether-based electrolyte of 5 M NaFSI in DME in Na metal batteries [129]. Similar to previous reports, the increase in the NaFSI concentration in DME gave rise to strong interactions between Na+ ions and DME solvent, as confirmed by the appearance of a new peak at 1 085 cm−1 in the Fourier transform infrared (FTIR) spectrum (Fig. 10b). The morphologies of Na deposited on Cu substrates in the electrolytes of 1 M NaPF6 in EC/PC, 1 M NaFSI in DME, and ultraconcentrated 5 M NaFSI in DME were investigated by SEM (Fig. 10c). When the highly concentrated electrolyte (5 M NaFSI in DME) was employed, the Na metal deposition layer was much smoother than that obtained in the dilute electrolytes, attesting to excellent plating/stripping processes. The superior performance of the Na anode in the highly concentrated NaFSI/DME electrolyte was explained by the stable passivation layer generated on the surface of the Na anode, which suppressed electrolyte decomposition [130].

a Schematic representation of the three main solvated species in NaFSI-triglyme binary mixtures: solvent-separated ion pairs (SSIPs) (left), contact ion pairs (CIP I and CIP II) (middle), and aggregate structures (AGG I and AGG II) (right). Reprinted with permission from Ref. [127]. Copyright © 2018, American Chemical Society. b FTIR spectra of xM NaFSI-DME (x = 0, 1, 2, 3, 4, or 5) and pure NaFSI salt. c Photographs of a pristine Na metal electrode and Cu substrates after the initial Na plating at a rate of C/10. The utilized capacity of the Na metal electrode was 5.0 mAh. Reprinted with permission from Ref. [129]. Copyright © 2017, American Chemical Society.
Although increasing the salt concentration is beneficial for stabilizing Na metal anodes, it also results in high viscosity and low ionic conductivity, which causes large polarization and limited performance in high current rate cycling. Interestingly, the ionic conductivity has been reported to remain high in highly concentrated NaFSI/glyme electrolytes owing to two different charge transport mechanisms (Fig. 11a) [131]. The first mechanism is mobility through diffusion of free ions, which is found in electrolytes with long-chain glymes (e.g., triglyme and tetraglyme). The second mechanism is based on FSI− hopping through making and breaking of ion pairs and aggregates, which is observed in short-chain glymes (e.g., monoglyme and diglyme). Molecular dynamics simulations further confirmed the proposed diffusion and hopping mechanisms. In general, the ionic conductivity and viscosity are intimately linked. Rheology measurements were performed, which revealed that the viscosity of all four NaFSI/glyme electrolytes linearly increased with the salt concentration (Fig. 11b). Thus, the unusual ionic conductivity behavior of highly concentrated electrolytes cannot be only caused by the viscosity properties, which further confirms two different mechanisms related to the chain length of glyme solvents.

a Representation of hopping and shuttling molecular mechanisms. b Conductivity (the upper panel) and viscosity (the lower panel) as a function of the molality of sodium triflate in different glymes: monoglyme (black), diglyme (red), triglyme (green), and tetraglyme (blue). Reprinted with permission from Ref. [131]. Copyright © 2018, American Chemical Society. c Schematic illustration of dilution from a highly concentrated electrolyte (HCE) to a localized highly concentrated electrolyte (LHCE). d Coulombic efficiency of Na metal plating/stripping on Cu current collectors at 1 mA cm−2 after two formation cycles at 0.2 mA cm−2 with an areal capacity of 1 mAh cm−2. e Atomic ratios of C/N/O/F/Na/S elements detected at the surface (0 nm, the upper panel) and 10 nm depth (the lower panel) of the SEI layer after the 10th stripping of Na∥Cu cells using a dilute electrolyte (1.7 M NaFSI/DME), an HCE (5.2 M NaFSI/DME), and an LHCE (2.1 M NaFSI/DME-BTFE (1:2)). Reprinted with permission from Ref. [132]. Copyright © 2018, American Chemical Society
Highly concentrated electrolytes provide wide electrochemical windows, high thermal resistance and low volatility. However, they usually suffer from high viscosity, low ionic conductivity and high cost. Recently, a new type of electrolyte, namely, localized highly concentrated electrolytes, has been developed for Li metal anodes to resolve the abovementioned challenges. These electrolytes consist of highly concentrated electrolytes and diluents. The diluent is miscible with the solvent but interacts very weakly with the metal salt (Fig. 11c); therefore, the localized highly concentrated electrolyte preserves the same Na+ ion solvation structure as the corresponding highly concentrated electrolyte. Meanwhile, the addition of the diluent improves the ionic conductivity and reduces the viscosity and cost of the electrolyte. Zheng et al. introduced bis(2,2,2-trifluoroethyl) ether (BTFE) into a highly concentrated electrolyte of NaFSI in DME [132]. They demonstrated that the as-prepared electrolyte displayed higher ionic conductivity and lower viscosity than its highly concentrated counterpart. Furthermore, similar Coulombic efficiencies of the Na plating and stripping processes on Cu substrates were obtained in localized highly concentrated electrolytes and highly concentrated electrolytes (Fig. 11d). As seen from the X-ray photoelectron spectroscopy (XPS) depth profiling in Fig. 11e, f, the atomic ratios of the SEIs formed on Na metal anodes in highly concentrated electrolytes and localized highly concentrated electrolytes are similar, which proves that the diluent has a negligible impact on the solvation shell of Na+. In contrast, the SEI formed in the dilute electrolyte contains fewer fluoride species and more oxygen species, confirming a more fragile SEI. Later, another group reported hydrofluoroether (HFE) solvent as a diluent for the electrolytes of sodium bis(trifluoromethanesulfonyl)amide (NaTFSA) in glymes (tetraglyme and pentaglyme) [133]. They discovered that the HFE solvent did not participate in the Na+ ion solvation shell, and therefore, the physical properties of the localized electrolyte could be improved. The key challenge for the development of localized highly concentrated electrolytes is to find an appropriate diluent to alleviate the drawbacks faced by highly concentrated electrolytes. A better understanding of the impact of the diluent on SEI formation is also required.
4.2.3 Developing IL Electrolytes for Na Metal Anodes
The ultradiverse available combination of ILs is particularly interesting for the development of liquid electrolytes for rechargeable batteries. Many ILs are safe (e.g., nonflammable, low volatility, low toxicity) and electrochemically and thermally stable and have recently been applied in Na metal batteries to reduce safety risks. However, ILs suffer from high cost, high viscosity and low ionic conductivity due to the strong interaction between the Na+ ions and the anions, causing the formation of anion aggregates Na[anion]x−y. Tuanan et al. theoretically investigated the Na+ ion transport mechanism of 25 different ILs by molecular dynamics simulations (Fig. 12a) [134]. They demonstrated that the ionic concentration is closely related to the Na+ ion transport behavior. An increase in the salt concentration resulted in an increase in the size of anion aggregates and facilitated the Na+ ion hopping diffusion mechanism, thereby improving the Na+ ion apparent transport number in the system. Furthermore, increasing the concentration of Na salt in ILs allowed more reversible Na metal plating and stripping. This observation was further explored by Forsyth’s group, who intensively investigated the influence of the NaFSI salt concentration in an N-methyl-N-propylpyrrolidinium bis(fluorosulfonyl)imide (C3mpyrFSI) IL electrolyte [135]. They employed radial distribution function analysis to simulate the molecular structure evolution when the NaFSI concentration increases. If the concentration of Na salt becomes higher than that of the IL, then large and interconnected Na+-anion aggregates are formed. Meanwhile, an increased coordination number of the Na+ ion solvation sheath with the FSI anion makes the denticity equal to one. As a result, the Na+ ion diffusion dynamics increase, while the mobility of the anions and cations of the IL decreases (Fig. 12b). When applied with a Na metal anode, the superconcentrated NaFSI/C3mpyrFSI electrolyte (i.e., 50 mol% (mol% means the molar percentage) NaFSI) demonstrated a highly aggregated Nax(FSI)y structure near the anode, which excluded the [C3mpyr]+ cation from the interface [136]. Therefore, such electrolytes could benefit the formation of a stable SEI and enhance the Na metal plating and stripping processes. By applying a high-voltage pretreatment, an increased concentration of aggregates was observed near the anode, thereby achieving better cycling performance. The effect of the Na salt concentration was also reported with other systems, which confirmed the benefit of increasing the salt concentration in ILs [137]. However, increasing the NaFSI salt concentration results in a sharp increase in the cost of the electrolyte. The high cost of ILs can be alleviated by employing the dicyanamide (DCA) family of ILs, such as the methylpropyl pyrrolidinium ([C3mpyr]) DCA IL [138]. Several fluorinated Na salts (NaFSI, NaTFSI and NaFTFSI) or nonfluorinated salts (NaDCA) were introduced at high concentrations into DCA ILs. The battery performance followed the trend of NaTFSI < NaFTFSI < NaDCA < NaFSI owing to the improved SEI formation ability of FSI anions. In addition, several reports demonstrated that less expensive IL electrolytes could be formulated by introducing additives.

a Atomic structures of ILs obtained by DFT calculations. The C, O, H, S, F, and N atoms are indicated by silver, red, white, yellow, gold, and blue spheres, respectively. Reprinted with permission from Ref. [134]. Copyright © 2021, American Chemical Society. b Mean square displacements of N from both the IL cation and anion (the left panel) and the Na+ ion (the right panel) at 393 K for several salt concentrations. Reprinted with permission from Ref. [135]. Copyright © 2018, American Chemical Society. c 23Na MAS-NMR spectra of the SEI products on the surface of Na metal (the left panel), and high-resolution F 1s and S 2p XPS curves of the plated Na surfaces comparing the SEI composition on plated Na electrodes for dry and wet electrolytes after 100 cycles at a current density of 1.0 mA cm–2 and 1 h polarization (the right panel). Reprinted with permission from Ref. [140]. Copyright © 2021, American Chemical Society
In most nonaqueous electrolytes, the presence of water degrades the electrochemical performance of alkali metal batteries. Furthermore, Na metal reacts extremely violently with water. However, several publications affirm the beneficial effect of an extremely small amount of water added into ILs [139,140,141]. Ferdousi et al. investigated the Na metal stability in a 50 mol% NaFSI-[C3mpyrFSI] IL electrolyte with different water concentrations (up to 5 000 μmol mol−1)] [139]. The elemental characterization of the Na metal anode after cycling in these electrolytes demonstrated the formation of an SEI containing NaF, Na2O and FSI-related species. The benefit of scarce water molecules in the electrolyte for SEI formation may be related to additional bond formation between water molecules, FSI anions, and Na+ ions, along with the formation of other inorganic compounds, as demonstrated by magic-angle spinning nuclear magnetic resonance (MAS-NMR) and XPS analysis (Fig. 12c).
Sun et al. developed for the first time a nonflammable chloroaluminate IL-based electrolyte for Na metal batteries (Fig. 13a, b) [142]. They employed an aluminum chloride/1-methyl-3-ethylimidazolium chloride/sodium chloride IL, into which two additives were introduced, namely, ethylaluminum dichloride and 1-ethyl-3-methylimidazolium bis(fluorosulfonyl)imide. Such an electrolyte composition contributes to high ionic conductivity (~ 9.2 × 10−3 S cm−1 at 25 °C) and robust SEI owing to the passivation effect from chloroaluminate molecules and the presence of a small amount of FSI anions. The synergistic effect of chloroaluminate and FSI species enables Na||NVP@rGO and Na||NVPF@rGO to achieve long-term cycling stability even at high current rates (Fig. 13c).

a Schematic illustration of the battery configuration and electrolyte composition of IL electrolytes. b Flammability tests using a buffered IL (the left picture) and conventional electrolytes (1.0 M NaClO4 in EC:DEC with 5 wt% FEC) (the right panel). Scale bars = 1 cm. c Rate performance (at various current densities from 20 to 500 mA g−1) and cycling stability (current density of 150 mA g−1) of a Na|NVP@rGO cell using a buffered IL electrolyte (the left panel), and cycling stability of a Na|NVPF@rGO cell using a buffered IL electrolyte at 300 mA g−1 (the right panel). Reprinted with permission from Ref. [142]. Copyright © 2019, Nature. d Electrochemical performance of Na||Na symmetric cells at various low temperatures with a cycling capacity of 1 mAh cm−2 at 0.5 mA cm−2 (the left panel), and voltage–time profiles of Na||Na symmetric cells with a cycling capacity of 50 mAh cm−2 at 2 mA cm−2 (the right panel). Reprinted with permission from Ref. [144]. Copyright © 2020, John Wiley & Sons
Other approaches consisted of introducing cosolvents into IL-based electrolytes. A carbonate-based cosolvent (i.e., a mixture of EC:PC) was introduced into an C3mpyrTFSI IL containing 1 M NaFSI Na salt [143]. Later, Hu et al. introduced an ether cosolvent, namely, diglyme, into a 1-butyl-3-methylimidazolium tetrafluoroborate IL (vol. ratio 4:1) with 1 M NaPF6 salt [144]. As seen in Fig. 13d, this hybrid electrolyte can stabilize the Na metal anode at a low temperature (i.e., − 40 °C) in lean electrolyte conditions (i.e., 1.0 μL mAh−1), which contributes to achieving high energy density. Furthermore, the ether IL possesses a high ionic conductivity of 42 × 10−3 S cm−1, good desolvation and an SEI formation capability, which allowed stable cycling of Na||NVP batteries at RT and at − 20 °C.
4.2.4 Employing Additives in an Electrolyte for Na Metal Anodes
Additives are chemical compounds that are introduced in a small amount (generally < 10%) into the electrolyte with the purpose of stabilizing the electrode/electrolyte interface through the formation of a robust SEI layer. During the first charge process, additives react and protect the Na anode, thereby avoiding further electrolyte decomposition. In particular, fluorinated additives such as FEC assist in the creation of a strong SEI layer on Li metal anodes. Therefore, many publications have focused on understanding the impact of such additives on Na metal anodes. The morphologies and gas evolution during sodium electrodeposition in Na||Na batteries were investigated with several carbonate-based electrolyte systems (e.g., PC, EC, DEC) [145]. As previously discussed in Sect. 4.2.1, carbonate-based electrolytes generate a large amount of gas with poor Coulombic efficiency of Na plating and stripping. Upon FEC addition, the gassing is reduced. Meanwhile, the cycling performance of the Na||Na symmetric cell is improved, even though the morphology of the SEI remains porous. To maximize the benefits, the concentration of FEC in the electrolyte should be carefully controlled, as a small amount of FEC in PC-based electrolytes improves the Na battery performance. In contrast, an increase in the concentration of the fluorinated additive has an adverse impact [146].
Recently, Han et al. employed sophisticated cryo-TEM analysis to carefully observe the SEI composition and morphology on Na metal anodes in carbonate electrolytes (NaPF6 in EC/DMC) with and without an FEC additive [147]. When FEC is absent, the SEI layer is mainly composed of Na2CO3 and Na3PO4, which cannot efficiently protect the Na metal anode. The electrolyte continuously reacts with the anode upon cycling, as schematically illustrated in Fig. 14a. In contrast, the cryo-TEM images of the anodes retrieved from the cells containing the FEC additive displayed a compact SEI layer composed of a Na3PO4 inner layer and an amorphous NaF-rich outer layer (Fig. 14b). The NaF species in the outer layer of the SEI prevent dissolution of Na3PO4 and other amorphous compounds, avoiding direct contact of fresh Na metal with the electrolytes, which results in effective protection of the Na metal anode. Reducing the solubility of the SEI species in the electrolyte is more important in Na-based batteries than in Li-based batteries. Fluorinated additives, such as FEC or difluoroethylene carbonate (DFEC), were confirmed to be able to similarly limit, to some extent, the dissolution of the SEI layer [148]. Zhu et al. proposed a nonflammable electrolyte for Na metal batteries based on TMP solvent with FEC and 1,3,2-dioxathiolane 2,2-dioxide (DTD) additives (Fig. 14c) [149]. TMP solvent has been previously reported as a nonflammable alternative to flammable carbonate solvents owing to the phosphorous functional groups that can scavenge hydrogens [11]. As explained above, FEC facilitates the creation of a strong SEI layer containing NaF species. Meanwhile, the contribution of DTD to the SEI was identified with S-containing compounds (e.g., Na2S, Na2SO3 and organic S-containing salt), as observed from the XPS depth profiling spectra (Fig. 14d, e).

a Schematic of SEI formation in the FEC-free EC/DMC electrolyte in the 1st cycle and after 10 cycles, and the corresponding cryo-TEM image after 10 cycles. b Schematic of SEI formation in the EC/DMC/FEC electrolyte in the 1st cycle and after 10 cycles, and the corresponding cryo-TEM image after 10 cycles. Reprinted with permission from Ref. [147]. Copyright © 2021, Nature. c Flammability tests for the EC/PC carbonate electrolyte (the red panel) and TMP/FEC/DTD-E electrolyte (the purple panel). High-resolution F 1s, P 2p and S 2p XPS depth profiling of the Na metal anode retrieved from cells after 50 cycles in d TMP/FEC/DTD-E and e TMP/FEC-E electrolytes. Reprinted with permission from Ref. [149]. Copyright © 2021, Elsevier. f Atomic structure and binding energy between the Na+ cation and the SEI components (Na-EDC—sodium ethylene decarbonate; Na-EG—sodium ethylene glycol; Na-DMDT, Na2-DMDT+, Na3-DMDT-CH3—organic sodium sulfide salts from the decomposition of TMTD). g Nyquist plots of the symmetric cells in the TMTD-added electrolyte (the upper panel) and in the blank electrolyte (the lower panel) after 10, 20 and 40 cycles (red, orange and blue lines, respectively). Reprinted with permission from Ref. [151]. Copyright © 2020, American Chemical Society
Although adding FEC into the electrolyte efficiently enhances the performance of Na metal batteries, a higher polarization of the Na metal anode was observed in half-cells, which encouraged the search for other additives [150]. To reduce the interfacial resistance, an organosulfur compound, namely, tetramethylthiuram disulfide (TMTD), was introduced into 1 M NaPF6 in EC:PC [151]. The SEI layer formed in this system rich in organic sulfide salts can efficiently protect the Na anode; meanwhile, the Na+ cation can easily penetrate the SEI layer owing to the lower binding energy between SEI compounds (e.g., sodium dimethyldithiocarbamate (DMDT)) and Na+ (Fig. 14f, g). Wang et al. introduced Na2S6 as an additive into an ether-based electrolyte, which contributed to S-containing SEI layer formation on the Na anode [152]. Interestingly, when the Na2S6-NaNO3 coadditive was mixed with the electrolyte, the Na||Na symmetric battery performance degraded, unlike in the Li metal system.
Another approach of exploring cation additives to stabilize Na metal anodes through electrostatic shielding has been demonstrated to be effective in inhibiting electrolyte decomposition on Na metal anodes. This method has been applied with cations such as Li+ and K+, where the presence of cation additives near the tip of the Na dendrites forces Na+ ions to deposit on a smoother area of the Na anode (Fig. 15a, b) [153, 154]. Recently, Chen et al. reviewed this strategy by proposing three principles for cation additive design [155]. First, the newly formed cation-solvent complex must be stabler than the Na+-solvent complex to suppress electrolyte decomposition. Second, the cation additive should present a lower redox potential than that of Na/Na+ (i.e., avoid reaction with the Na anode). However, the reduction of certain cation additives creates an alloy protective layer on Na metal, which will be discussed in the next paragraph. Third, to ensure suppression of cation additive reduction on the Na metal anode, the cation additives should have a larger binding energy than Na+. As shown in Fig. 15c, d, cation additives with these properties are Li+, K+ and Ca2+, of which the Li+ cation additive is the most promising cation additive.

a Simulated electric field intensity distribution and schematic of Li/Na plating mechanisms. Reprinted with permission from Ref. [153]. Copyright © 2018, Wiley–VCH. b Schematics of the stabilization effects on the Na metal anode of the cation additives in the NaOTf electrolyte without any additives (the upper left panel), with the TFSI− additive (the lower left panel), with the K+ additive (the upper right panel), and with both the K+ and TFSI− additives (the lower right panel). Reprinted with permission from Ref. [154]. Copyright © 2018, Wiley–VCH. c Comparison of the LUMO energy level decreases and binding energy of ion-DME complexes. d Calculated and experimental electrode potentials of the considered metal/cation pairs. Reprinted with permission from Ref. [155]. Copyright © 2020, Elsevier. e Schematics of SEI formation on a Na metal anode cycled in a regular carbonate electrolyte (the upper panel) and in a SnCl2-added electrolyte with the formation of a Na–Sn alloy layer plus a NaCl-rich SEI (the lower panel). f Pictures of Na metal before and after reaction with the SnCl2-added carbonate electrolyte. Cross-sectional SEM and energy-dispersive X-ray spectroscopy (EDX) mapping images of SnCl2-treated Na metal with Na, Sn, and Cl elemental signals. Reprinted with permission from Ref. [156]. Copyright © 2019, American Chemical Society
Na metal anodes can also be protected by introducing reduction/alloy-type additives. The formation of an alloy phase on the surface of the Na metal anode was demonstrated to reduce the Na+ ion diffusion barrier, thereby lowering the interfacial impedance and achieving smoother Na metal plating [156,157,158]. Zheng et al. introduced SnCl2 into an EC/PC carbonate electrolyte. The reduction/alloying compound was chemically reduced on the surface of the Na metal anode, forming an SEI with a Sn-containing inner layer and a NaCl-rich outer layer (Fig. 15e, f) [156].
4.3 Gel Polymer Electrolytes (Quasi-Solid-State Electrolytes)
Gel polymer electrolytes (GPEs), also known as quasi-solid-state electrolytes (QSEs), are a class of electrolytes with an intermediate phase between liquid and solid. With the application of liquid electrolytes as a plasticizer, a polymer matrix as a mechanical scaffold, and sodium salt and additives, GPEs can minimize the safety risks by locking the liquid solvent inside the polymer matrix to form a quasi-solid-state structure and maintain a high ionic conductivity at the same time [159]. Furthermore, GPEs offer several advantages, including a high concentration of charge carriers, superior interfacial contact between the electrolytes and electrodes, free-standing consistency, and good mechanical properties (e.g., strength, flexibility) [160, 161]. Moreover, GPEs have been reported to be beneficial when applied with Na metal anodes in terms of safety assurance owing to their ability to prevent leakage and reduce Na dendrite growth [162].
However, there are also some challenges that need to be solved for the future practical application of GPEs. For instance, to ensure high ionic conductivity, a high liquid electrolyte content is inevitably required, which will reduce the mechanical strength of the GPE and increase the safety risk due to leakage [161]. Some other challenges, including interfacial instability on the cathode side, poor dimensional stability, and poor liquid retention capacity, are also nonnegligible in GPE development [160]. Exploring new preparation methods and optimizing the composition and structures of GPEs have been demonstrated to be promising pathways to overcome the abovementioned challenges.
In 2004, Tian et al. reported a type of gel electrolytes from a copolymer of polyvinylidene fluoride and hexafluoropropylene (PVDF-HFP), which achieved a good sodium ion conductivity of 1 × 10−4 S cm−1 at RT [163]. After that, Ryu et al. applied this type of GPE in Li–S batteries, achieving 72% sulfur utilization with a high capacity of 1 206 mAh g−1 during the first discharge [164]. In 2006, Park et al., for the first time, reported the application of a GPE combined with a sulfur cathode and a Na anode in quasi-solid-state RT Na–S batteries [165]. They used PVDF-HFP comprising a glyme solvent and NaCF3SO3 salt to synthesize a GPE, which offered a good sodium ion conductivity of 5.1 × 10−4 S cm−1 and provided an initial capacity of 489 mAh g−1 at RT. However, significant cycling decay could not be avoided, and only 40 mAh g−1 remained after 20 cycles at 0.144 mAh cm−2 [165]. The potential of using PVDF-HFP-based GPEs in RT Na–S batteries was further demonstrated by Kim et al. [166]. However, there is a suggestion that the nucleophilic polysulfides decompose the polymer via hydrodefluorination.
Later, many improvement strategies were applied to enhance the performance of GPEs: (1) manipulating the morphology of GPEs by introducing fillers, new types of liquid electrolytes, or novel polymer matrices, (2) in situ formation of GPEs to offer higher binding energy, (3) SEI formation optimization by introducing additives into the liquid part of the electrolyte, (4) ion flux regulation by creating ionic channels in the electrolyte, and (5) fabrication of a robust electrolyte with enhanced mechanical properties (i.e., high Young’s modulus) in which dendrite penetration is inhibited.
Kumar et al. optimized the PVDF-HFP GPE by introducing SiO2 NPs (15 wt%) into it, which induced a more porous morphology (Fig. 16a) and thus offered a better capacity to hold the liquid solvent, free-standing consistency and a highly transparent character [167]. Due to the change in morphology, the ionic conductivity of PVDF-HFP-15% SiO2 was also significantly higher than that of conventional PVDF-HFP GPEs (Fig. 16b), which provided a satisfactory electrochemical potential window of ~ 4.0 V (from − 2.0 to 2.0 V). However, the Na–S battery with the PVDF-HFP-15% SiO2 GPE only delivered an initial capacity of ~ 165 mAh g−1 in the first discharge, which remained at ~ 21 mAh g−1 after 8 cycles due to the formation of irreversible short-chain Na polysulfides. Kumar et al. subsequently continuously improved the GPE by introducing an IL (1-ethyl-3-methylimidazolium trifluoromethanesulfonate (EMITf)) with EC-PC as a liquid solvent and NaCF3SO3 as a Na salt [160]. The NaCF3SO3/EC:PC:EMITf/PVDF-HFP gel electrolyte delivered an excellent ionic conductivity of ~ 8.4 × 10−3 S cm−1 at RT and a wide electrochemical stability window (ESW) of ~ 4.6 V (from − 2.3 to 2.3 V). However, this design can only provide a capacity of ~ 267 mAh g−1 in the first discharge, and rapid cycling decay is still observed after the first charge/discharge cycle.

a SEM images of the PVDF-HFP GPE compared with PVDF-HFP with 15% SiO2. Reprinted with permission from Ref. [167]. Copyright © 2011, Elsevier B.V. b Ionic conductivity of the PVDF-HFP GPE with 0%, 15%, and 25% SiO2. Reprinted with permission from Ref. [167]. Copyright © 2011, Elsevier B.V. c SEM image of the porous PAN membrane. Reprinted with permission from Ref. [168]. Copyright © 2018, Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim. d Cycling performance of the Na–S battery with the PAN membrane GPE compared to the Na–S battery with a commercial Whatman™ separator. Reprinted with permission from Ref. [168]. Copyright © 2018, Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim. e Schematic of the in situ formation of the cross-linked (PETEA-THEICTA)-based GPE combined with the poly(S-PETEA)@C cathode for the quasi-solid-state RT Na–S battery. Reprinted with permission from Ref. [176]. Copyright © 2018, Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim. f High binding energy between the GPE components (THEICTA monomer and ester group-rich PETEA) and Na2S6. Reprinted with permission from Ref. [176]. Copyright © 2018, Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim
In addition to using PVDF-HFP as the polymer matrix, other polymer materials, such as polyacrylonitrile (PAN) [168, 169], poly(methyl methacrylate) (PMMA) [170], poly(ethylene glycol) divinyl ether with flame-retardant polysulfonamide (PDE/PSA) [171], poly(vinylene carbonate) and polymeric sodium tartaric acid borate with polyethylene terephthalate (PVCA/PSTB/PET) [172], poly(ethylene glycol) diglycidyl ether with diamino-poly(propylene oxide) (PEGDE/DPPO) [173], poly(vinyl chloride) (PVC) [174], poly(dimethyldiallylammonium) (polyDADMA) [175], and pentaerythritol tetraacrylate-tris[2(acryloyloxy)ethyl] isocyanurate (PETEA-THEICTA) [176], can also be applied to fabricate GPEs for RT Na–S batteries. Lim et al. immersed a PAN electrospun nanofiber membrane (ES-NFM) into poly(ethylene glycol) dimethyl ether (PEGDME) with sodium triflate (NaCF3SO3) salt, forming a GPE for use in the RT Na–S battery system [168]. Due to the high porosity of the nanofiber membrane (Fig. 16c), the PAN membrane GPE provided a good ionic conductivity of ~ 6.0 × 10−4 S cm−1 at 30 °C, and the Na–S battery with the GPE electrolyte delivered a much higher capacity and a lower overvoltage polarization than the Na–S battery using the commercial separator (Fig. 16d).
The previous GPEs for RT Na–S batteries were mostly prepared by using the conventional solution casting method [160, 167]. Recently, Zhou et al. prepared a new type of (PETEA-THEICTA)-based GPE by using an in situ radiation-initiated synthesis route. This remarkable GPE used the PETEA and THEICTA monomers combined with the 2-hydroxy-2-methylpropiophenone (HMPP) photoinitiator dissolved in PC-FEC with 1 M NaTFSI as a precursor solution. The precursor solution was irradiated by ultraviolet (UV) light to trigger C = C bond radical polymerization of the monomers in a glass fiber separator. Subsequently, the (PETEA-THEICTA)-based GPE with a cross-linked structure was formed in situ (Fig. 16e). The formed GPE delivered an excellent ionic conductivity of ~ 3.85 × 10−3 S cm−1 at 25 °C, and it could also strongly suppress dissolution and diffusion of Na polysulfides due to the higher binding energy between the inner components (THEICTA monomer and ester group-rich PETEA) and Na2S6 compared to FEC-Na2S6 (− 1.22 eV) and PC-Na2S6 (− 1.57 eV) (Fig. 16f). By combining this GPE with a novel poly(S-PETEA)@C cathode, the RT Na–S battery system avoided significant capacity fading and offered good cycling stability with a high capacity of 877 mAh g−1 in the first discharge, which remained at 736 mAh g−1 after 100 cycles at 0.1 C [176].
Similar to liquid electrolytes, SEI formation in Na metal battery systems with GPEs can prevent side reactions and dendrite growth. A series of additives have been introduced into the liquid part of GPEs to generate a protective SEI on the Na metal anode [177, 178]. Xu et al. synthesized a multifunctional GPE by an in situ polymerization process for rechargeable dual-ion sodium metal batteries [177]. The electrolyte precursor solution contained 1,3-propanesultone (PS) as an additive in a 0.5 M NaPF6 in PC/EMC/FEC solvent (1:1:1 by volume) with 1.5 wt% ethoxylated pentaerythritol tetraacrylate (EPTA) monomer and 0.1 wt% AIBN initiator. Subsequently, the precursor solution was polymerized in situ at 60 °C. FEC and PS molecules exhibit a lower LUMO energy level than PC and EMC molecules. Thus, FEC and PS will be preferentially reduced on the Na metal anode, forming a protective SEI (Fig. 17a, b). Moreover, the gel structure (i.e., matrix arrangement) contributes to regulating the ion flux, thereby achieving smooth plating and stripping of Na+ ions and Na.

a Schematic illustration of the full cell using a 0.5 M NaPF6 in PC/EMC liquid electrolyte (left) or a GPE (right). b HOMO and LUMO energy comparison of PC, EMC, FEC, and PS molecules. Reprinted with permission from Ref. [177]. Copyright © 2020, Elsevier. c Schematic of Na||NVP batteries using the AN-GPE (the upper panel) and glass fiber (GF)-GPE (the lower panel) with the structure and Na-ion transport mechanism in the electrolytes. d SEM images of Na metal deposition on a Cu current collector using the GF-liquid electrolyte (LE), GPE, GF-GPE and AN-GPE at current densities of 0.5 mA cm−2 and 60 °C. Areal capacity of 3 mAh cm−2. e Cycling performance of Na||NVP batteries using the GF-LE, GPE, GF-GPE, and AN-GPE at 1 C under 25 °C (the left panel) and 60 °C (the right panel). Reprinted with permission from Ref. [181]. Copyright © 2019, Springer Nature. f SEM images of 2-GO + PVDF-HFP (upper left) and PVDF-HFP (lower left), surface roughness of 2-GO + PVDF-HFP (upper middle) and PVDF-HFP (lower middle) and Young’s modulus of 2-GO + PVDF-HFP (upper right) and PVDF-HFP (lower right) gel electrolytes. Reprinted with permission from Ref. [182]. Copyright © 2021, Wiley–VCH
Depending on the monomer chemical structure, the gel matrix can also facilitate ionic dissociation and provide anionic anchoring points through chemical interactions, thus improving the ionic conductivity of the gel electrolyte [179, 180]. Lei et al. confirmed that the creation of ionic channels in a GPE enabled smooth Na deposition on the anode [181]. They synthesized a PVDF-HFP-containing cross-linked beta alumina nanowire membrane, which allowed ion transport along and through the nanowires (Fig. 17c). To prepare the GPE, the synthesized porous membrane was soaked in a liquid electrolyte (1 M NaClO4 in EC:DEC with 5% FEC). The as-prepared GPE displayed smooth Na deposition on the Cu current collector (Fig. 17d), as well as high cycling performance in a Na||NVP full cell (Fig. 17e). Notably, the commonly employed glass fiber separator was replaced by the cross-linked beta alumina nanowire gel polymer electrolyte (AN-GPE), which played the roles of both the electrolyte and separator, proving the structural robustness of the QSE.
Fabricating a mechanically robust (i.e., high Young’s modulus) GPE is another strategy to reduce dendrite generation by mechanically preventing dendrite growth [182]. Glass fiber filters are largely used as separators owing to their high electrolyte intake capability and inexpensive feature. However, their low Young’s modulus and large pores benefit dendrite propagation. Glass fiber separators can be strengthened by copolymerization inside the glass fiber network, thereby allowing better electrode/electrolyte contact, inhibiting dendrite growth and easing smooth Na plating and stripping processes [173]. Luo et al. significantly increased the Young’s modulus of a PVDF-HFP membrane by simply tailoring the amount of graphene oxide (GO) in the PVDF-HFP-GO membrane [182]. Subsequently, the membrane was immersed in a 1.0 M NaClO4 in EC/PC (with a volumetric ratio of 1:1) electrolyte to form the GPE. The high structural strength of the GPEs enabled smooth Na plating and stripping (Fig. 17f).
In general, the properties of GPEs can be easily tailored by introducing additives, manipulating the preparation methods, or adapting the monomer/polymer structure. In particular, GPEs constitute great candidates for flexible batteries owing to their superior safety and high flexibility [183].
4.4 Solid-State Electrolytes
The solid-state electrolytes (SSEs) applied in Na metal batteries are identified as two classes: solid inorganic electrolytes (SIEs) and solid polymer electrolytes (SPEs). SSEs require the following main properties to be competitive with other electrolyte systems in Na metal battery applications: (1) high ionic conductivity (> 10−3 S cm−1) with minor anionic and electronic contributions, (2) high mechanical strength to suppress Na dendrite growth, (3) superior interfacial contact with electrodes, (4) easy and inexpensive manufacturing, and (5) excellent electrochemical and thermal stability [184, 185].
4.4.1 SIEs
The SIEs applied in Na metal batteries include Na superionic conductor (NASICON) electrolytes, the β-alumina (β″-Al2O3) solid electrolyte (BASE), and sulfide-based electrolytes.
NASICON belongs to the rhombohedral system. The octahedral ZrO6 and tetrahedral (Si,P)O4 inside NASICON are connected by their apex angles to build up the 3D network [186]. As the bottlenecks formed by ZrO6 octahedra and (Si,P)O4 tetrahedra are larger than 4.8 Å (1 Å = 1 × 10−10 m), the Na+ ions at the interstitial sites of the 3D network can pass through the bottlenecks and diffuse in this material (Fig. 18a) [187]. β″-Al2O3 belongs to the hexagonal system. As one of the sodium aluminates, β″-Al2O3 has a layered structure consisting of tightly packed Al-O spinel blocks and loosely packed sodium conduction planes that allow diffusion of Na+ ions (Fig. 18b) [188]. Sulfide-based electrolytes such as Na3SbS4 belong to the tetragonal system. The tetrahedral SbS43− inside Na3SbS4 is connected with the adjacent tetrahedra by Na+ ions. According to the bond valence sum mapping (BVSM) (Fig. 18c), the 3D framework built-up by tetrahedral SbS43− offers Na+ ion conduction routes in all three dimensions (along the a-, b- and c-axes), and this type of structure connotes good ionic conductivity for the solid electrolyte [189]. The structural analysis of a sulfide-based electrolyte (Na3SbS4) is shown in Fig. 18d.

a Schematic of the Na+ ion transport path inside the NASICON structure, where the rhombohedral symmetry represents the ZrO6 and (Si,P)O4 polyhedra, circles with dark shading and circles with light shading represent the Na(1) and Na(2) positions, respectively, and the curved line with an arrow is the conduction route of sodium ions. Reprinted with permission from Ref. [187]. Copyright © 1999, Elsevier Science B.V. b Schematic of the crystal structure of BASE. Reprinted with permission from Ref. [188]. Copyright © 2016, Elsevier. c 3D bond valence map isosurfaces of Na3SbS4 (isovalue: ± 0.3 v.u.). d Crystal structure with the unit cell outlined of Na3SbS4. Reprinted with permission from Ref. [189]. Copyright © 2016, Wiley–VCH Verlag GmbH & Co. e Schematic of the solid-state RT Na–S batteries applying the NASICON electrolyte coated with the PIN. Reprinted with permission from Ref. [193]. Copyright © 2019. Elsevier Inc. f Comparison between the electrolyte with and without BASE in RT Na–S batteries. Reprinted with permission from Ref. [194]. Copyright © 2013, Elsevier B.V. g Schematic of applying an in situ formation method to prepare Na3PS4 SIEs and the Na2S–Na3PS4-CMK-3 cathode. Reprinted with permission from Ref. [78]. Copyright © 2018, American Chemical Society. h Combination of Na3SbS4 with an organic electrolyte to form a thin and transferable hybrid electrolyte layer between the solid electrolyte and electrode to stabilize the interface. Reprinted with permission from Ref. [199]. Copyright © 2020, American Chemical Society
4.4.1.1 NASICON-Type SIEs
The first NASICON structure (Na1+xZr2P3−xSixO12, 0 < x < 3) was discovered by Hong and Goodenough, and it displayed a decent sodium ion conductivity of ~ 1.0 × 10−4 S cm−1 [190]. However, its conductivity is still more than one order of magnitude lower than that of conventional liquid electrolytes. Furthermore, the high interfacial resistance between NASICON-type electrolytes and metallic Na anodes is an issue limiting the performance of NASICON-type electrolytes in Na metal batteries. Heteroatom doping can improve the ionic conductivity of NASICON-type electrolytes. For example, Guin et al. reported a type of NASICON structure material by replacing Zr with Sc (Na1+xSc2P3−xSixO12), and it gave an enhanced sodium ion conductivity of 6.9 × 10−4 S cm−1 at 25 °C for x = 0.4 [191]. Song et al. synthesized a novel NASICON-type electrolyte by doping Mg into NASICON (Na3.1Zr1.95Mg0.05Si2PO12), and this electrolyte offered an excellent conductivity of 3.5 × 10−3 S cm−1 with a wide ESW of 4.5 V at RT [192].
Introducing an interlayer between the NASICON electrolytes and electrodes can be a pathway toward alleviating the large interfacial resistance. Yu et al. applied a conventional NASICON-type SIE (Na3Zr2Si2PO12) coated by a layer of a polymer with intrinsic nanoporosity (PIN) that significantly enhanced the interfacial conduction between the NASICON-type SIE and the sodium metal anode (Fig. 18e) [193]. Moreover, this NASICON-type SIE with the PIN retained the advantages of SSEs, such as suppressing the shuttle effect and inhibiting Na dendrite growth. Thus, it effectively improved the cycling stability of RT Na–S batteries compared to the RT Na–S cell with a Celgard separator and offered a good ionic conductivity of 1 × 10−3 S cm−1 at RT. Combining the Na metal anode with the carbon nanofiber (CNF)/sulfur cathode, the NaǁPIN-Na3Zr2Si2PO12ǁCNF/S RT Na–S cell delivered an initial discharge capacity of 700 mAh g−1, which remained at 550 mAh g−1 after 100 cycles at 0.2 C.
4.4.1.2 BASE-Type SIEs
Another type of SIE that has been applied in Na metal batteries is BASE. The BASE already widely applied in HT Na–S batteries can offer a comparably high ionic conductivity of ~ 2 × 10−3 S cm−1 at RT [194]. However, the limited wettability of BASE with Na is still a problem under RT conditions, which makes BASE mainly used in combination with liquid electrolytes. Wenzel et al. reported an RT Na–S battery with a liquid/solid electrolyte by applying BASE with 1 M sodium triflate (NaCF3SO3) in DOL/DME (with a volumetric ratio of 1:1) and compared it with the liquid electrolyte (LE) without BASE, as shown in Fig. 18f [194]. Compared with the electrolyte without BASE (starting from 450 mAh g−1 and remaining at ~ 80 mAh g−1 after 40 cycles at 0.148 mA cm−2), the electrolyte with BASE gives better capacity and cycling performance by suppressing shuttling of Na polysulfides and thus preventing the formation of an irreversible layer with increased resistivity on the Na anode surface (starting from 475 mAh g−1 and remaining at ~ 200 mAh g−1 after 40 cycles at 0.148 mA cm−2). Kim et al. applied BASE with TEGDME, which also provided better cycling stability and higher capacity (855 mAh g−1 to 521 mAh g−1) than the cell using the porous separator with the LE (starting from 350 mAh g−1 and remaining at 100 mAh g−1 after 2 cycles at 1/64 C) [195]. BASE provides total physical shielding to inhibit shuttling of Na polysulfides compared to the bare LEs and conventional porous separators.
4.4.1.3 Sulfide-Based SIEs
The last type of SIE is sulfide-based electrolytes, which can also provide an acceptable ionic conductivity at RT (e.g., Na3SbS4, 10−4–10−3 S cm−1). However, the interfacial resistance, parasitic reaction when in contact with the metallic sodium anode, and potential brittle cracking due to volume change during charge/discharge are still challenges that need to be solved for the application of sulfide-based SIEs. Several methods, such as using an in situ formation method [196,197,198] or a novel cathode material [198] or combining sulfide-based SIEs with other electrolytes [199], were adopted to solve these issues, which improved the electrochemical performance of sulfide-based SIEs in Na metal batteries.
The conventional cold-pressing process to fabricate sulfide-based SIEs induces poor interfacial contact between the electrolyte and electrodes. The in situ fabrication of a sulfide solid electrolyte with cathode active materials can ensure high ionic and electronic conductivity and sufficient interfacial contact. Yue et al. used the Na3PS4–Na2S-C nanocomposite as the cathode and contained Na3PS4 SIEs inside the nanocomposite to solve the contact problem between the electrodes and SSE [196]. The RT Na–S batteries with Na3PS4 as sulfide-based SIEs offered good cycling stability and a high capacity at 60 °C (starting from 869.2 mAh g−1, which remained at 438.4 mAh g−1 after 50 cycles at 50 mA g−1). Tanibata et al. employed an amorphous S-P2S5 electrode on the cathode side, which was in situ reacted with the Na metal anode to form a nanosized Na3PS4 and Na2S crystal mixture [197]. The crystal mixture precipitated on the surface of the cathode and endowed the Na–S battery with a high initial discharge capacity of approximately 1 250 mAh g−1 at 0.013 mA cm−2. Recently, a type of Na3SbS4-based SIEs reported by Wan et al. could provide an excellent conductivity of 1.14 × 10−3 S cm−1 at RT along with a S-Na3SbS4-C cathode and a Na metal anode [198]. They used a mechanochemical process (liquid-phase reaction + mechanical milling) to trigger in situ formation of S-Na3SbS4, which can provide both microscale primary and nanoscale secondary pathways for electronic/ionic transformation. In addition to offering stabler interfacial contact between the SSE and cathode, the in situ formation method also reduces the cathode material stress/strain and leads to superior rate and cycling performance of the Na–S battery (showing a high initial capacity of 1 504.3 mAh g−1 at 50 mA g−1, which remains at 468.1 mAh g−1 after 100 cycles at 1 000 mA g−1).
More recently, Ren et al. applied Na3SbS4 combined with an organic electrolyte (pentaerythritol tetraacrylate (PETEA)) by using an in situ cross-linking method and generated a thin and transferable hybrid electrolyte that can offer a stable interface with sodium during cycling (Fig. 18h) [199]. This inorganic/organic hybrid sulfide-based solid electrolyte provides a stabler electrochemical performance by preventing parasitic reactions between the SIEs and electrode, and the discharge capacity gradually increases from 0.32 to 0.52 mAh cm−2 over 90 cycles due to potential pore formation and delamination. However, the ionic conductivity of Na3SbS4/PETEA is only 0.47 × 10−4 S cm−1 at RT, which is ~ 22% of that of Na3SbS4 and requires further improvement in future research.
4.4.2 SPEs
SPEs are commonly assembled by dissolving metal salts in a polymer matrix. SPEs can be easily achieved by using hot molding, solvent casting, or extrusion techniques without the participation of liquid solvents as plasticizers [200]. For the polymer matrix in SPEs, poly(ethylene oxide) (PEO) is the most widely researched material, and it is a type of semicrystalline polymers with both crystalline regions and amorphous regions (Fig. 19a) [201]. The stable interfacial contact with electrodes and the excellent solubility for sodium salts make PEO quite attractive in sodium metal battery research. However, the operation temperature range for PEO is quite limited since the crystalline regions of PEO offer very limited ionic conductivity (the amorphous regions of the PEO matrix are the main ion transport pathways) [202]. On one hand, when the temperature is below 65 °C, the PEO chains will crystallize and deliver a very poor ionic conductivity of 10−8–10−7 S cm−1. On the other hand, when the temperature is higher than 80 °C, the mechanical properties of PEO may be degraded, limiting the ESW of the solid electrolyte, which should also be noted [161, 200].

a Schematic of the ion transport mechanism in the amorphous parts of PEO-based SPEs. Reprinted with permission from Ref. [201]. Copyright © 2016, The Royal Society of Chemistry. b Schematic illustration of the introduction of non-ion-conductive filler SiO2 into the PEO matrix. c TEM image of PEO combined with SiO2 fillers. Reprinted with permission from Ref. [204]. Copyright © 2015, American Chemical Society. d Schematic drawing of the Na+ ion transport mechanism inside PEO with the addition of NASICON. Reprinted with permission from Ref. [207]. Copyright © 2017, Elsevier B.V. e SEM images of the Na metal surface after 50 cycles in Na–SnS2 cells with the Na3PS4 and Na3PS4-PEO-NaClO4 electrolytes. f Comparison of the cycling performance of the Na–SnS2 batteries with the Na3PS4 electrolyte and with the Na3PS4-PEO-NaClO4 electrolyte after 40 cycles at 20 mA g−1. Reprinted with permission from Ref. [208]. Copyright © 2019, Elsevier B.V. and Science Press
Modification strategies, including using optimized concentrated sodium salts, introducing inorganic fillers, and employing novel cathode materials in PEO, need to be applied to ensure satisfactory mechanical properties, decent ionic conductivities (> 1.0 × 10−4 S cm−1), and wide ESWs. Park et al. embedded NaCF3SO3 salt into the PEO matrix and first tested it in an all-solid-state RT Na–S battery system [203]. The PEO/NaCF3SO3 electrolyte delivered a high ionic conductivity of 3.38 × 10−4 S cm−1 at 90 °C, and the Na–S battery with this SPE provided a high initial capacity of ~ 505 mAh g−1, which remained at 166 mAh g−1 after 10 cycles at 0.144 mA cm−2.
There are two classes of fillers that can be introduced into the PEO matrix. The first type is non-ion-conductive fillers (e.g., SiO2, TiO2), which aim to suppress the crystallization of PEO and increase cation transport (Fig. 19b) [204,205,206]. The surface groups on the SiO2 particles can incur a local structural change in the PEO matrix (Fig. 19c), which effectively prevents the formation of crystalline regions and accelerates cation transport by increasing the free cations. Zhu et al. introduced 1% TiO2 filler into the PEO electrolyte with NaFSI salt, and the PEO-NaFSI-1% TiO2 solid electrolyte achieved a high conductivity of 4.89 × 10−4 S cm−2 at 60 °C because of the extension of the amorphous regions [205].
The other type of filler is ion-conductive fillers (e.g., NASICON, Na3PS4, MIL-53(Al)), which can offer additional ion diffusion pathways to improve the ionic conductivity of PEO-based SPEs (Fig. 19d) [207,208,209]. Zhang et al. applied NASICON as an ion-conductive filler for the PEO matrix [207]. PEO can increase the Na vacancies on the NASICON surface and accelerate ion transport along the surface region. Meanwhile, the NASICON ceramic bulk can provide a competing ionic conductivity. Therefore, the composite SSE resulted in a high ionic conductivity of approximately 2.8 × 10−3 S cm−1 at 80 °C. Xu et al. coated PEO with NaClO4 salt onto Na3PS4 particles to form a solid electrolyte, which provided a high ionic conductivity of 9.4 × 10−5 S cm−1 at RT and 2.5 × 10−4 S cm−2 at 60 °C [208]. This coating strategy effectively stabilized the surface contact between the solid electrolyte and electrodes (Fig. 19e). Thereby, the all-solid-state RT Na–SnS2 battery offered an enhanced cycling performance of approximately 230 mAh g−1 after 40 cycles at 20 mA g−1 (Fig. 19f). The addition of ion-conductive fillers is similar to combining SIEs and SPEs to form composite electrolytes, which can synergize the advantages of both SIEs (e.g., sufficient mechanical strength, good ionic conductivity) and SPEs (e.g., high flexibility, good interfacial contact between electrodes and the electrolyte) to improve the electrochemical performance of the all-solid-state RT Na–S battery system. However, these types of composite SSEs still have not attracted enough interest, and this field can be a promising pathway for future SSE development for RT Na–S batteries.
In general, the ionic conductivity, mechanical strength, interfacial conduction, and electrochemical and thermal stability for both SIEs and SPEs can be manipulated by introducing other electrolytes or fillers, adapting the synthesis method and cathode structure, or coating the surface of SSEs. However, the Na metal electrode, which is much more unstable than Li metal electrodes, will incur more serious contact problems when using SSEs. SSEs were first thought to be able to suppress dendrite formation owing to the high Young’s modulus. However, battery failure due to short circuit was observed, attesting to dendrite penetration through the SSE. The mechanism of dendrite formation in SSEs is still under investigation, with two predominant mechanisms being mostly accepted. (1) Preferential Na plating where the local current density is higher (similar to dendrite formation in LEs). The dendrites grow through the SSE and propagate via defects or voids, which present higher energy. Usually, such dendrites are bulky with a large diameter, which can generate cracks in the SSE and lead to battery failure. (2) Dendrite growth through the grain boundaries due to electronic leakage. In this situation, the dendrites are very thin and follow the grain boundaries. The very small diameter of these dendrites results in a stronger electric field at the tip of the dendrites, which in turn further promotes their growth. Therefore, dendrite growth is even severer than that in LEs or GPEs [210]. Due to the above issues, the contact stability between the Na metal and SSEs needs to be given more focus along with enhancement strategies in the all-solid-state Na metal battery system, which will be investigated in the next section.
4.4.3 Enhancement of the Interfacial Contact of Na Metal/SSEs
The Na metal/SSE interface can be categorized into three types: (1) a thermodynamically stable interface, (2) a mixed ionic and electronic conducting interphase, and (3) an SEI with ionic conductivity and electronic insulation [211, 212]. Only the former and the latter type of interfaces can withstand long cycling.
SIEs usually suffer from poor electrode/electrolyte contact, which further deteriorates during the plating/stripping processes due to the creation of voids and accumulation of dead Na at the interface. The void formation is even more pronounced at high current densities, where larger voids are created during stripping and harder to fill during plating [213]. Applying an external pressure to the cell enhances the electrode/electrolyte contact and efficiently reduces the creation of voids during cycling. However, the external force applied must be carefully chosen to prevent mechanical failure [214]. Generally, Na metal is directly pressed on an SSE to achieve high solid‒solid contact. However, undesirable functional groups with high resistance can form on the surface of solid electrolytes, thereby leading to poor contact with the Na anode.
To improve the Na metal electrode/SSE contact, several advanced approaches have been investigated, such as surface polishing, heat treatment and the introduction of additives. Heat treatments have been employed to clean the solid electrolyte surface. For instance, by thermal treatment of the Na3Zr2Si2PO12 surface, the Na wettability was improved, and the interfacial resistance decreased [215]. Recently, an annealing process of a Na3Zr2Si2PO12 NASICON solid electrolyte surface enabled the removal of the side products on the surface of the solid electrolyte as well as partial reduction, thereby reducing the interfacial impedance [216]. Zhao et al. proposed another method: the introduction of the sintering additive Na2B4O7 into a NASICON solid, where NaO− and BO3− would reside on the surface of the sample (Fig. 20a, b) [217]. The as-prepared electrolyte allowed a high ionic conductivity of 1.72 × 10−3 S cm−1 at RT owing to the facile Na+ ion migration across the grain boundaries. Furthermore, the additive enabled the formation of a Na+-rich interface on the Na metal anode, which blocked electronic conductivity but was permeable to Na+ ions, thereby promoting high cycling performance (Fig. 20c, d).

a Time-of-flight secondary-ion mass spectrometry 3D illustration of NaO−, BO3− and PO3− along the depth of the sample, and b corresponding depth profiles of SiO4−, PO4−, NaO− and BO3− signals. c SEM image of the cross-section and d EDS mapping of NASICON with a Na2B4O7 additive/Na interface disassembled from a symmetric cell cycled at 0.3 mA cm−2 for approximately 2 500 h. Reprinted with permission from Ref. [217]. Copyright © 2021, Elsevier. e Schematic illustration of the Na+ ion mobility in PEO and optimized PEO-PPC-NASICON electrolytes. f Cycling performance of the Na|PEO-PPC-NASICON|Na symmetric cell at a current density of 0.1 mA cm−2. g SEM images of the surface morphologies of Na anodes after cycling in PEO-PPC-NASICON (the upper panel) and PEO (the lower panel) electrolytes. Reprinted with permission from Ref. [218]. Copyright © 2021, Royal Society of Chemistry
SPE systems can also be tailored to further enhance the interfacial solid‒solid contact stability. The polypropylene carbonate (PPC) polymer undergoes nucleophilic attacks in which Na+ randomly breaks C–O bonds in the PPC backbone; therefore, PPC is not a suitable SPE for Na metal batteries. However, the reacted PPC could be employed as a highly conductive interfacial wetting layer on the Na metal anode, which would be formed in situ on the anode (Fig. 20e). The interfacial resistance observed between Na metal and an SPE mixture of PEO and PPC was smaller than that of Na/PEO owing to the decomposition of PPC on the Na metal, which enhanced the electrode/electrolyte contact [218]. The Na|Na symmetric cell was cycled for more than 1 000 h, and the surface morphology of Na after cycling remained smooth in comparison with the cell cycled in the PEO electrolyte (Fig. 20f, g). PPC was also introduced into amorphous phases in a polymer blend structure, thereby contributing to higher ionic conductivity. The ionic conductivity of this electrolyte was further improved by introducing a small amount of NASICON as an inorganic filler. As a result, the optimized PEO-PPC-NASICON (48-37-15 wt%) SPE exhibited high chemical stability, high ionic conductivity and resistance to dendrite growth owing to the superior tensile strength and Young’s modulus of the polymer electrolyte.
5 Nanostructured Na Metal Anodes
To enable the practical application of Na–S and Na–Se batteries, we need to achieve high-performance Na metal anodes with long-term cyclability and good safety. As mentioned in the previous section, Na metal is more reactive than Li metal. The severe reactions between Na metal and the organic electrolyte generate a thick SEI between them, which causes a significant increase in the charge transfer resistance, inducing large voltage hysteresis and low energy efficiency. In addition, the infinite volume change of the Na metal anode during repeated plating and stripping is another major challenge that needs to be resolved to achieve a stable Na metal anode. Therefore, nanostructure engineering, including protective layer engineering and skeleton design, has been a focus of this research area, and tremendous outcomes have been achieved thus far.
5.1 Artificial Protective Layers for Na Metal Anodes
Constructing protective layers on Na metal anodes is beneficial for stabilizing the SEI during the repeated plating/stripping process [219]. The protective layers can isolate the highly reactive Na metals from the LEs. In addition, a protective layer with high ionic conductivity can regulate the Na+ ion flux on the anode surface, leading to uniform metallic Na plating. The in situ formation of protective layers or an SEI via electrolyte engineering has been summarized in the previous section. Therefore, in this section, we will focus on the recently developed ex situ artificial protective layers. The ex situ artificial layers should possess superior ionic conductivity and abundant Na+ ion transport channels to ensure that electrochemical deposition of Na metal occurs underneath the artificial layers. In addition, the protective layers should possess good flexibility to adapt to the large volume change and sufficient mechanical strength to withstand dendrite penetration.
5.1.1 Constructing Protective Layers via Solution Methods
Traditional solution methods have been used to form artificial protective layers with controlled composition on Na metal anodes. By exposing Na metal anodes to solutions containing certain chemicals, an artificial protective layer can be formed on Na anodes via chemical reactions (Fig. 21a, b). Tian et al. successfully constructed a NaI layer via a Wurtz reaction. The Na metal anodes were first exposed to a solution containing 2-iodopropane or 1-iodopropane. Then, a NaI layer was formed on the Na metal anode after one electron from the Na metal was transferred to the iodine, which endowed the Na metal anodes in Na-I2 batteries with significantly enhanced electrochemical performance [220]. By using the same reaction mechanism, a thin layer of NaBr was constructed on a Na metal anode through the reaction of metallic Na with 1-bromopropane solution [221]. A joint DFT study of the SEI demonstrated that the energy barrier for adatom diffusion on NaBr is as low as that of magnesium, which is known to form spherical nuclei upon charging. Compared to pristine Na metal, the reduced energy barrier for ion transport (0.02 eV per atom) of NaBr promoted uniform Na plating and prevented unwanted parasitic reactions between the electrode and electrolyte. Electrochemical testing in symmetric cells showed that NaBr-coated Na metal anodes achieved an almost 10 times longer cycle life than unprotected Na metal anodes.

a Schematic illustration of the formation of NaI-protected Na metal anodes. b SEM image of NaI-coated Na metal anodes. Reprinted with permission from Ref. [220]. Copyright © 2021, Elsevier. c Schematic illustration of the preparation of an MOF (MOF-199)-coated Cu electrode. d Photographs of pristine Cu foil and MOF-199-coated Cu foil. Reprinted with permission from Ref. [224]. Copyright © 2021, Elsevier. e Synthesis process of graphene film-protected Na metal anodes. f Cycling performance of Na metal anodes with and without a graphene film at different current densities. Reprinted with permission from Ref. [226]. Copyright © 2017, American Chemical Society. g Schematic illustration of the ALD of Al2O3 on Na metal anodes. Reprinted with permission from Ref. [13]. Copyright © 2020, Wiley–VCH. h Schematic illustration of the Na plating/stripping behavior with and without an ALD-Al2O3 protective layer. Reprinted with permission from Ref. [227]. Copyright © 2017, Wiley–VCH
Slurries containing organics and/or inorganics have been coated on the surface of Na metal anodes through a variety of methods, such as tape casting and spin coating. After evaporation of the solvents, uniform protective layers can be conformally coated on the surfaces of Na metal anodes. Hou et al. prepared PVDF@Cu, PVDF-HFP@Cu, and PEO@Cu current collectors by casting solutions with the corresponding polymers on Cu current collectors [222]. Among all the as-prepared current collectors, the PVDF@Cu current collector exhibited the best electrochemical performance, with an ultralong cycling life (1 200 h) and high average Coulombic efficiency (approximately 99.91%) at 1 mA cm−2. By using a similar preparation method, an inorganic‒organic hybrid protective layer (NaF–PVDF) was coated on commercial Cu current collectors [223]. The organic component, a flexible PVDF matrix, can maintain the integrity of the protective interface against volume change. Meanwhile, the inorganic component, NaF particles, can enhance the mechanical strength and increase the Na+ ion conductivity, which is beneficial for suppressing dendrite growth. As a result, the hybrid interface-coated Cu current collector showed a tenfold higher lifespan than the bare Cu current collector. Qian et al. also fabricated several MOF-coated Cu electrodes using the tape casting method. Slurries containing MOF particles and PVDF binders in N-methyl-2-pyrrolidone (NMP) solvent were cast on Cu foil by a doctor blade. After drying in an oven, MOF-coated Cu electrodes were obtained [224]. The porous and robust MOF protective layer acted as a resilient shield and a homogenized metal ion concentration layer, which could physically suppress Li/Na dendrite growth.
5.1.2 Interface Engineering via Advanced Nanotechnologies
Highly uniform and compact artificial protective layers constructed by nanomaterials, especially ultrathin 2D materials, can be realized via the chemical vapor deposition (CVD) method. The thickness of the protective layer based on 2D materials can be controlled at the atomic scale while maintaining high mechanical strength. However, many factors influence the physical and chemical properties of the 2D material-based protective layer, such as the defects, bond length and metal proximity of the 2D materials [225]. Increasing the defects and reducing the thickness of the 2D materials benefit the ionic conductivity but show negative effects on the mechanical strength of the protective layer. Therefore, finding a balance between the electrochemical performance and the mechanical properties is essential for the design of an effective artificial protective layer with 2D materials. For instance, free-standing graphene films with different thicknesses were synthesized via the CVD method and used as the artificial protective layer for Na metal anodes (Fig. 21e) [226]. A few-layer graphene (FLG) film with a thickness of approximately 2.3 nm favors good electrochemical performance at a lower current density (Fig. 21f). In contrast, a multilayer graphene (MLG) film with a thickness of approximately 5 nm shows the best performance at a higher current density (Fig. 21f). Other 2D materials, such as hexagonal boron nitride (h-BN), have already been used in Li metal anodes, have achieved significantly improved performance, and could be potential candidates for Na metal anode protection.
Other advanced thin film fabrication nanotechnologies, including atomic layer deposition (ALD) and molecular layer deposition (MLD) technologies, were introduced to form atomic-scale artificial protective layers on Na metal anodes (Fig. 21g). Through ALD and MLD technologies, the thickness and composition of the protective film can be precisely controlled. For instance, an inorganic layer, such as an Al2O3 layer, was directly coated on Na metal anodes via plasma-enhanced ALD (PE-ALD), which demonstrated a significant positive effect on the cycling stability of Na metal anodes in carbonate electrolytes [13, 227, 228]. To further enhance the mechanical properties of the artificial protective layer (i.e., alucone), an inorganic and organic protective layer was prepared by using the MLD method [229, 230]. The inorganic and organic layer (MLD-alucone)-coated Na metal anodes showed much-improved cycling stability compared to the inorganic layer (ALD-Al2O3)-coated electrode and the pristine electrode. The enhanced flexibility has been assigned as the major contributor to maintaining the integrity of the protective film against repeated volume changes during the Na plating/stripping process.
In brief, employing nanotechnology has been demonstrated to be an effective strategy to fabricate uniform artificial protective layers at the atomic scale. The good chemical stability, high ionic conductivity, and enhanced mechanical properties facilitate uniform Na plating. The major challenges of using these sophisticated methods are how to reduce the processing cost and simplify the fabrication process for large-scale applications.
5.2 Skeleton Design for Sodium Metal Anodes
As “hostless” anodes, Na metal anodes suffer from severe volume change during repeated plating/stripping processes. Designing a suitable 3D “host” has been demonstrated to be a promising strategy to avoid very large volume change and guide homogeneous Na metal deposition [231, 232]. To meet the requirements of practical applications, some selection criteria, including the mechanical strength, electronic conductivity, surface area, chemical stability and cost, need to be carefully considered to achieve a suitable “host” for metallic Na to achieve high electrochemical performance. To date, a variety of skeletons have been developed to form composite anodes with Na metal, such as porous metal skeletons, alloy-based skeletons, carbon-based nanostructure skeletons, and polymer skeletons. Therefore, in this section, we will discuss the recent progress in skeleton design for sodium metal anodes.
5.2.1 Metallic Skeletons
Metallic materials, such as Cu or Al foils, have been used as standard substrates to investigate Na metal plating/stripping behavior. Severe dendrite formation has been observed on planar metallic substrates due to their low surface area and poor sodiophilic property. The first approach to improve the Na metal plating/stripping behavior is to increase the surface areas of the dense metallic foils to reduce the local current density [233]. The lower local current density can facilitate homogenous Na metal plating owing to the minimal Na-ion concentration gradient near the surface of Na metal anodes. For instance, constructing a porous Al skeleton to increase the surface area of the substrate has been demonstrated to be an effective strategy to suppress Na dendrite formation and enhance the cycling stability of Na metal anodes [234]. The interconnected porous structure of porous Al foils increases the available sites for Na nucleation and regulates the Na+ ion flux distribution, which facilitates uniform Na deposition without obvious dendrite formation (Fig. 22a). For the Li|Al half-cell with porous Al foil as the working electrode, a low and stable voltage hysteresis and an average plating/stripping Coulombic efficiency of above 99.9% were achieved for over 1 000 cycles, which is a significant improvement compared to planar Al foil. In addition, a Na-O2 full cell with a low amount of metallic Na predeposited on porous Al foil as the anode showed superb cycling stability for 200 cycles.

a Schematic illustration of Na deposition on planar and porous Al foils, and corresponding SEM observation at different magnifications. Reprinted with permission from Ref. [234]. Copyright © 2017, American Chemical Society. b Schematic illustration of the fabrication process of in situ growth of Cu nanowires on Cu foam. c Long-term time vs. voltage curves of Na plating and stripping on pristine Cu foam and CuNW-Cu foam. Reprinted with permission from Ref. [236]. Copyright © 2021, Elsevier. d Schematic illustration of the Na nucleation and deposition behavior on pristine Cu foam and ZnO nanorod-decorated Cu foam. e COMSOL simulation results of the local current density distribution and Na+ flux at the interfaces of ZnO nanorod-decorated Cu foam (CF@ZnO) and bare Cu foam. Reprinted with permission from Ref. [237]. Copyright © 2021, Elsevier
Constructing 3D conductive frameworks on Cu foils to increase the surface area of the current collectors has also been reported to develop high-performance Na metal anodes. Lu et al. in situ grew Cu nanowires on commercial Cu foil through a hydrothermal process in absolute alcohol solvent [233]. The obtained Cu nanowires showed much thinner diameters of 40 nm compared with previously reported Cu nanowires (> 300 nm) for Li metal anodes. The significantly increased surface area of the 3D conductive skeleton with Cu nanowires effectively regulated the electric field distribution and thus facilitated reversible Na plating and stripping with high Coulombic efficiencies. The full cells with the Na predeposited 3D Cu nanowires as the anode and Na3V2(PO4)3@C as the cathode showed extremely stable cycling performance and enhanced high-rate capabilities, with an energy density approaching 314.5 Wh kg−1. In addition, Xu et al. electrodeposited porous Ni on Cu foil via a facile hydrogen bubble dynamic template method [235]. The 3D Ni@Cu substrate showed a unique honeycomb-like porous structure, which could accommodate both metallic Li and Na deposition in the porous networks and prevent large volume expansion. In the half-cell Coulombic efficiency testing, the 3D Ni@Cu substrate achieved excellent stability of Na reversible deposition without dendrite formation under high current density and capacity limit.
Modifying commercial Cu foams as conductive substrates for Na metal anodes has also attracted significant attention recently. As shown in Fig. 22b, through in situ growth of Cu nanowires on 3D Cu form, a hierarchical porous Cu substrate (CuNW-Cu) showed an excellent confinement effect to reduce the volume change during Na plating/stripping [236]. The enlarged surface area and nanoporous architecture facilitated homogenous Na+ ion flux, which promoted uniform Na metal deposition without dendrite formation. The controlled half-cell cycling test results demonstrated repeated and uniform Na metal deposition with a capacity limit of up to 12 mAh cm−2 for over 1 000 h (Fig. 22c). In addition, Yang et al. reported in situ growth of ZnO nanorods on Cu foam via a simple chemical precipitation method and used this material as a 3D skeleton for a dendrite-free Na metal anode (Fig. 22d) [237]. The excellent sodiophilic property of ZnO nanorods provided abundant nucleation sites during the initial Na deposition process, which was reflected by the reduced nucleation overpotential compared with the pristine Cu foam skeleton. The COMSOL Multiphysics simulation results in Fig. 22e indicated that the rationally designed core–shell cylindrical structure of the nanorod arrays could reduce the local current density and facilitate uniform Na deposition. Furthermore, Wang et al. treated a commercial Cu foam through a facile chemical engineering process (e.g., oxidation or sulfurization) to form a cylindrical core–shell skeleton with a good sodiophilic property [238]. The chemical interaction between Na and chemically processed Cu foam, especially oxygen-treated Cu foam, facilitated uniform molten Na impregnation during the infusion process and confined metallic Na within the matrix pores during the repeated plating/stripping process. The galvanostatic cycling tests in symmetric cells with two Na-infused O-treated Cu foam (Na@O-CF) electrodes showed exciting cycling stability in widely used carbonate electrolytes, which promotes their future practical application.
For the future development of metallic skeletons as the current collectors for Na metal batteries, there are still several challenges that need to be resolved. The optimization of the multiscale porous structure design needs to effectively accommodate the Na metal while maintaining the robust mechanical strength. Meanwhile, how to reduce the cost for feasible mass production needs to be considered.
5.2.2 Alloy Skeletons
Alloying Na metal with other metals (e.g., Li, Na, K, Sn and Sb) to form Na–M alloy anodes is another strategy to improve the electrochemical performance of Na metal anodes. The alloying process will change the physical and chemical properties of Na metal-based anodes, such as the melting points, mechanical properties, chemical reactivities, and plating behavior.
Li–Na alloy anodes have been used in oxygen batteries and have achieved improved cycling stability [239]. A robust and flexible SEI was formed on Li–Na anodes via an in situ reaction with a 1,3-dioxolane additive, which could buffer the volume change of the alloy anodes and thus prevent cracking and further electrolyte consumption. In addition, the continuous release of Li+ ions into the electrolyte will help form a strong electrostatic shield that can effectively suppress dendrite formation. The Li–Na alloy-oxygen cells with optimized Li–Na alloy anodes achieved good cycling stability up to 137 cycles at a current density of 200 mA g−1 with a capacity limit of 1 000 mAh g−1. Similarly, Yu et al. infused Li–Na alloy into a carbon cloth (CC) host and used it as the anode in Li–Na alloy-iodine batteries [240]. The Li–Na alloy-CC anodes showed not only excellent cycling stability in symmetric cells but also significantly improved cycling stability in Li–Na alloy-iodine batteries.
Owing to their liquid state at RT, Na–K alloys have also been employed in rechargeable batteries. Replacing the solid‒liquid interface with a liquid‒liquid interface completely avoided dendrite formation. Na–K alloys can be used as the anodes for both Na-based and K-based batteries, depending on the reaction mechanism of the counter cathode electrode. If Na+ ions are involved in the redox reaction of the cathode materials, then the alloys can be used as the anodes for Na-ion batteries. Otherwise, they serve as the anodes for K-ion batteries.
5.2.3 Carbonaceous Skeletons
Carbonaceous materials are considered alternative candidates for synthesizing nanostructure frameworks due to their high electronic conductivity, low cost, low density, and diverse nanostructures. Many nanostructure frameworks have been developed to improve the electrochemical performance and safety of alkali metal anodes, including 1D nanotubes and nanofibers, 2D graphene, and 3D porous carbon materials.
5.2.3.1 Carbon Nanotube (CNT) Skeletons
As one of the most used carbon materials, CNTs have been massively produced in industry and comprehensively used to obtain 3D conductive skeletons due to their excellent conductivity, unique 1D structure, and good mechanical robustness. The interconnected conductive networks of CNT skeletons have large specific surface areas, which can reduce the local current density and facilitate homogenous Na plating. Meanwhile, the abundant voids formed between the CNTs effectively alleviate the volume change during the metallic Na plating/stripping process. Through a facile mechanical mixing process, a CNT-Na composite was fabricated and showed significantly reduced nucleation overpotential during Na plating (Fig. 23a). The CNT content in the CNT-Na composite significantly influenced the nucleation overpotential; therefore, the optimized 11.8CNT-Na electrode exhibited the best cycling performance, which was approximately 5 times longer than that of the pristine Na metal anodes in symmetric cell testing [241].

a Schematic illustration of the fabrication processes of CNT-Na electrodes, and mechanisms of Na nucleation and stripping on CNT-Na and bare Na electrodes. Reprinted with permission from Ref. [241]. Copyright © 2021, Elsevier. b Schematic diagram of the fabrication procedure of Na@CP-NCNTs and the Na plating/stripping behavior on the surface of pristine Na foil and Na@CP-NCNT skeletons. Reprinted with permission from Ref. [242]. Copyright © 2018, Wiley–VCH. c Schematic illustration of uniform Na deposition on an oxygen-functionalized CNT skeleton. d Na growth behavior on oxygen-functionalized CNT and conventional Cu substrates obtained via DFT calculations. Reprinted with permission from Ref. [243]. Copyright © 2019, Wiley–VCH. e Schematic illustration of Na deposition on the pristine Na metal anode and Na/NSCNT anode. f Digital photos of bent NSCNT paper. g SEM observation of the Na anode and Na/NSCNT anode before and after 20 cycles. h Reprinted with permission from Ref. [244]. Copyright © 2018, Wiley–VCH
Due to the “sodiophobic” property of pristine CNTs, a variety of surface modification strategies have been reported to enhance their “sodiophilic” property. Nitrogen-doped carbon nanotubes (NCNTs) rationally grown on carbon paper (CP) have been used as a 3D porous host for Na metal anodes (Fig. 23b). The vertical NCNTs serve as “sodiophilic” sites, which effectively decrease the contact angle between CP and liquid metal Na. The interconnected conductive network of CNTs on CP increases the surface area and reduces the local current density, facilitating smooth Na nucleation and deposition. After the Na infusion process, the as-prepared Na@CP-NCNT composite anodes demonstrate excellent cycling stability at different current densities with a capacity limit of up to 3 mAh cm−2 [242]. Introducing oxygen-functional groups onto CNTs via oxygen microwave plasma is another method to enhance their “sodiophilic” property (Fig. 23c, d) [243]. DFT calculations showed that the binding energy between Na and oxygen-functionalized CNTs is stronger than that with pristine CNTs and Cu current collectors. Therefore, the oxygen-functionalized CNT matrix exhibited homogeneous Na nucleation and dendrite-free growth behavior with a remarkable electrochemical performance (e.g., a stable Coulombic efficiency of over 99.5% for more than 680 cycles at 10 mA h cm−2 with a current density of 5 mA cm−2). In addition, Sun et al. introduced N and S-codoped CNT (NSCNT) paper as a highly “sodiophilic’’ matrix for dendrite-free Na metal anodes (Fig. 23e) [244]. The NSCNT paper showed very good mechanical strength with excellent flexibility and could be bent and punched into different shapes (Fig. 23f). DFT calculations proved that the codoped CNTs showed a better “sodiophilic’’ property than the singly doped and pristine CNTs. As a result, the NSCNT paper can guide uniform Na+ ion flux and facilitate dendrite-free Na plating at high current densities and capacity limits. Furthermore, the Na/NSCNT composite anodes can be used to improve the cycling performance of RT Na-O2 batteries, showing great potential for use in RT Na–S and Na–Se batteries.
Although great improvements have been achieved through the advantages of CNT skeletons, further in-depth studies must still be conducted with the help of advanced characterization and testing methods, such as cryogenic electron microscopy, in situ optical microscopy, and titration gas chromatography, to study the mechanism of the enhanced electrochemical performance of Na metal anodes.
5.2.3.2 Graphene Skeletons
As one of the most popular 2D materials for rechargeable batteries, graphene attracts great attention in alkali metal anode research owing to its high electronic conductivity, good chemical stability, and excellent mechanical properties at the atomic scale. Graphene nanosheets have been used as building blocks to construct a variety of porous structures, which can be used as promising hosts for metallic Na. A reduced graphene oxide aerogel (rGa) was synthesized via hydrothermal reduction followed by an oriented freeze-drying treatment (Fig. 24a) [245]. After infusing molten sodium into the porous rGa, a Na/rGa composite anode was obtained and showed excellent flexibility (Fig. 24a). The large specific surface area and uniform pore structure of the rGa can effectively reduce the local current density and inhibit Na dendrite formation (Fig. 24b). Due to the ultralight weight of the porous graphene host, the Na/rGa anodes obtained a high specific capacity of 1 064 mAh g−1. In symmetric cell testing, the Na@rGa anode exhibited a low overpotential of approximately 50 mV and a stable cycling performance for 1 000 cycles at a high current density of 5 mA cm−2 in a carbonate electrolyte (1.0 M NaClO4 in EC/DEC). For full cell application, Na@rGa presented enhanced cycling stability when paired with the Na0.67Ni0.25Mn0.75O2 cathode. Free-standing molten Na-infused reduced graphene oxide (Na@rGO) was also demonstrated to be a promising anode for high-energy Na-based batteries [246]. A porous GO film was first synthesized via a freeze-drying method (Fig. 24c). Then, the gray GO film was immediately reduced to dark black rGO after parallel connection with molten Na, resulting in a more porous structure. The final Na@rGO was obtained after fast Na infusion by simply placing the edge of the rGO film into the molten Na. The Na@rGO anode showed excellent mechanical strength and could be easily processed to assemble flexible batteries. No obvious Na dendrites were observed after cycling for 300 h at a high current density (5 mA cm−2) and a high capacity (5 mAh cm−2) in a carbonate electrolyte.

a, b Schematic illustration of the fabrication and plating/stripping behavior of Na@rGa anodes. Reprinted with permission from Ref. [245]. Copyright © 2019, Elsevier. c SEM images of GO film, rGO film and rGO-Na anodes. Reprinted with permission from Ref. [246]. Copyright © 2017, National Academy of Sciences, USA. d Schematic illustrations of Na plating on different matrices, including Cu foil, rGO and partially reduced GO (PrGO). e Tension illustrations of Na plating on planar rGO and PrGO films obtained through mechanical simulation. Reprinted with permission from Ref. [249]. Copyright © 2020, American Chemical Society. f Schematic illustration of the Na nucleation, growth, and further plating process on BG. Reprinted with permission from Ref. [247]. Copyright © 2019, Wiley–VCH. g Schematic of CVD growth of N-doped graphene microcubes on cubic NaCl crystals, and schematic of the Na stripping/plating process with PN-G microcubes as solid additives. Reprinted with permission from Ref. [250]. Copyright © 2020, Elsevier. h Schematic illustrations of Na deposition on NaF/SnO2@rGO. Reprinted with permission from Ref. [248]. Copyright © 2020, Elsevier
The doping strategy can also be used to enhance the “sodiophilic” property of graphene toward Na plating. For instance, boron-doped graphene (BG) was synthesized via the CVD method [247]. DFT calculation results showed that the binding energy of BG is higher than that of N-doped graphene, bare Cu and pristine graphene. The enhanced “sodiophilic” property through heteroatom doping greatly reduced the Na nucleation barrier and facilitated uniform Na plating. The obtained BG/Na anode showed a very good cycling stability up to 300 h at a current density of 5 mA cm−2 with a capacity limit of 2 mAh cm−2. Later, by employing lightweight N-doped graphene microcubes in Na metal anodes, super-stable cycling stability and ultrahigh-rate capability were achieved. With only 3 wt% of the N-doped graphene microcube additive, excellent cycling stability was realized at a high current density of 20 mA cm−2 with 10 mAh cm−2 in additive-free carbonate electrolytes. When paired with the Na3V2(PO4)3 cathode, the full cell could maintain a high capacity of approximately 95.2 mAh g−2 after 1 000 cycles at 5 C (1 C = 110 mA g−2).
Introducing metal oxides into graphene skeletons to form composites is another effective way to increase the “sodiophilic” property of porous hosts. Through theoretical modeling, Jin et al. identified the driving force of the “sodiophilic” property-guided Na plating and the influence of interfacial interactions on the cyclability of the sodiated NaF/SnO2@rGO system [248]. Furthermore, through experimental investigation, they discovered that the intermediate phases of Na2O and/or Na15Sn4 reduced the nucleation overpotentials and guided uniform Na plating owing to the strong binding energy to metallic Na. In addition, the strong interaction between the SnO2 nanodots and graphene skeletons reduced the loss of the sodiophilic sites, benefiting the improved cycling stability of Na plating/stripping. This interfacial design strategy provides an effective way to enhance Na metal anodes in Na-based batteries.
6 Separator Modification and Interlayer Design
Commercialized polyolefin separators produced by Celgard® and glass fiber (GF) separators are commonly used for RT Na–S and Na–Se batteries. The long-chain sodium polysulfide and polyselenide intermediates formed during discharging are prone to dissolve in conventional LEs and diffuse across both types of separators due to their micron-sized porous structures (Fig. 25a). Polysulfide and polyselenide shuttling leads to severe self-discharge, deterioration of the metal anode and loss of active materials at the cathode, manifesting as a low Coulombic efficiency and rapid capacity decay [251]. Kaskel et al. first achieved an enhanced cycling performance in RT Na–S batteries by adopting a sodiated Nafion (Na-Nafion) coating on a polypropylene (PP) backbone as a cation-selective separator [252]. Compared to the porous PP membrane, the nonporous Na-Nafion separator with sufficient Na+ conductivity has much smaller channels that can efficiently block polysulfide permeation (Fig. 25b) [105]. As shown in Fig. 25c, a Nafion membrane is composed of a hydrophobic poly(tetrafluoroethylene) (PTFE) backbone and a hydrophilic region (4–5 nm) with fixed SO3− clusters, which are connected to each other through a hydrophilic channel (1–2 nm in diameter) [253]. These sulfonic groups provide a negatively charged environment, enabling the counter Na+ to transfer through the hydrophilic pores and blocking the negatively charged polysulfides due to the “charge repulsion” of the Donnan potential (Fig. 25d). Therefore, the RT Na–S batteries using the Na-Nafion separator showed improved cycling stability.

a, b SEM images of a porous Celgard separator and a Nafion membrane. Reprinted with permission from Ref. [105]. Copyright © 2015, John Wiley & Sons. c, d Architecture and schematic of the ion selectivity of the Nafion membrane. Reprinted with permission from Ref. [253] Copyright © 2016, American Chemical Society. e Schematic of the S/C composite (top) and S@HCS/MoS2 electrode with modified GFs (bottom) during discharging. Reprinted with permission from Ref. [255]. Copyright © 2019, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. f Illustration of the fabrication of PMTFSINa-grafted|DN-MXene-coated Janus separators for RT Na–S batteries. Reprinted with permission from Ref. [103]. Copyright © 2020, John Wiley & Sons
Additionally, most of the inorganic host materials introduced above can be employed to modify separators to improve the electrochemical performance of Na–S and Na–Se batteries. In general, functional materials are directly coated on one side of PP or GF separators via a drop-coating or filtration-assisted method (the obtained coating layer is denoted as an interlayer). The additional interlayers cover the large pores of commercial separators and can directly block the dissolved sodium polysulfides or polyselenides from transferring across the separators. Furthermore, some interlayers containing polar materials can chemically trap intermediates and enable them to be reutilized as active materials [254]. For example, Xu et al. prepared hollow carbon spheres decorated with MoS2 (HCS/MoS2) as both a sulfur host and a separator modifier [255]. The dual cathode-electrolyte defense system efficiently prevents polysulfide shuttling (Fig. 25e). A similar tactic has also been applied to improve the performance of Na–Se batteries [256, 257].
To maintain the electronic insulating property of separators, the strategies mentioned above are all focused on modification of the cathode side of batteries. The exploitation of the anode side of the separator to further enhance the electrochemical performance has been overlooked. Recently, our group designed and prepared a Janus separator with a single-ion-conducting polymer-grafted side and a functional low-dimensional material coated side by a grafting-filtering route for Na–S batteries [103]. A poly(1-[3-(methacryloyloxy)propylsulfonyl]-1-(trifluoromethanesulfonyl)imide sodium) (PMTFSINa) layer was first grafted on one side of the PP separator by a UV-induced surface polymerization method. Subsequently, a defective Ti3−xCNTy MXene (DN-MXene) was adhered to the other side of the separator through filtration (Fig. 25f). PMTFSINa remarkably enhances the electrolyte wettability, prevents polysulfide diffusion via electrostatic repulsion, and suppresses Na dendrite growth. Meanwhile, the DN-MXene next to the cathode electrocatalytically boosts the polysulfide conversion kinetics. In brief, an ideal separator for Na–S and Na–Se batteries should possess excellent Na+ selectivity and conductivity, superior isolation of polysulfides and polyselenides and good sodiophilicity (which is discussed in detail in Sect. 6).
7 Conclusions and Perspectives
7.1 Conclusions
We have comprehensively reviewed the recent progress regarding RT Na–S and Na–Se battery systems. These include the reaction mechanisms, the design of the electrode materials, i.e., sulfur cathodes and Na metal anodes, the optimization of the LEs, and the exploration of novel (quasi-) SSEs. Although significant achievements have been reported, many issues still need to be overcome to realize future commercialization.
The slow reaction kinetics and the shuttle effect of soluble sodium polysulfides/polyselenides are the major challenges in obtaining high-performance cathodes. The rational design of cathode hosts with multifunctionalities can overcome these challenges, including physical and chemical confinement to capture dissolved high-order polysulfides/polyselenides, superior electrocatalytic activity to accelerate the conversion from long-chain to short-chain Na polysulfide/selenide species, and high conductivity to remedy the low conductivity drawback of the sulfur/selenium element. Several cathode engineering strategies have been demonstrated to be effective for realizing this purpose: (1) constructing a hierarchical porous architecture to tackle the volume change and provide excellent physical confinement, (2) loading precious metal NPs or SAs onto a highly conductive carbon-based host to offer an electrocatalytic function, (3) utilizing polar functional or polymeric sulfur composites to provide strong chemical confinement, and (4) using other sulfur-based composites, which can bypass the use of elemental S to increase the structural stability of the electrode. Notably, a combination of the strategies mentioned above that can synergize individual advantages is highly desirable for achieving the enhanced performance of RT Na–S and Na–Se batteries.
Electrolyte modulation is also key to realizing high-performance Na–S/Se batteries, and here, we generalize the challenges in electrolyte exploration and offer guidelines for future research directions as follows (1) The understanding of the SEI formation on Na metal in LEs should be deepened, and more compatible solvents, electrolyte additives, or Na salts that can assist the formation of a resistant SEI should be identified. In this way, Na dendrites would be largely suppressed, leading to long cycling stability and high Coulombic efficiency. (2) The cathode species dissolution into the LE should be reduced. Several strategies can be further explored, such as developing catholytes and electrolytes in which polysulfides/polyselenides present a low solubility and form a cathode-electrolyte interphase (CEI) layer. (3) Some LEs, i.e., highly concentrated ILs, exhibit high cycling performance and Coulombic efficiency; however, these LEs are usually too expensive for the industrial scale. Reducing the cost of the electrolyte is key for commercialization. (4) An LE can be gelled into a quasi-solid electrolyte to lower cathode species dissolution and dendrite growth propagation. However, polymer synthesis is sometimes complex and costly. Alternative synthesis routes should be explored to enable polymer electrolyte synthesis at a large scale and at an adequate cost. (5) Ultimately, SSEs can resolve the above issues but face other challenges, such as low ionic conductivity at RT, which prevents a high cycling rate, narrow electrochemical stability, which impedes the utilization of high-voltage cathodes, and/or high cost. (6) Additional effort should also be devoted to the safety and eco-friendliness of electrolyte synthesis by prioritizing nonflammable, nontoxic and nonenvironmentally hazardous chemicals. (7) With specific and advanced experiments, such as XPS depth profiling and cryo-TEM, as well as theoretical calculations, further progress is expected to support an in-depth understanding of the electrode interfacial chemistry and electrolyte solvation structure, thereby facilitating electrolyte design.
Reducing parasitic reactions and suppressing dendrite formation are still great challenges in achieving stable and high-performance Na metal anodes. More fundamental and experimental studies are critical to reveal the reaction mechanisms on Na metal anodes, including, practically, the Na plating/stripping behavior on different substrates and the reactions with the dissolved byproducts from the cathodes. Constructing an artificial protective layer on Na metal anodes has attracted significant attention and will become even more popular via coating other innovative materials with high ionic conductivity and good mechanical strength. Advanced surface coating technologies such as ALD and MLD will provide more options to improve the coating quality of the artificial protective layers. In addition, designing suitable hosts to accommodate metallic Na is also important to reduce the volume change during continuous Na plating/stripping. Rational structure design combined with surface functionalization will facilitate Na plating inside these hosts to minimize the volume change.
The low-cost and high-energy properties of Na metal-based rechargeable batteries hold great promise for large-scale renewable energy storage. The currently developed sulfur cathodes, modified separators, optimized electrolytes and rationally designed Na anodes show significant improvement toward high-performance and long-cycle-life Na RT Na–S and Na–Se batteries. Although there is still a long way to go to before the practical application of RT Na–S and Na–Se batteries, the fast-developing advanced technologies would definitively accelerate this process.
7.2 Perspectives
According to the above discussion, significant progress has been made in the development of high-performance RT Na–S(Se) batteries for grid-scale SEES systems. However, there are still many challenges that need to be overcome before their practical application. Here, we propose a few potential research directions to promote the development of these systems.
-
(A)
Increasing the sulfur loading and sulfur utilization.
Currently, the sulfur loading and cycling performance of RT Na–S batteries are contradictory. Normally, the sulfur-based cathode used in RT Na–S batteries can only have an approximately 35%–50% sulfur loading to achieve high electrochemical performance. With a higher sulfur loading, the cycling performance will be worse for the RT Na–S batteries because the state-of-the-art cathode hosts still cannot confine a higher amount of sulfur to prevent the shuttling of Na polysulfides. Many cathode hosts for sulfur-based cathodes used in RT Na–S batteries were taken from the Li–S battery system. These hosts may be good enough to confine the Li polysulfides. However, they cannot effectively limit the long-chain soluble Na polysulfides that have much more active chemical characteristics. The cathode hosts, whether based on 3D porous carbon or a 2D material, for RT Na–S batteries need to be further developed in terms of the sulfur confinement function to realize a high sulfur loading, which is very important for reducing the cost and increasing the volumetric energy density of the batteries. Another aspect that needs to be mentioned is sulfur utilization. Some cathode hosts with good physical or chemical confinement of sulfur will combine with sulfur or Na polysulfides and no longer release them. This factor directly leads to loss of the active material (sulfur) and low initial Coulombic efficiency of the RT Na–S batteries. Hence, the development of a cathode host for RT Na–S battery cathodes in future will need to have the ability to confine sulfur and Na polysulfide as well as the ability to release them during the charging process.
-
(B)
Boosting the conversion reaction kinetics of sodium polysulfides.
Another research path is increasing the reaction kinetics of short-chain sodium polysulfides. Unlike active long-chain Na polysulfides (e.g., Na2S4, Na2S6), short-chain solid-state Na polysulfides (e.g., Na2S2, Na2S) are very passive and hard to transform back to long-chain Na polysulfides, which will also incur the loss of the specific capacity. If the redox kinetics of short-chain sodium polysulfides can be increased, especially Na2S2 ↔ Na2S, then the reaction overpotential of the RT Na–S batteries can be greatly reduced and the capacity and cycling performance thus significantly improved. This improvement could be realized with further development of cathode hosts with effective electrocatalytic functions. Moreover, catalytic functions to accelerate the reaction kinetics of short-chain Na polysulfides can also be developed by adding additives or optimizing the composition of the electrolyte, which will also be a promising direction in future.
-
(C)
Developing solid-state RT Na–S batteries.
The development of solid-state RT Na–S batteries will be a promising strategy to fulfill the safety expectations of stationary energy storage. The safety concerns arising from LEs, such as leakage, fire, or even explosion, which may contaminate the environment or threaten communities, require configuration optimization of the SEES system. RT Na–S batteries with SSEs can prevent all these risks and effectively enhance the practical application of RT Na–S batteries in different extreme environments, such as at extremely high temperatures or under abuse usage. However, many challenges, such as the sluggish ion transport inside the SSE and the uneven solid-to-solid interfacial contact between the solid electrolytes and electrodes, still need to be overcome to ensure the stable operation of solid-state RT Na–S batteries before real application.
-
(D)
Optimization of the separator, interlayer and binder.
Compared with Li-based batteries, research on other components, including separators, interlayers, and binders, in RT Na–S(Se) batteries is still in its infancy. Currently, separators based on GFs are mainly used in the RT Na–S battery system. To prevent the shuttle effect and enhance the confinement of sulfur, novel types of separators or interlayers based on 2D materials or 3D frameworks with electrocatalytic functions can be potential alternatives to GFs. Moreover, the solvent NMP used with the PVDF binder is toxic and nonenvironmentally friendly. The development of biocompatible and aqueous binders derived from, for example, wood or fertilizer that are based on a water-based slurry can effectively reduce the cost, confine the Na polysulfides on the cathode side, act as a flame retardant to reduce the safety risks, and be environmentally benign, which is worth further investigation in future. These research paths of separators, interlayers, and binders all have a bright future to further enhance the performance of the RT Na–S(Se) battery system.
Although there are still many challenges that need to be overcome for RT Na–S(Se) batteries, considering that research of RT Na–S(Se) batteries has already become one of the hottest topics and the extremely inexpensive and abundant raw materials, as well as the high operating safety, RT Na–S(Se) batteries are predicted to have significant breakthroughs in the near future and obtain a considerable part of the market share in the grid-scale energy storage field.
References
Goikolea, E., Palomares, V., Wang, S.J., et al.: Na-ion batteries: approaching old and new challenges. Adv. Energy Mater. 10, 2002055 (2020). https://doi.org/10.1002/aenm.202002055
Wang, Y.Z., Zhou, D., Palomares, V., et al.: Revitalising sodium–sulfur batteries for non-high-temperature operation: a crucial review. Energy Environ. Sci. 13, 3848–3879 (2020). https://doi.org/10.1039/d0ee02203a
Rai, A., Esplin, R., Nunn, O., et al.: The times they are a changin’: current and future trends in electricity demand and supply. Electr. J. 32, 24–32 (2019). https://doi.org/10.1016/j.tej.2019.05.017
Lachuriya, A., Kulkarni, R.: Stationary electrical energy storage technology for global energy sustainability: a review. In: 2017 international conference on nascent technologies in engineering (ICNTE), Vashi, India (2017)
Papaefthymiou, G., Dragoon, K.: Towards 100% renewable energy systems: uncapping power system flexibility. Energy Policy 92, 69–82 (2016). https://doi.org/10.1016/j.enpol.2016.01.025
Hameer, S., van Niekerk, J.L.: A review of large-scale electrical energy storage. Int. J. Energy Res. 39, 1179–1195 (2015). https://doi.org/10.1002/er.3294
Kousksou, T., Bruel, P., Jamil, A., et al.: Energy storage: applications and challenges. Sol. Energy Mater. Sol. Cells 120, 59–80 (2014). https://doi.org/10.1016/j.solmat.2013.08.015
Liu, A.M., Liang, X.Y., Ren, X.F., et al.: Recent progress in MXene-based materials for metal-sulfur and metal-air batteries: potential high-performance electrodes. Electrochem. Energy Rev. 5, 112–144 (2022). https://doi.org/10.1007/s41918-021-00110-w
Wang, H., Matios, E., Luo, J.M., et al.: Combining theories and experiments to understand the sodium nucleation behavior towards safe sodium metal batteries. Chem. Soc. Rev. 49, 3783–3805 (2020). https://doi.org/10.1039/d0cs00033g
Wang, Y.X., Zhang, B.W., Lai, W.H., et al.: Room-temperature sodium–sulfur batteries: a comprehensive review on research progress and cell chemistry. Adv. Energy Mater. 7, 1602829 (2017). https://doi.org/10.1002/aenm.201602829
Jaumaux, P., Wu, J.R., Shanmukaraj, D., et al.: Non-flammable liquid and quasi-solid electrolytes toward highly-safe alkali metal-based batteries. Adv. Funct. Mater. 31, 2008644 (2021). https://doi.org/10.1002/adfm.202008644
Liang, Y.L., Dong, H., Aurbach, D., et al.: Current status and future directions of multivalent metal-ion batteries. Nat. Energy 5, 646–656 (2020). https://doi.org/10.1038/s41560-020-0655-0
Sun, B., Xiong, P., Maitra, U., et al.: Design strategies to enable the efficient use of sodium metal anodes in high-energy batteries. Adv. Mater. 32, 1903891 (2020). https://doi.org/10.1002/adma.201903891
Perveen, T., Siddiq, M., Shahzad, N., et al.: Prospects in anode materials for sodium ion batteries: a review. Renew. Sustain. Energy Rev. 119, 109549 (2020). https://doi.org/10.1016/j.rser.2019.109549
Kumar, D., Kuhar, S.B., Kanchan, D.K.: Room temperature sodium–sulfur batteries as emerging energy source. J. Energy Storage 18, 133–148 (2018). https://doi.org/10.1016/j.est.2018.04.021
Hueso, K.B., Armand, M., Rojo, T.: High temperature sodium batteries: status, challenges and future trends. Energy Environ. Sci. 6, 734 (2013). https://doi.org/10.1039/c3ee24086j
Duan, J., Tang, X., Dai, H.F., et al.: Building safe lithium-ion batteries for electric vehicles: a review. Electrochem. Energy Rev. 3, 1–42 (2020). https://doi.org/10.1007/s41918-019-00060-4
US Geological Survey. Mineral Commodity Summaries 2022. Reston, VA (2022). https://doi.org/10.3133/mineral2022
Hans Wedepohl, K.: The composition of the continental crust. Geochim. Cosmochim. Acta 59, 1217–1232 (1995). https://doi.org/10.1016/0016-7037(95)00038-2
Haynes, W.M., Lide, D.R., Bruno, T.J.: CRC handbook of chemistry and physics. CRC Press, Boca Raton (2016). https://doi.org/10.1201/9781315380476
Ma, D.T., Li, Y.L., Yang, J.B., et al.: New strategy for polysulfide protection based on atomic layer deposition of TiO2 onto ferroelectric-encapsulated cathode: toward ultrastable free-standing room temperature sodium–sulfur batteries. Adv. Funct. Mater. 28, 1705537 (2018). https://doi.org/10.1002/adfm.201705537
Shiraz, M.H.A., Zhao, P., Liu, J.: High-performance sodium–selenium batteries enabled by microporous carbon/selenium cathode and fluoroethylene carbonate electrolyte additive. J. Power Sources 453, 227855 (2020). https://doi.org/10.1016/j.jpowsour.2020.227855
Wang, Y.X., Wang, Y.X., Wang, Y.X., et al.: Developments and perspectives on emerging high-energy-density sodium-metal batteries. Chem 5, 2547–2570 (2019). https://doi.org/10.1016/j.chempr.2019.05.026
Hwang, J.Y., Myung, S.T., Sun, Y.K.: Sodium-ion batteries: present and future. Chem. Soc. Rev. 46, 3529–3614 (2017). https://doi.org/10.1039/c6cs00776g
Kumar, D., Rajouria, S.K., Kuhar, S.B., et al.: Progress and prospects of sodium–sulfur batteries: a review. Solid State Ion. 312, 8–16 (2017). https://doi.org/10.1016/j.ssi.2017.10.004
Manthiram, A., Yu, X.W.: Ambient temperature sodium–sulfur batteries. Small 11, 2108–2114 (2015). https://doi.org/10.1002/smll.201403257
Wen, Z.Y., Hu, Y.Y., Wu, X.W., et al.: Main challenges for high performance NAS battery: materials and interfaces. Adv. Funct. Mater. 23, 1005–1018 (2013). https://doi.org/10.1002/adfm.201200473
Zhang, F., Xiong, P., Guo, X., et al.: A nitrogen, sulphur dual-doped hierarchical porous carbon with interconnected conductive polyaniline coating for high-performance sodium–selenium batteries. Energy Storage Mater. 19, 251–260 (2019). https://doi.org/10.1016/j.ensm.2019.03.019
Li, Q.Q., Liu, H.G., Yao, Z.P., et al.: Electrochemistry of selenium with sodium and lithium: kinetics and reaction mechanism. ACS Nano 10, 8788–8795 (2016). https://doi.org/10.1021/acsnano.6b04519
Huang, X.L., Zhou, C.F., He, W.D., et al.: An emerging energy storage system: advanced Na–Se batteries. ACS Nano 15, 5876–5903 (2021). https://doi.org/10.1021/acsnano.0c10078
Parant, J.P., Olazcuaga, R., Devalette, M., et al.: Sur quelques nouvelles phases de formule NaxMnO2 (x ⩽ 1). J. Solid State Chem. 3, 1–11 (1971). https://doi.org/10.1016/0022-4596(71)90001-6
Nagelberg, A.S., Worrell, W.L.: A thermodynamic study of sodium-intercalated TaS2 and TiS2. J. Solid State Chem. 29, 345–354 (1979). https://doi.org/10.1016/0022-4596(79)90191-9
Braconnier, J.J., Delmas, C., Fouassier, C., et al.: Comportement electrochimique des phases NaxCoO2. Mater. Res. Bull. 15, 1797–1804 (1980). https://doi.org/10.1016/0025-5408(80)90199-3
Yoshino, A.: The birth of the lithium-ion battery. Angew. Chem. Int. Ed. 51, 5798–5800 (2012). https://doi.org/10.1002/anie.201105006
Zubi, G., Dufo-López, R., Carvalho, M., et al.: The lithium-ion battery: state of the art and future perspectives. Renew. Sustain. Energy Rev. 89, 292–308 (2018). https://doi.org/10.1016/j.rser.2018.03.002
Yang, C.P., Xin, S., Yin, Y.X., et al.: An advanced selenium-carbon cathode for rechargeable lithium-selenium batteries. Angew. Chem. Int. Ed. 52, 8363–8367 (2013). https://doi.org/10.1002/anie.201303147
Kummer, J. T., Neill, W.: Google Patents (1968)
Xin, S., Yin, Y.X., Guo, Y.G., et al.: A high-energy room-temperature sodium–sulfur battery. Adv. Mater. 26, 1261–1265 (2014). https://doi.org/10.1002/adma.201304126
NGK Insulators (2021) About NGK history. https://www.ngk-insulators.com/en/info/history/. Accessed 23 Feb 2023
NGK Insulators (2009) The vendors’ perspective on barriers & issues encountered in U.S. deployment. https://efiling.energy.ca.gov/GetDocument.aspx?tn=50786&DocumentContentId=9147. Accessed 23 Feb 2023
Abraham, K., Rauh, R., Brummer, S.: A low temperature Na–S battery incorporating a soluble S cathode. Electrochim. Acta 23, 501–507 (1978). https://doi.org/10.1016/0013-4686(78)85027-0
Sciamanna, S.F., Lynn, S.: Sulfur solubility in pure and mixed organic solvents. Ind. Eng. Chem. Res. 27, 485–491 (1988). https://doi.org/10.1021/ie00075a019
Lu, X.C., Kirby, B.W., Xu, W., et al.: Advanced intermediate-temperature Na–S battery. Energy Environ. Sci. 6, 299–306 (2013). https://doi.org/10.1039/c2ee23606k
Xu, X.F., Zhou, D., Qin, X.Y., et al.: A room-temperature sodium–sulfur battery with high capacity and stable cycling performance. Nat. Commun. 9, 3870 (2018). https://doi.org/10.1038/s41467-018-06443-3
Yang, C.P., Yin, Y.X., Guo, Y.G.: Elemental selenium for electrochemical energy storage. J. Phys. Chem. Lett. 6, 256–266 (2015). https://doi.org/10.1021/jz502405h
Ding, J., Zhou, H., Zhang, H.L., et al.: Exceptional energy and new insight with a sodium–selenium battery based on a carbon nanosheet cathode and a pseudographite anode. Energy Environ. Sci. 10, 153–165 (2017). https://doi.org/10.1039/c6ee02274j
Chawla, N., Safa, M.E.: Sodium batteries: a review on sodium–sulfur and sodium–air batteries. Electronics 8, 1201 (2019). https://doi.org/10.3390/electronics8101201
Wang, N.N., Wang, Y.X., Bai, Z.C., et al.: High-performance room-temperature sodium–sulfur battery enabled by electrocatalytic sodium polysulfides full conversion. Energy Environ. Sci. 13, 562–570 (2020). https://doi.org/10.1039/c9ee03251g
Lee, B., Paek, E., Mitlin, D., et al.: Sodium metal anodes: emerging solutions to dendrite growth. Chem. Rev. 119, 5416–5460 (2019). https://doi.org/10.1021/acs.chemrev.8b00642
Ye, H.L., Ma, L., Zhou, Y., et al.: Amorphous MoS3 as the sulfur-equivalent cathode material for room-temperature Li–S and Na–S batteries. PNAS 114, 13091–13096 (2017). https://doi.org/10.1073/pnas.1711917114
Wang, Y.X., Yang, J.P., Lai, W.H., et al.: Achieving high-performance room-temperature sodium–sulfur batteries with S@interconnected mesoporous carbon hollow nanospheres. J. Am. Chem. Soc. 138, 16576–16579 (2016). https://doi.org/10.1021/jacs.6b08685
Guo, Q.B., Li, S., Liu, X.J., et al.: Ultrastable sodium–sulfur batteries without polysulfides formation using slit ultramicropore carbon carrier. Adv. Sci. 7, 1903246 (2020). https://doi.org/10.1002/advs.201903246
Carter, R., Oakes, L., Douglas, A., et al.: A sugar-derived room-temperature sodium sulfur battery with long term cycling stability. Nano Lett. 17, 1863–1869 (2017). https://doi.org/10.1021/acs.nanolett.6b05172
Zhou, J.H., Xu, S.M., Yang, Y.: Strategies for polysulfide immobilization in sulfur cathodes for room-temperature sodium–sulfur batteries. Small 17, 2100057 (2021). https://doi.org/10.1002/smll.202100057
Eng, A.Y.S., Wang, Y., Nguyen, D.T., et al.: Tunable nitrogen-doping of sulfur host nanostructures for stable and shuttle-free room-temperature sodium–sulfur batteries. Nano Lett. 21, 5401–5408 (2021). https://doi.org/10.1021/acs.nanolett.1c01763
Xiao, F.P., Yang, X.M., Wang, H.K., et al.: Covalent encapsulation of sulfur in a MOF-derived S, N-doped porous carbon host realized via the vapor-infiltration method results in enhanced sodium–sulfur battery performance. Adv. Energy Mater. 10, 2000931 (2020). https://doi.org/10.1002/aenm.202000931
Qiang, Z., Chen, Y.M., Xia, Y.F., et al.: Ultra-long cycle life, low-cost room temperature sodium–sulfur batteries enabled by highly doped (N, S) nanoporous carbons. Nano Energy 32, 59–66 (2017). https://doi.org/10.1016/j.nanoen.2016.12.018
Yao, Y., Chen, M.L., Xu, R., et al.: CNT interwoven nitrogen and oxygen dual-doped porous carbon nanosheets as free-standing electrodes for high-performance Na–Se and K–Se flexible batteries. Adv. Mater. 30, 1805234 (2018). https://doi.org/10.1002/adma.201805234
Liu, H.W., Pei, W., Lai, W.H., et al.: Electrocatalyzing S cathodes via multisulfiphilic sites for superior room-temperature sodium–sulfur batteries. ACS Nano 14, 7259–7268 (2020). https://doi.org/10.1021/acsnano.0c02488
Aslam, M.K., Seymour, I.D., Katyal, N., et al.: Metal chalcogenide hollow polar bipyramid prisms as efficient sulfur hosts for Na–S batteries. Nat. Commun. 11, 5242 (2020). https://doi.org/10.1038/s41467-020-19078-0
Zhang, B.W., Li, S.N., Yang, H.L., et al.: Atomically dispersed S-Fe-N4 for fast kinetics sodium–sulfur batteries via a dual function mechanism. Cell Rep. Phys. Sci. 2, 100531 (2021). https://doi.org/10.1016/j.xcrp.2021.100531
Xiao, F.P., Wang, H.K., Xu, J., et al.: Generating short-chain sulfur suitable for efficient sodium–sulfur batteries via atomic copper sites on a N, O-codoped carbon composite. Adv. Energy Mater. 11, 2100989 (2021). https://doi.org/10.1002/aenm.202100989
Mou, J.R., Li, Y.J., Liu, T., et al.: Metal–organic frameworks-derived nitrogen-doped porous carbon nanocubes with embedded Co nanoparticles as efficient sulfur immobilizers for room temperature sodium–sulfur batteries. Small Methods 5, 2100455 (2021). https://doi.org/10.1002/smtd.202100455
Zhang, B.W., Sheng, T., Liu, Y.D., et al.: Atomic cobalt as an efficient electrocatalyst in sulfur cathodes for superior room-temperature sodium–sulfur batteries. Nat. Commun. 9, 4082 (2018). https://doi.org/10.1038/s41467-018-06144-x
Lai, W.H., Wang, H., Zheng, L.R., et al.: General synthesis of single-atom catalysts for hydrogen evolution reactions and room-temperature Na–S batteries. Angew. Chem. Int. Ed. 59, 22171–22178 (2020). https://doi.org/10.1002/anie.202009400
Sajjad, M., Hussain, T., Singh, N., et al.: Superior anchoring of sodium polysulfides to the polar C2N 2D material: a potential electrode enhancer in sodium–sulfur batteries. Langmuir 36, 13104–13111 (2020). https://doi.org/10.1021/acs.langmuir.0c02616
Zhang, T., Zhang, L., Hou, Y.L.: MXenes: synthesis strategies and lithium–sulfur battery applications. eScience 2, 164–182 (2022). https://doi.org/10.1016/j.esci.2022.02.010
Reddy, R.C.K., Lin, X.M., Zeb, A., et al.: Metal–organic frameworks and their derivatives as cathodes for lithium-ion battery applications: a review. Electrochem. Energy Rev. 5, 312–347 (2022). https://doi.org/10.1007/s41918-021-00101-x
Reddy, B., Premasudha, M., Oh, K.M., et al.: Hydrothermal synthesis of MoS2/rGO composite as sulfur hosts for room temperature sodium–sulfur batteries and its electrochemical properties. J. Energy Storage 39, 102660 (2021). https://doi.org/10.1016/j.est.2021.102660
Bao, W.Z., Shuck, C.E., Zhang, W.X., et al.: Boosting performance of Na–S batteries using sulfur-doped Ti3C2Tx MXene nanosheets with a strong affinity to sodium polysulfides. ACS Nano 13, 11500–11509 (2019). https://doi.org/10.1021/acsnano.9b04977
Ye, C., Jiao, Y., Chao, D.L., et al.: Electron-state confinement of polysulfides for highly stable sodium–sulfur batteries. Adv. Mater. 32, 1907557 (2020). https://doi.org/10.1002/adma.201907557
Wang, Y.X., Lai, Y.Y., Chu, J., et al.: Tunable electrocatalytic behavior of sodiated MoS2 active sites toward efficient sulfur redox reactions in room-temperature Na–S batteries. Adv. Mater. 33, 2100229 (2021). https://doi.org/10.1002/adma.202100229
Wang, J.L., Yang, J., Nuli, Y.N., et al.: Room temperature Na/S batteries with sulfur composite cathode materials. Electrochem. Commun. 9, 31–34 (2007). https://doi.org/10.1016/j.elecom.2006.08.029
Hwang, T.H., Jung, D.S., Kim, J.S., et al.: One-dimensional carbon–sulfur composite fibers for Na–S rechargeable batteries operating at room temperature. Nano Lett. 13, 4532–4538 (2013). https://doi.org/10.1021/nl402513x
Ghosh, A., Shukla, S., Monisha, M., et al.: Sulfur copolymer: a new cathode structure for room-temperature sodium–sulfur batteries. ACS Energy Lett. 2, 2478–2485 (2017). https://doi.org/10.1021/acsenergylett.7b00714
Yu, Y., Wang, Z., Hou, Z., et al.: 3D printing of hierarchical graphene lattice for advanced Na metal anodes. ACS Appl. Energy Mater. 2, 3869–3877 (2019). https://doi.org/10.1021/acsaem.9b00540
Bloi, L.M., Pampel, J., Dörfler, S., et al.: Sodium sulfide cathodes superseding hard carbon pre-sodiation for the production and operation of sodium–sulfur batteries at room temperature. Adv. Energy Mater. 10, 1903245 (2020). https://doi.org/10.1002/aenm.201903245
Fan, X.L., Yue, J., Han, F.D., et al.: High-performance all-solid-state Na–S battery enabled by casting–annealing technology. ACS Nano 12, 3360–3368 (2018). https://doi.org/10.1021/acsnano.7b08856
Wang, C.L., Wang, H., Hu, X.F., et al.: Frogspawn-coral-like hollow sodium sulfide nanostructured cathode for high-rate performance sodium–sulfur batteries. Adv. Energy Mater. 9, 1803251 (2019). https://doi.org/10.1002/aenm.201803251
Ghosh, A., Kumar, A., Das, T., et al.: Lewis acid–base interactions between polysulfides and boehmite enables stable room-temperature sodium–sulfur batteries. Adv. Funct. Mater. 30, 2005669 (2020). https://doi.org/10.1002/adfm.202005669
Ghosh, A., Kumar, A., Roy, A., et al.: Three-dimensionally reinforced freestanding cathode for high-energy room-temperature sodium–sulfur batteries. ACS Appl. Mater. Interfaces 11, 14101–14109 (2019). https://doi.org/10.1021/acsami.9b00203
Ponrouch, A., Monti, D., Boschin, A., et al.: Non-aqueous electrolytes for sodium-ion batteries. J. Mater. Chem. A 3, 22–42 (2015). https://doi.org/10.1039/c4ta04428b
Cresce, A.V., Russell, S.M., Borodin, O., et al.: Solvation behavior of carbonate-based electrolytes in sodium ion batteries. Phys. Chem. Chem. Phys. 19, 574–586 (2017). https://doi.org/10.1039/c6cp07215a
Abraham, K.M.: How comparable are sodium-ion batteries to lithium-ion counterparts? ACS Energy Lett. 5, 3544–3547 (2020). https://doi.org/10.1021/acsenergylett.0c02181
Eshetu, G.G., Elia, G.A., Armand, M., et al.: Electrolytes and interphases in sodium-based rechargeable batteries: recent advances and perspectives. Adv. Energy Mater. 10, 2000093 (2020). https://doi.org/10.1002/aenm.202000093
Ponrouch, A., Marchante, E., Courty, M., et al.: In search of an optimized electrolyte for Na-ion batteries. Energy Environ. Sci. 5, 8572 (2012). https://doi.org/10.1039/c2ee22258b
Zhao, X.M., Zhu, Q., Xu, S.D., et al.: Fluoroethylene carbonate as an additive in a carbonates-based electrolyte for enhancing the specific capacity of room-temperature sodium–sulfur cell. J. Electroanal. Chem. 832, 392–398 (2019). https://doi.org/10.1016/j.jelechem.2018.11.021
Lee, H.G., Lee, J.T., Eom, K.: Improving the stability of an RT-NaS battery via in situ electrochemical formation of protective SEI on a sulfur-carbon composite cathode. Adv. Sustainable Syst. 2, 1800076 (2018). https://doi.org/10.1002/adsu.201800076
Zhao, X.M., Yan, Y.W., Ren, X.X., et al.: Trimethyl phosphate for nonflammable carbonate-based electrolytes for safer room-temperature sodium–sulfur batteries. ChemElectroChem 6, 1229–1234 (2019). https://doi.org/10.1002/celc.201801833
Wu, J.X., Liu, J.P., Lu, Z.H., et al.: Non-flammable electrolyte for dendrite-free sodium–sulfur battery. Energy Storage Mater. 23, 8–16 (2019). https://doi.org/10.1016/j.ensm.2019.05.045
Chen, W.J., Zhao, C.X., Li, B.Q., et al.: A mixed ether electrolyte for lithium metal anode protection in working lithium–sulfur batteries. Energy Environ. Mater. 3, 160–165 (2020). https://doi.org/10.1002/eem2.12073
Zhang, B.H., Wu, J.F., Gu, J.K., et al.: The fundamental understanding of lithium polysulfides in ether-based electrolyte for lithium-sulfur batteries. ACS Energy Lett. 6, 537–546 (2021). https://doi.org/10.1021/acsenergylett.0c02527
di Lecce, D., Minnetti, L., Polidoro, D., et al.: Triglyme-based electrolyte for sodium-ion and sodium–sulfur batteries. Ionics 25, 3129–3141 (2019). https://doi.org/10.1007/s11581-019-02878-w
Eng, A.Y.S., Kumar, V., Zhang, Y.W., et al.: Room-temperature sodium–sulfur batteries and beyond: realizing practical high energy systems through anode, cathode, and electrolyte engineering. Adv. Energy Mater. 11, 2003493 (2021). https://doi.org/10.1002/aenm.202003493
Zhang, J., Wang, D.W., Lv, W., et al.: Ethers illume sodium-based battery chemistry: uniqueness, surprise, and challenges. Adv. Energy Mater. 8, 1801361 (2018). https://doi.org/10.1002/aenm.201801361
Li, P.R., Ma, L., Wu, T.P., et al.: Chemical immobilization and conversion of active polysulfides directly by copper current collector: a new approach to enabling stable room-temperature Li–S and Na–S batteries. Adv. Energy Mater. 8, 1800624 (2018). https://doi.org/10.1002/aenm.201800624
Zhang, H., Diemant, T., Qin, B.S., et al.: Solvent-dictated sodium sulfur redox reactions: investigation of carbonate and ether electrolytes. Energies 13, 836 (2020). https://doi.org/10.3390/en13040836
Ryu, H., Kim, T., Kim, K., et al.: Discharge reaction mechanism of room-temperature sodium–sulfur battery with tetra ethylene glycol dimethyl ether liquid electrolyte. J. Power Sources 196, 5186–5190 (2011). https://doi.org/10.1016/j.jpowsour.2011.01.109
Liu, H.W., Lai, W.H., Yang, Q.R., et al.: Understanding sulfur redox mechanisms in different electrolytes for room-temperature Na–S batteries. Nano Micro Lett. 13, 1–14 (2021). https://doi.org/10.1007/s40820-021-00648-w
Kohl, M., Borrmann, F., Althues, H., et al.: Hard carbon anodes and novel electrolytes for long-cycle-life room temperature sodium–sulfur full cell batteries. Adv. Energy Mater. 6, 1502185 (2016). https://doi.org/10.1002/aenm.201502185
Lin, Z., Liu, Z.C., Fu, W.J., et al.: Phosphorous pentasulfide as a novel additive for high-performance lithium–sulfur batteries. Adv. Funct. Mater. 23, 1064–1069 (2013). https://doi.org/10.1002/adfm.201200696
Yu, X.W., Manthiram, A.: Room-temperature sodium–sulfur batteries with liquid-phase sodium polysulfide catholytes and binder-free multiwall carbon nanotube fabric electrodes. J. Phys. Chem. C 118, 22952–22959 (2014). https://doi.org/10.1021/jp507655u
Zhou, D., Tang, X., Guo, X., et al.: Polyolefin-based Janus separator for rechargeable sodium batteries. Angew. Chem. Int. Ed. 59, 16725–16734 (2020). https://doi.org/10.1002/anie.202007008
Medenbach, L., Hartmann, P., Janek, J., et al.: A sodium polysulfide battery with liquid/solid electrolyte: improving sulfur utilization using P2S5 as additive and tetramethylurea as catholyte solvent. Energy Technol. 8, 1901200 (2020). https://doi.org/10.1002/ente.201901200
Yu, X.W., Manthiram, A.: Ambient-temperature sodium–sulfur batteries with a sodiated nafion membrane and a carbon nanofiber-activated carbon composite electrode. Adv. Energy Mater. 5, 1500350 (2015). https://doi.org/10.1002/aenm.201500350
Kumar, A., Ghosh, A., Roy, A., et al.: High-energy density room temperature sodium–sulfur battery enabled by sodium polysulfide catholyte and carbon cloth current collector decorated with MnO2 nanoarrays. Energy Storage Mater. 20, 196–202 (2019). https://doi.org/10.1016/j.ensm.2018.11.031
Kumar, A., Ghosh, A., Forsyth, M., et al.: Free-radical catalysis and enhancement of the redox kinetics for room-temperature sodium–sulfur batteries. ACS Energy Lett. 5, 2112–2121 (2020). https://doi.org/10.1021/acsenergylett.0c00913
Basile, A., Hilder, M., Makhlooghiazad, F., et al.: Sodium energy storage: ionic liquids and organic ionic plastic crystals: advanced electrolytes for safer high performance sodium energy storage technologies (adv. energy mater. 17/2018). Adv. Energy Mater. 8, 1870078 (2018). https://doi.org/10.1002/aenm.201870078
Stettner, T., Balducci, A.: Protic ionic liquids in energy storage devices: past, present and future perspective. Energy Storage Mater. 40, 402–414 (2021). https://doi.org/10.1016/j.ensm.2021.04.036
Yang, Q.W., Zhang, Z.Q., Sun, X.G., et al.: Ionic liquids and derived materials for lithium and sodium batteries. Chem. Soc. Rev. 47, 2020–2064 (2018). https://doi.org/10.1039/c7cs00464h
Nozawa, R., Harimoto, R., Tsuchiya, M., et al.: Sodium-sulfur batteries with room-temperature ionic liquid electrolytes. Electrochem Soc Meeting Abstr 222. 2, 22 (2012)
Wei, S.Y., Xu, S.M., Agrawral, A., et al.: A stable room-temperature sodium–sulfur battery. Nat. Commun. 7, 11722 (2016). https://doi.org/10.1038/ncomms11722
Wang, D., Hwang, J., Chen, C.Y., et al.: A β-alumina/inorganic ionic liquid dual electrolyte for intermediate-temperature sodium–sulfur batteries. Adv. Funct. Mater. 31, 2105524 (2021). https://doi.org/10.1002/adfm.202105524
Ruiz-Martínez, D., Gómez, R.: The liquid ammoniate of sodium iodide as an alternative electrolyte for sodium ion batteries: the case of titanium dioxide nanotube electrodes. Energy Storage Mater. 22, 424–432 (2019). https://doi.org/10.1016/j.ensm.2019.07.036
Ruiz-Martínez, D., Kovacs, A., Gómez, R.: Development of novel inorganic electrolytes for room temperature rechargeable sodium metal batteries. Energy Environ. Sci. 10, 1936–1941 (2017). https://doi.org/10.1039/c7ee01735a
Jeong, G., Kim, H., Sug Lee, H., et al.: A room-temperature sodium rechargeable battery using an SO2− based nonflammable inorganic liquid catholyte. Sci. Rep. 5, 12827 (2015). https://doi.org/10.1038/srep12827
Iermakova, D.I., Dugas, R., Palacín, M.R., et al.: On the comparative stability of Li and Na metal anode interfaces in conventional alkyl carbonate electrolytes. J. Electrochem. Soc. 162, A7060–A7066 (2015). https://doi.org/10.1149/2.0091513jes
Pan, K.H., Lu, H.Y., Zhong, F.P., et al.: Understanding the electrochemical compatibility and reaction mechanism on Na metal and hard carbon anodes of PC-based electrolytes for sodium-ion batteries. ACS Appl. Mater. Interfaces 10, 39651–39660 (2018). https://doi.org/10.1021/acsami.8b13236
Chen, X., Shen, X., Li, B., et al.: Ion–solvent complexes promote gas evolution from electrolytes on a sodium metal anode. Angew. Chem. Int. Ed. 57, 734–737 (2018). https://doi.org/10.1002/anie.201711552
Zhou, L., Cao, Z., Zhang, J., et al.: Engineering sodium-ion solvation structure to stabilize sodium anodes: universal strategy for fast-charging and safer sodium-ion batteries. Nano Lett. 20, 3247–3254 (2020). https://doi.org/10.1021/acs.nanolett.9b05355
Liu, Q., Wu, F., Mu, D.B., et al.: A theoretical study on Na+ solvation in carbonate ester and ether solvents for sodium-ion batteries. Phys. Chem. Chem. Phys. 22, 2164–2175 (2020). https://doi.org/10.1039/c9cp05636j
Westman, K., Dugas, R., Jankowski, P., et al.: Diglyme based electrolytes for sodium-ion batteries. ACS Appl. Energy Mater. 1, 2671–2680 (2018). https://doi.org/10.1021/acsaem.8b00360
Seh, Z.W., Sun, J., Sun, Y.M., et al.: A highly reversible room-temperature sodium metal anode. ACS Cent. Sci. 1, 449–455 (2015). https://doi.org/10.1021/acscentsci.5b00328
Goktas, M., Bolli, C., Buchheim, J., et al.: Stable and unstable diglyme-based electrolytes for batteries with sodium or graphite as electrode. ACS Appl. Mater. Interfaces 11, 32844–32855 (2019). https://doi.org/10.1021/acsami.9b06760
Le, P.M.L., Vo, T.D., Pan, H.L., et al.: Excellent cycling stability of sodium anode enabled by a stable solid electrolyte interphase formed in ether-based electrolytes. Adv. Funct. Mater. 30, 2001151 (2020). https://doi.org/10.1002/adfm.202001151
Kamysbayev, V., Filatov, A.S., Hu, H.C., et al.: Covalent surface modifications and superconductivity of two-dimensional metal carbide MXenes. Science 369, 979–983 (2020). https://doi.org/10.1126/science.aba8311
Geysens, P., Rangasamy, V.S., Thayumanasundaram, S., et al.: Solvation structure of sodium bis(fluorosulfonyl)imide-glyme solvate ionic liquids and its influence on cycling of Na-MNC cathodes. J. Phys. Chem. B 122, 275–289 (2018). https://doi.org/10.1021/acs.jpcb.7b10158
Schafzahl, L., Hanzu, I., Wilkening, M., et al.: An electrolyte for reversible cycling of sodium metal and intercalation compounds. Chemsuschem 10, 401–408 (2017). https://doi.org/10.1002/cssc.201601222
Lee, J., Lee, Y., Lee, J., et al.: Ultraconcentrated sodium bis(fluorosulfonyl)imide-based electrolytes for high-performance sodium metal batteries. ACS Appl. Mater. Interfaces 9, 3723–3732 (2017). https://doi.org/10.1021/acsami.6b14878
Cao, R.G., Mishra, K., Li, X.L., et al.: Enabling room temperature sodium metal batteries. Nano Energy 30, 825–830 (2016). https://doi.org/10.1016/j.nanoen.2016.09.013
Galle Kankanamge, S.R., Li, K., Fulfer, K.D., et al.: Mechanism behind the unusually high conductivities of high concentrated sodium ion glyme-based electrolytes. J. Phys. Chem. C 122, 25237–25246 (2018). https://doi.org/10.1021/acs.jpcc.8b06991
Zheng, J.M., Chen, S.R., Zhao, W.G., et al.: Extremely stable sodium metal batteries enabled by localized high-concentration electrolytes. ACS Energy Lett. 3, 315–321 (2018). https://doi.org/10.1021/acsenergylett.7b01213
Terada, S., Susa, H., Tsuzuki, S., et al.: Dissociation and diffusion of glyme-sodium bis(trifluoromethanesulfonyl)amide complexes in hydrofluoroether-based electrolytes for sodium batteries. J. Phys. Chem. C 120, 23339–23350 (2016). https://doi.org/10.1021/acs.jpcc.6b06804
Lourenço, T.C., Dias, L.G., da Silva, J.L.F.: Theoretical investigation of the Na+ transport mechanism and the performance of ionic liquid-based electrolytes in sodium-ion batteries. ACS Appl. Energy Mater. 4, 4444–4458 (2021). https://doi.org/10.1021/acsaem.1c00059
Chen, F.F., Howlett, P., Forsyth, M.: Na-ion solvation and high transference number in superconcentrated ionic liquid electrolytes: a theoretical approach. J. Phys. Chem. C 122, 105–114 (2018). https://doi.org/10.1021/acs.jpcc.7b09322
Rakov, D.A., Chen, F.F., Ferdousi, S.A., et al.: Engineering high-energy-density sodium battery anodes for improved cycling with superconcentrated ionic-liquid electrolytes. Nat. Mater. 19, 1096–1101 (2020). https://doi.org/10.1038/s41563-020-0673-0
Yang, H., Hwang, J., Wang, Y.S., et al.: N-ethyl-N-propylpyrrolidinium bis(fluorosulfonyl)amide ionic liquid electrolytes for sodium secondary batteries: effects of Na ion concentration. J. Phys. Chem. C 123, 22018–22026 (2019). https://doi.org/10.1021/acs.jpcc.9b05941
Forsyth, M., Hilder, M., Zhang, Y.F., et al.: Tuning sodium interfacial chemistry with mixed-anion ionic liquid electrolytes. ACS Appl. Mater. Interfaces 11, 43093–43106 (2019). https://doi.org/10.1021/acsami.9b12913
Basile, A., Ferdousi, S.A., Makhlooghiazad, F., et al.: Beneficial effect of added water on sodium metal cycling in super concentrated ionic liquid sodium electrolytes. J. Power Sources 379, 344–349 (2018). https://doi.org/10.1016/j.jpowsour.2018.01.044
Ferdousi, S.A., O’Dell, L.A., Hilder, M., et al.: SEI formation on sodium metal electrodes in superconcentrated ionic liquid electrolytes and the effect of additive water. ACS Appl. Mater. Interfaces 13, 5706–5720 (2021). https://doi.org/10.1021/acsami.0c18119
Ferdousi, S.A., Hilder, M., Basile, A., et al.: Water as an effective additive for high-energy-density Na metal batteries? Studies in a superconcentrated ionic liquid electrolyte. Chemsuschem 12, 1700–1711 (2019). https://doi.org/10.1002/cssc.201802988
Sun, H., Zhu, G.Z., Xu, X.T., et al.: A safe and non-flammable sodium metal battery based on an ionic liquid electrolyte. Nat. Commun. 10, 3302 (2019). https://doi.org/10.1038/s41467-019-11102-2
Manohar, C.V., Raj, K.A., Kar, M., et al.: Stability enhancing ionic liquid hybrid electrolyte for NVP@C cathode based sodium batteries. Sustain. Energy Fuels 2, 566–576 (2018). https://doi.org/10.1039/c7se00537g
Hu, X.F., Matios, E., Zhang, Y.W., et al.: Deeply cycled sodium metal anodes at low temperature and in lean electrolyte conditions. Angew. Chem. Int. Ed. 60, 5978–5983 (2021). https://doi.org/10.1002/anie.202014241
Rodriguez, R., Loeffler, K.E., Nathan, S.S., et al.: In situ optical imaging of sodium electrodeposition: effects of fluoroethylene carbonate. ACS Energy Lett. 2, 2051–2057 (2017). https://doi.org/10.1021/acsenergylett.7b00500
Bouibes, A., Takenaka, N., Fujie, T., et al.: Concentration effect of fluoroethylene carbonate on the formation of solid electrolyte interphase layer in sodium-ion batteries. ACS Appl. Mater. Interfaces 10, 28525–28532 (2018). https://doi.org/10.1021/acsami.8b07530
Han, B., Zou, Y.C., Zhang, Z., et al.: Probing the Na metal solid electrolyte interphase via cryo-transmission electron microscopy. Nat. Commun. 12, 3066 (2021). https://doi.org/10.1038/s41467-021-23368-6
Mogensen, R., Brandell, D., Younesi, R.: Solubility of the solid electrolyte interphase (SEI) in sodium ion batteries. ACS Energy Lett. 1, 1173–1178 (2016). https://doi.org/10.1021/acsenergylett.6b00491
Zhu, M., Li, L.L., Zhang, Y.J., et al.: An in situ formed stable interface layer for high-performance sodium metal anode in a non-flammable electrolyte. Energy Storage Mater. 42, 145–153 (2021). https://doi.org/10.1016/j.ensm.2021.07.012
Dugas, R., Ponrouch, A., Gachot, G., et al.: Na reactivity toward carbonate-based electrolytes: the effect of FEC as additive. J. Electrochem. Soc. 163, A2333–A2339 (2016). https://doi.org/10.1149/2.0981610jes
Zhu, M., Zhang, Y., Yu, F., et al.: Stable sodium metal anode enabled by an interface protection layer rich in organic sulfide salt. Nano Lett. 21, 619–627 (2020). https://doi.org/10.1021/acs.nanolett.0c04158
Wang, H., Wang, C.L., Matios, E., et al.: Facile stabilization of the sodium metal anode with additives: unexpected key role of sodium polysulfide and adverse effect of sodium nitrate. Angew. Chem. Int. Ed. 57, 7734–7737 (2018). https://doi.org/10.1002/anie.201801818
Zhang, Q., Lu, Y.Y., Miao, L.C., et al.: An alternative to lithium metal anodes: non-dendritic and highly reversible sodium metal anodes for Li–Na hybrid batteries. Angew. Chem. Int. Ed. 57, 14796–14800 (2018). https://doi.org/10.1002/anie.201808592
Shi, Q.W., Zhong, Y.R., Wu, M., et al.: High-performance sodium metal anodes enabled by a bifunctional potassium salt. Angew. Chem. Int. Ed. 57, 9069–9072 (2018). https://doi.org/10.1002/anie.201803049
Chen, X., Shen, X., Hou, T.Z., et al.: Ion-solvent chemistry-inspired cation-additive strategy to stabilize electrolytes for sodium-metal batteries. Chem 6, 2242–2256 (2020). https://doi.org/10.1016/j.chempr.2020.06.036
Zheng, X.Y., Fu, H.Y., Hu, C.C., et al.: Toward a stable sodium metal anode in carbonate electrolyte: a compact, inorganic alloy interface. J. Phys. Chem. Lett. 10, 707–714 (2019). https://doi.org/10.1021/acs.jpclett.8b03536
Fang, W., Jiang, R., Zheng, H., et al.: Stable sodium metal anode enhanced by advanced electrolytes with SbF3 additive. Rare Met. 40, 433–439 (2021). https://doi.org/10.1007/s12598-020-01576-1
Fang, W., Jiang, H., Zheng, Y., et al.: A bilayer interface formed in high concentration electrolyte with SbF3 additive for long-cycle and high-rate sodium metal battery. J. Power Sources 455, 227956 (2020). https://doi.org/10.1016/j.jpowsour.2020.227956
Cheng, X.L., Pan, J., Zhao, Y., et al.: Gel polymer electrolytes for electrochemical energy storage. Adv. Energy Mater. 8, 1702184 (2018). https://doi.org/10.1002/aenm.201702184
Kumar, D.: Effect of organic solvent addition on electrochemical properties of ionic liquid based Na+ conducting gel electrolytes. Solid State Ion. 318, 65–70 (2018). https://doi.org/10.1016/j.ssi.2017.09.006
Zhou, D., Shanmukaraj, D., Tkacheva, A., et al.: Polymer electrolytes for lithium-based batteries: advances and prospects. Chem 5, 2326–2352 (2019). https://doi.org/10.1016/j.chempr.2019.05.009
Ma, L.B., Cui, J., Yao, S.S., et al.: Dendrite-free lithium metal and sodium metal batteries. Energy Storage Mater. 27, 522–554 (2020). https://doi.org/10.1016/j.ensm.2019.12.014
Tian, L.Y., Huang, X.B., Tang, X.Z.: Single-ionic gel polymer electrolyte based on polyvinylidene fluoride and fluorine-containing ionomer. Eur. Polym. J. 40, 735–742 (2004). https://doi.org/10.1016/j.eurpolymj.2003.12.006
Ryu, H., Ahn, H., Kim, K., et al.: Self-discharge of lithium-sulfur cells using stainless-steel current-collectors. J. Power Sources 140, 365–369 (2005). https://doi.org/10.1016/j.jpowsour.2004.08.039
Park, C.W., Ahn, J.H., Ryu, H.S., et al.: Room-temperature solid-state sodium/sulfur battery. Electrochem. Solid-State Lett. 9, A123 (2006). https://doi.org/10.1149/1.2164607
Kim, J.S., Ahn, H.J., Kim, I.P., et al.: The short-term cycling properties of Na/PVdF/S battery at ambient temperature. J. Solid State Electrochem. 12, 861–865 (2008). https://doi.org/10.1007/s10008-008-0504-8
Kumar, D., Suleman, M., Hashmi, S.A.: Studies on poly(vinylidene fluoride-co-hexafluoropropylene) based gel electrolyte nanocomposite for sodium–sulfur batteries. Solid State Ion. 202, 45–53 (2011). https://doi.org/10.1016/j.ssi.2011.09.001
Lim, D.H., Agostini, M., Ahn, J.H., et al.: An electrospun nanofiber membrane as gel-based electrolyte for room-temperature sodium–sulfur batteries. Energy Technol. 6, 1214–1219 (2018). https://doi.org/10.1002/ente.201800170
Manuel, J., Zhao, X.H., Cho, K.K., et al.: Ultralong life organic sodium ion batteries using a polyimide/multiwalled carbon nanotubes nanocomposite and gel polymer electrolyte. ACS Sustain. Chem. Eng. 6, 8159–8166 (2018). https://doi.org/10.1021/acssuschemeng.7b04561
Yi, Q., Zhang, W.Q., Li, S.Q., et al.: Durable sodium battery with a flexible Na3Zr2Si2PO12–PVDF–HFP composite electrolyte and sodium/carbon cloth anode. ACS Appl. Mater. Interfaces 10, 35039–35046 (2018). https://doi.org/10.1021/acsami.8b09991
Zhang, J.J., Wen, H.J., Yue, L.P., et al.: In situ formation of polysulfonamide supported poly(ethylene glycol) divinyl ether based polymer electrolyte toward monolithic sodium ion batteries. Small 13, 1601530 (2017). https://doi.org/10.1002/smll.201601530
Wang, P., Zhang, H.R., Chai, J.C., et al.: A novel single-ion conducting gel polymer electrolyte based on polymeric sodium tartaric acid borate for elevated-temperature sodium metal batteries. Solid State Ion. 337, 140–146 (2019). https://doi.org/10.1016/j.ssi.2019.04.022
Yu, Q.P., Lu, Q.W., Qi, X.G., et al.: Liquid electrolyte immobilized in compact polymer matrix for stable sodium metal anodes. Energy Storage Mater. 23, 610–616 (2019). https://doi.org/10.1016/j.ensm.2019.03.011
Rani, M.A.A., Hwang, J., Matsumoto, K., et al.: Poly(vinyl chloride) ionic liquid polymer electrolyte based on bis(fluorosulfonyl)amide for sodium secondary batteries. J. Electrochem. Soc. 164, H5031–H5035 (2017). https://doi.org/10.1149/2.0221708jes
Fdz de Anastro, A., Lago, N., Berlanga, C., et al.: Poly(ionic liquid) iongel membranes for all solid-state rechargeable sodium battery. J. Membr. Sci. 582, 435–441 (2019). https://doi.org/10.1016/j.memsci.2019.02.074
Zhou, D., Chen, Y., Li, B.H., et al.: A stable quasi-solid-state sodium–sulfur battery. Angew. Chem. Int. Ed. 57, 10168–10172 (2018). https://doi.org/10.1002/anie.201805008
Xu, X.F., Lin, K., Zhou, D., et al.: Quasi-solid-state dual-ion sodium metal batteries for low-cost energy storage. Chem 6, 902–918 (2020). https://doi.org/10.1016/j.chempr.2020.01.008
Yang, J.F., Zhang, M., Chen, Z., et al.: Flame-retardant quasi-solid polymer electrolyte enabling sodium metal batteries with highly safe characteristic and superior cycling stability. Nano Res. 12, 2230–2237 (2019). https://doi.org/10.1007/s12274-019-2369-9
Gao, Y., Chen, G., Wang, X., et al.: PY13FSI-infiltrated SBA-15 as nonflammable and high ion-conductive ionogel electrolytes for quasi-solid-state sodium-ion batteries. ACS Appl. Mater. Interfaces 12, 22981–22991 (2020). https://doi.org/10.1021/acsami.0c04878
Kim, J.I., Choi, Y., Chung, K.Y., et al.: A structurable gel-polymer electrolyte for sodium ion batteries. Adv. Funct. Mater. 27, 1701768 (2017). https://doi.org/10.1002/adfm.201701768
Lei, D.N., He, Y.B., Huang, H.J., et al.: Cross-linked beta alumina nanowires with compact gel polymer electrolyte coating for ultra-stable sodium metal battery. Nat. Commun. 10, 4244 (2019). https://doi.org/10.1038/s41467-019-11960-w
Luo, C.Z., Shen, T., Ji, H.Q., et al.: Mechanically robust gel polymer electrolyte for an ultrastable sodium metal battery. Small 16, 1906208 (2020). https://doi.org/10.1002/smll.201906208
Mendes, T.C., Zhang, X.M., Wu, Y.T., et al.: Supported ionic liquid gel membrane electrolytes for a safe and flexible sodium metal battery. ACS Sustain. Chem. Eng. 7, 3722–3726 (2019). https://doi.org/10.1021/acssuschemeng.8b06212
Ji, X., Hou, S., Wang, P.F., et al.: Solid-state electrolyte design for lithium dendrite suppression. Adv. Mater. 32, 2002741 (2020). https://doi.org/10.1002/adma.202002741
Wu, J.H., Shen, L., Zhang, Z.H., et al.: All-solid-state lithium batteries with sulfide electrolytes and oxide cathodes. Electrochem. Energy Rev. 4, 101–135 (2021). https://doi.org/10.1007/s41918-020-00081-4
Liu, Y.K., Li, J., Shen, Q.Y., et al.: Advanced characterizations and measurements for sodium-ion batteries with NASICON-type cathode materials. eScience 2, 10–31 (2022). https://doi.org/10.1016/j.esci.2021.12.008
Bohnke, O.: Conductivity measurements on nasicon and nasicon-modified materials. Solid State Ion. 122, 127–136 (1999). https://doi.org/10.1016/s0167-2738(99)00062-4
Chi, C., Katsui, H. and Goto, T.: Effect of Li addition on the formation of Na-β/βʹʹ-alumina film by laser chemical vapor deposition. Ceram. Int. 43, 1278–1283 (2017). https://doi.org/10.1016/j.ceramint.2016.10.077
Banerjee, A., Park, K.H., Heo, J.W., et al.: Na3SbS4: a solution processable sodium superionic conductor for all-solid-state sodium-ion batteries. Angew. Chem. Int. Ed. 55, 9634–9638 (2016). https://doi.org/10.1002/anie.201604158
Goodenough, J.B., Hong, H.Y.P., Kafalas, J.A.: Fast Na+-ion transport in skeleton structures. Mater. Res. Bull. 11, 203–220 (1976). https://doi.org/10.1016/0025-5408(76)90077-5
Guin, M., Tietz, F., Guillon, O.: New promising NASICON material as solid electrolyte for sodium-ion batteries: correlation between composition, crystal structure and ionic conductivity of Na3+xSc2SixP3–xO12. Solid State Ion. 293, 18–26 (2016). https://doi.org/10.1016/j.ssi.2016.06.005
Song, S.F., Duong, H.M., Korsunsky, A.M., et al.: A Na+ superionic conductor for room-temperature sodium batteries. Sci. Rep. 6, 32330 (2016). https://doi.org/10.1038/srep32330
Yu, X.W., Manthiram, A.: Sodium–sulfur batteries with a polymer-coated NASICON-type sodium-ion solid electrolyte. Matter 1, 439–451 (2019). https://doi.org/10.1016/j.matt.2019.03.008
Wenzel, S., Metelmann, H., Raiß, C., et al.: Thermodynamics and cell chemistry of room temperature sodium/sulfur cells with liquid and liquid/solid electrolyte. J. Power Sources 243, 758–765 (2013). https://doi.org/10.1016/j.jpowsour.2013.05.194
Kim, I., Park, J.Y., Kim, C.H., et al.: A room temperature Na/S battery using a β″ alumina solid electrolyte separator, tetraethylene glycol dimethyl ether electrolyte, and a S/C composite cathode. J. Power Sources 301, 332–337 (2016). https://doi.org/10.1016/j.jpowsour.2015.09.120
Yue, J., Han, F.D., Fan, X.L., et al.: High-performance all-inorganic solid-state sodium–sulfur battery. ACS Nano 11, 4885–4891 (2017). https://doi.org/10.1021/acsnano.7b01445
Tanibata, N., Tsukasaki, H., Deguchi, M., et al.: Characterization of sulfur nanocomposite electrodes containing phosphorus sulfide for high-capacity all-solid-state Na/S batteries. Solid State Ion. 311, 6–13 (2017). https://doi.org/10.1016/j.ssi.2017.08.022
Wan, H.L., Weng, W., Han, F.D., et al.: Bio-inspired nanoscaled electronic/ionic conduction networks for room-temperature all-solid-state sodium–sulfur battery. Nano Today 33, 100860 (2020). https://doi.org/10.1016/j.nantod.2020.100860
Ren, Y.X., Hortance, N., McBride, J., et al.: Sodium–sulfur batteries enabled by a protected inorganic/organic hybrid solid electrolyte. ACS Energy Lett. 6, 345–353 (2021). https://doi.org/10.1021/acsenergylett.0c02494
Yue, L.P., Ma, J., Zhang, J.J., et al.: All solid-state polymer electrolytes for high-performance lithium ion batteries. Energy Storage Mater. 5, 139–164 (2016). https://doi.org/10.1016/j.ensm.2016.07.003
Chen, R.J., Qu, W.J., Guo, X., et al.: The pursuit of solid-state electrolytes for lithium batteries: from comprehensive insight to emerging horizons. Mater. Horiz. 3, 487–516 (2016). https://doi.org/10.1039/c6mh00218h
Meyer, W.H.: Polymer electrolytes for lithium-ion batteries. Adv. Mater. 10, 439–448 (1998). https://doi.org/10.1002/(sici)1521-4095(199804)10:6439:aid-adma439%3e3.0.co;2-i
Park, C.W., Ryu, H.S., Kim, K.W., et al.: Discharge properties of all-solid sodium–sulfur battery using poly (ethylene oxide) electrolyte. J. Power Sources 165, 450–454 (2007). https://doi.org/10.1016/j.jpowsour.2006.11.083
Lin, D.C., Liu, W., Liu, Y.Y., et al.: High ionic conductivity of composite solid polymer electrolyte via in situ synthesis of monodispersed SiO2 nanospheres in poly(ethylene oxide). Nano Lett. 16, 459–465 (2016). https://doi.org/10.1021/acs.nanolett.5b04117
Zhu, T.C., Dong, X.L., Liu, Y., et al.: An all-solid-state sodium–sulfur battery using a sulfur/carbonized polyacrylonitrile composite cathode. ACS Appl. Energy Mater. 2, 5263–5271 (2019). https://doi.org/10.1021/acsaem.9b00953
Quartarone, E., Mustarelli, P.: Electrolytes for solid-state lithium rechargeable batteries: recent advances and perspectives. Chem. Soc. Rev. 40, 2525 (2011). https://doi.org/10.1039/c0cs00081g
Zhang, Z.Z., Xu, K.Q., Rong, X.H., et al.: Na3.4Zr1.8Mg0.2Si2PO12 filled poly(ethylene oxide)/Na(CF3SO2)2N as flexible composite polymer electrolyte for solid-state sodium batteries. J. Power Sources 372, 270–275 (2017). https://doi.org/10.1016/j.jpowsour.2017.10.083
Xu, X.Y., Li, Y.Y., Cheng, J., et al.: Composite solid electrolyte of Na3PS4-PEO for all-solid-state SnS2/Na batteries with excellent interfacial compatibility between electrolyte and Na metal. J. Energy Chem. 41, 73–78 (2020). https://doi.org/10.1016/j.jechem.2019.05.003
Ge, Z., Li, J., Liu, J.: Enhanced electrochemical performance of all-solid-state sodium–sulfur batteries by PEO-NaCF3SO3-MIL-53(Al) solid electrolyte. Ionics 26, 1787–1795 (2020). https://doi.org/10.1007/s11581-020-03513-9
Tang, X., Liu, H., Guo, X., et al.: A novel lithium-ion hybrid capacitor based on an aerogel-like MXene wrapped Fe2O3 nanosphere anode and a 3D nitrogen sulphur dual-doped porous carbon cathode. Mater. Chem. Front. 2, 1811–1821 (2018). https://doi.org/10.1039/c8qm00232k
Wenzel, S., Leichtweiss, T., Weber, D.A., et al.: Interfacial reactivity benchmarking of the sodium ion conductors Na3PS4 and sodium β-alumina for protected sodium metal anodes and sodium all-solid-state batteries. ACS Appl. Mater. Interfaces 8, 28216–28224 (2016). https://doi.org/10.1021/acsami.6b10119
Wenzel, S., Randau, S., Leichtweiß, T., et al.: Direct observation of the interfacial instability of the fast ionic conductor Li10GeP2S12 at the lithium metal anode. Chem. Mater. 28, 2400–2407 (2016). https://doi.org/10.1021/acs.chemmater.6b00610
Bay, M.C., Wang, M., Grissa, R., et al.: Sodium plating from Na-β″-alumina ceramics at room temperature, paving the way for fast-charging all-solid-state batteries. Adv. Energy Mater. 10, 1902899 (2020). https://doi.org/10.1002/aenm.201902899
Spencer Jolly, D., Ning, Z.Y., Darnbrough, J.E., et al.: Sodium/Na β″ alumina interface: effect of pressure on voids. ACS Appl. Mater. Interfaces 12, 678–685 (2020). https://doi.org/10.1021/acsami.9b17786
Gao, Z., Yang, J., Yuan, H., et al.: Stabilizing Na3Zr2Si2PO12/Na interfacial performance by introducing a clean and Na-deficient surface. Chem. Mater. 32, 3970–3979 (2020). https://doi.org/10.1021/acs.chemmater.0c00474
Oh, J.A.S., Wang, Y.M., Zeng, Q.B., et al.: Intrinsic low sodium/NASICON interfacial resistance paving the way for room temperature sodium-metal battery. J. Colloid Interface Sci. 601, 418–426 (2021). https://doi.org/10.1016/j.jcis.2021.05.123
Zhao, Y.J., Wang, C.Z., Dai, Y.J., et al.: Homogeneous Na+ transfer dynamic at Na/Na3Zr2Si2PO12 interface for all solid-state sodium metal batteries. Nano Energy 88, 106293 (2021). https://doi.org/10.1016/j.nanoen.2021.106293
Matios, E., Wang, H., Luo, J.M., et al.: Reactivity-guided formulation of composite solid polymer electrolytes for superior sodium metal batteries. J. Mater. Chem. A 9, 18632–18643 (2021). https://doi.org/10.1039/d1ta05490b
Peled, E., Menkin, S.: Review: SEI: past, present and future. J. Electrochem. Soc. 164, A1703–A1719 (2017). https://doi.org/10.1149/2.1441707jes
Tian, H.J., Shao, H.Z., Chen, Y., et al.: Ultra-stable sodium metal-iodine batteries enabled by an in situ solid electrolyte interphase. Nano Energy 57, 692–702 (2019). https://doi.org/10.1016/j.nanoen.2018.12.084
Choudhury, S., Wei, S.Y., Ozhabes, Y., et al.: Designing solid-liquid interphases for sodium batteries. Nat. Commun. 8, 898 (2017). https://doi.org/10.1038/s41467-017-00742-x
Hou, Z., Wang, W.H., Yu, Y.K., et al.: Poly(vinylidene difluoride) coating on Cu current collector for high-performance Na metal anode. Energy Storage Mater. 24, 588–593 (2020). https://doi.org/10.1016/j.ensm.2019.06.026
Hou, Z., Wang, W.H., Chen, Q.W., et al.: Hybrid protective layer for stable sodium metal anodes at high utilization. ACS Appl. Mater. Interfaces 11, 37693–37700 (2019). https://doi.org/10.1021/acsami.9b12059
Qian, J., Li, Y., Zhang, M.L., et al.: Protecting lithium/sodium metal anode with metal-organic framework based compact and robust shield. Nano Energy 60, 866–874 (2019). https://doi.org/10.1016/j.nanoen.2019.04.030
Tian, H.Z., Seh, Z.W., Yan, K., et al.: Theoretical investigation of 2D layered materials as protective films for lithium and sodium metal anodes. Adv. Energy Mater. 7, 1602528 (2017). https://doi.org/10.1002/aenm.201602528
Wang, H., Wang, C.L., Matios, E., et al.: Critical role of ultrathin graphene films with tunable thickness in enabling highly stable sodium metal anodes. Nano Lett. 17, 6808–6815 (2017). https://doi.org/10.1021/acs.nanolett.7b03071
Luo, W., Lin, C.F., Zhao, O., et al.: Ultrathin surface coating enables the stable sodium metal anode. Adv. Energy Mater. 7, 1601526 (2017). https://doi.org/10.1002/aenm.201601526
Zhao, Y., Goncharova, L.V., Lushington, A., et al.: Superior stable and long life sodium metal anodes achieved by atomic layer deposition. Adv. Mater. 29, 1606663 (2017). https://doi.org/10.1002/adma.201606663
Zhao, Y., Goncharova, L.V., Zhang, Q., et al.: Inorganic–organic coating via molecular layer deposition enables long life sodium metal anode. Nano Lett. 17, 5653–5659 (2017). https://doi.org/10.1021/acs.nanolett.7b02464
Zhao, Y., Sun, X.L.: Molecular layer deposition for energy conversion and storage. ACS Energy Lett. 3, 899–914 (2018). https://doi.org/10.1021/acsenergylett.8b00145
Ye, S.F., Wang, L.F., Liu, F.F., et al.: Integration of homogeneous and heterogeneous nucleation growth via 3D alloy framework for stable Na/K metal anode. eScience 1, 75–82 (2021). https://doi.org/10.1016/j.esci.2021.09.003
Xu, Z.X., Deng, W.J., Wang, X.L.: 3D hierarchical carbon-rich micro-/ nanomaterials for energy storage and catalysis. Electrochem. Energy Rev. 4, 269–335 (2021). https://doi.org/10.1007/s41918-021-00094-7
Lu, Y.Y., Zhang, Q., Han, M., et al.: Stable Na plating/stripping electrochemistry realized by a 3D Cu current collector with thin nanowires. Chem. Commun. 53, 12910–12913 (2017). https://doi.org/10.1039/c7cc07485a
Liu, S., Tang, S., Zhang, X.Y., et al.: Porous Al current collector for dendrite-free Na metal anodes. Nano Lett. 17, 5862–5868 (2017). https://doi.org/10.1021/acs.nanolett.7b03185
Xu, Y.L., Menon, A.S., Harks, P.P.R., et al.: Honeycomb-like porous 3D nickel electrodeposition for stable Li and Na metal anodes. Energy Storage Mater. 12, 69–78 (2018). https://doi.org/10.1016/j.ensm.2017.11.011
Wang, T.S., Liu, Y.C., Lu, Y.X., et al.: Dendrite-free Na metal plating/stripping onto 3D porous Cu hosts. Energy Storage Mater. 15, 274–281 (2018). https://doi.org/10.1016/j.ensm.2018.05.016
Yang, W., Yang, W., Dong, L.B., et al.: Hierarchical ZnO nanorod arrays grown on copper foam as an advanced three-dimensional skeleton for dendrite-free sodium metal anodes. Nano Energy 80, 105563 (2021). https://doi.org/10.1016/j.nanoen.2020.105563
Wang, C.L., Wang, H., Matios, E., et al.: A chemically engineered porous copper matrix with cylindrical core-shell skeleton as a stable host for metallic sodium anodes. Adv. Funct. Mater. 28, 1802282 (2018). https://doi.org/10.1002/adfm.201802282
Ma, J.L., Meng, F.L., Yu, Y., et al.: Prevention of dendrite growth and volume expansion to give high-performance aprotic bimetallic Li–Na alloy–O2 batteries. Nat. Chem. 11, 64–70 (2019). https://doi.org/10.1038/s41557-018-0166-9
Yu, D.L., Liu, D., Shi, L., et al.: High-performance metal–iodine batteries enabled by a bifunctional dendrite-free Li–Na alloy anode. J. Mater. Chem. A 9, 538–545 (2021). https://doi.org/10.1039/d0ta08072a
Kim, Y.J., Lee, J.H., Yuk, S., et al.: Tuning sodium nucleation and stripping by the mixed surface of carbon nanotube-sodium composite electrodes for improved reversibility. J. Power Sources 438, 227005 (2019). https://doi.org/10.1016/j.jpowsour.2019.227005
Zhao, Y., Yang, X.F., Kuo, L.Y., et al.: High capacity, dendrite-free growth, and minimum volume change Na metal anode. Small 14, 1703717 (2018). https://doi.org/10.1002/smll.201703717
Ye, L., Liao, M., Zhao, T.C., et al.: A sodiophilic interphase-mediated, dendrite-free anode with ultrahigh specific capacity for sodium-metal batteries. Angew. Chem. Int. Ed. 58, 17054–17060 (2019). https://doi.org/10.1002/anie.201910202
Sun, B., Li, P., Zhang, J.Q., et al.: Dendrite-free sodium-metal anodes for high-energy sodium-metal batteries. Adv. Mater. 30, 1801334 (2018). https://doi.org/10.1002/adma.201801334
Wu, F., Zhou, J.H., Luo, R., et al.: Reduced graphene oxide aerogel as stable host for dendrite-free sodium metal anode. Energy Storage Mater. 22, 376–383 (2019). https://doi.org/10.1016/j.ensm.2019.02.015
Hu, X.F., Li, Z.F., Zhao, Y.R., et al.: Quasi–solid state rechargeable Na-CO2 batteries with reduced graphene oxide Na anodes. Sci. Adv. 3, e1602396 (2017). https://doi.org/10.1126/sciadv.1602396
Hu, X.F., Joo, P.H., Wang, H., et al.: Nip the sodium dendrites in the bud on planar doped graphene in liquid/gel electrolytes. Adv. Funct. Mater. 29, 1807974 (2019). https://doi.org/10.1002/adfm.201807974
Jin, X., Zhao, Y., Shen, Z.H., et al.: Interfacial design principle of sodiophilicity-regulated interlayer deposition in a sandwiched sodium metal anode. Energy Storage Mater. 31, 221–229 (2020). https://doi.org/10.1016/j.ensm.2020.06.040
Yan, K., Zhao, S.Q., Zhang, J.Q., et al.: Dendrite-free sodium metal batteries enabled by the release of contact strain on flexible and sodiophilic matrix. Nano Lett. 20, 6112–6119 (2020). https://doi.org/10.1021/acs.nanolett.0c02215
Wang, H., Wang, C.L., Matios, E., et al.: Enabling ultrahigh rate and capacity sodium metal anodes with lightweight solid additives. Energy Storage Mater. 32, 244–252 (2020). https://doi.org/10.1016/j.ensm.2020.07.021
Wang, L., Wang, T., Peng, L.L., et al.: The promises, challenges and pathways to room-temperature sodium–sulfur batteries. Natl. Sci. Rev. 9, nwab050 (2022). https://doi.org/10.1093/nsr/nwab050
Bauer, I., Kohl, M., Althues, H., et al.: Shuttle suppression in room temperature sodium–sulfur batteries using ion selective polymer membranes. Chem. Commun. 50, 3208 (2014). https://doi.org/10.1039/c4cc00161c
Yu, X., Manthiram, A.: Performance enhancement and mechanistic studies of room-temperature sodium–sulfur batteries with a carbon-coated functional nafion separator and a Na2S/activated carbon nanofiber cathode. Chem. Mater. 28, 896–905 (2016). https://doi.org/10.1021/acs.chemmater.5b04588
Wang, H.M., Deng, C., Li, X.L., et al.: Designing dual-defending system based on catalytic and kinetic iron Pyrite@C hybrid fibers for long-life room-temperature sodium–sulfur batteries. Chem. Eng. J. 420, 129681 (2021). https://doi.org/10.1016/j.cej.2021.129681
Yang, T., Guo, B., Du, W., et al.: Design and construction of sodium polysulfides defense system for room‐temperature Na–S battery. Adv. Sci. 6, 1901557 (2019). https://doi.org/10.1002/advs.201901557
Wang, H., Jiang, Y., Manthiram, A.: Long cycle life, low self-discharge sodium–selenium batteries with high selenium loading and suppressed polyselenide shuttling. Adv. Energy Mater. 8, 1701953 (2018). https://doi.org/10.1002/aenm.201701953
Zhang, F., Guo, X., Xiong, P., et al.: Interface engineering of MXene composite separator for high-performance Li–Se and Na–Se batteries. Adv. Energy Mater. 10, 2000446 (2020). https://doi.org/10.1002/aenm.202000446
Acknowledgements
We would like to acknowledge the financial support from the Australian Research Council (ARC) through the ARC Discovery projects (DP200101249, DP210101389) and the ARC Research Hub for Integrated Energy Storage Solutions (IH180100020).
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Guoxiu Wang is an editorial board member for Electrochemical Energy Reviews and was not involved in the editorial review or the decision to publish this article. All authors declare that there are no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, Z., Jaumaux, P., Sun, B. et al. High-Energy Room-Temperature Sodium–Sulfur and Sodium–Selenium Batteries for Sustainable Energy Storage. Electrochem. Energy Rev. 6, 21 (2023). https://doi.org/10.1007/s41918-023-00182-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41918-023-00182-w