
Abstract
A composite cement made of strontium containing tricalcium silicate and hydroxyapatite (SrC3S/HAp) was prepared and studied through a two-stage sol–gel method. The elemental components are 75 wt% of strontium-doped tricalcium silicate (SrC3S) and 25 wt% of hydroxyapatite (HAp). According to previous studies, the SrC3S has good mechanical properties and hydraulic conductivity, and the HAp has good biocompatibility. Therefore, the 75SrC3S-25HAp powder with 10 wt% of NaH2PO4 solution was mixed to prepare the slurry material. This slurry was measured for the working time and setting time at 37 °C under saturated vapor pressure. The material properties were evaluated in terms of crystal structures, surface morphologies, and mechanical properties. The in vitro testing was conducted to determine the ion release rate and curing behavior under a simulated body fluid environment. Finally, the L929 murine fibroblast cells were cultured to study the biocompatibility of the material. The results indicated that the operating time was 15.2 min, and the setting time was 43.6 min. After being soaked into simulated body fluid for14 days of the experiment, the compressive strength of 75SrC3S-25HAp remained 22.1 MPa, and the release of calcium and strontium ions was 501.4 ppm and 49.5 ppm, respectively. The cell viabilities were higher than 70% in various concentrations. These results suggest the 75SrC3S-25HAp has excellent potential for bone cement application.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
For defect restoration and implant fixation in orthopedics, bone cement has been popularly applied in clinical. Several types of material have been developed for bone cement applications, including polymethylmethacrylate (PMMA) [1], bioactive glass (BG) [2, 3], and calcium phosphate cement. PMMA is an inert polymer-based material and lacks osteoconductivity. Although BG has excellent bioactive to form hydroxyapatite (HAp), a key component of bone, the BG demonstrates insufficient mechanical strength and brittleness [4,5,6]. On the other hand, bone cement based on calcium phosphate has caused extensive attention for the well self-setting property, good plasticity, biocompatibility, and bone conductivity [7,8,9,10]. However, the new composite material is still needed.
Tricalcium silicate is the main component of Portland cement. This material can provide sufficient mechanical strength after hydrolyzation [11] and excellent biocompatibility for bone cement application [12,13,14,15]. Furthermore, it can induce bone-like apatite formation, which can stimulate cell proliferation. It is also the leading resource of mineral trioxide aggregate (MTA), which is used in root canal therapy [15,16,17]. Strontium has been reported to benefit bone regeneration which can stimulate the activity of the osteoblasts on differentiation and inhibit osteoclast function from reducing bone resorption [18,19,20].
The strontium contained tricalcium silicate has shown the potential to serve as bone cement in our previous report; the previous report also suggested that 75 wt% of tricalcium silicate + 25 wt% of HAp has better compressive strength and higher cell viability [21]. To synthesize a novel bone cement to further improve the function compared with traditional bone cement. The strontium contained tricalcium silicate and HAp has been synthesized by a two-stage sol–gel process. The 75 wt% (1.75% SrC3S) of tricalcium silicate + 25 wt% of HAp is chosen in this study. The tricalcium phosphate can improve the mechanical properties of the composite and water absorption; the strontium can provide a function for bone healing and regeneration. Although, HAp can provide a similar function to strontium. The amount of HAp added to the cement can suppress water absorption, consequently changing hydraulic conductivity of tricalcium phosphate [22].
Materials and methods
Fabrication of SrC 3 S/HAp ceramics
All chemicals used in this study were purchased from Sigma-Aldrich®. This experiment aims to prepare strontium containing tricalcium silicate with HAp composite prepared through a sol–gel. The calcium nitrate tetrahydrate (Ca(NO3)2˙4H2O, 98%) was used as a calcium precursor; triethyl phosphate ((C2H5)3PO4, TEP, ≧ 99%) as a phosphorus precursor; strontium nitrate (Sr(NO3)2, 98%) as a strontium precursor; tetraethyl silicate (Si(OC2H5)4,≧ 99%) as silica precursor). The 2.86 mL of TEP and 0.11 mL of nitric acid (HNO3, 65%) (according to the molar ratio of 1:1.197:0.08) were well mixed in 5 mL of ethanol (99.9%) to hydrolyze for 30 min to get phosphate sol. And then mixed with 4.723 g calcium nitrate tetrahydrate and 6.027 g of as prepared Sr0.0525Ca2.9475SiO5 powder into the phosphate sol at 80 °C for 24 h. After 24 h of mixing, the material was placed in the 120 °C silicone oil bath for a few hours to get a viscous gel. The gel was moved to a platinum crucible and calcined in a high temperature furnace at 700 °C for 4 h. The calcined sample was ground and sieved through a screen of 400 mesh to control the particle size of the powder below 37 μm. This powder was composited by 75wt% Sr0.0525Ca2.9475SiO5 + 25wt% HAp which named 75SrC3S-25HAp powder. The crystal structure and functional groups of material were evaluated by XRD (Shimadzu XRD-6000, Japan) and FTIR (Spectrum GX, Perkin Elmer Application, USA).
Preparation of the paste
Ten weight percent of NaH2PO4(aq) and 75SrC3S-25HAp were mixed with water in a volume ratio of 2:1 (powder: water) for two minutes (sample 0.5 g, solution 0.25 mL) to prepare a slurry.
The slurry material was filled in a stainless-steel mold with an internal diameter of 12 mm and applied 10 kg/cm2 of pressure to make it into a compressed cylinder tablet by manual oil hydraulic press. The cylindrical materials were placed in the incubator at 37 °C for various days. The incubated samples were dried at 60 °C, then using XRD and SEM (S-3000H, Hitachi, Japan) to analyze the crystallization and surface morphologies of the material.
The self-setting properties of the paste
The slurry material was filled into a PMMA mold with a diameter of 10 mm and a height of 5 mm. The working time and setting time were measured using a Gilmore needle at room temperature and atmosphere. The working time was determined by placing the needle (with a diameter of 1/12 in and weight of 1/4 lb) on the surface of the test piece without dents. The setting time was determined by placing a needle (with a diameter of 1/24 in and weighing 1 lb on the surface) on the test piece without dents.
In vitro bioactive test
The simulated body fluid (SBF) was prepared according to KoKubo’s study, whose ion concentration is similar to human plasma [23]. The compressed tablet was wrapped by 1.5 wt% of ager and soaked into 45 mL of SBF at 37 °C for days to simulate the situation of material in the organization. The soaked samples were removed from the SBF and placed in a 60 °C oven to dry the material and removed ager.
We used XRD and SEM to analyze the crystallization and surface morphology of the material, ICP-AES (SPS 7800 Plasma Spectrometer, Japan) to investigate the concentrations of Ca, Sr, and P ions in the simulated body fluid, and pH meter (Eutech Instruments pH 510, UK) to measure the pH value.
Mechanical test
In order to understand the mechanical strength of the set material, the compressed tablet was made with 1 g of composite power and 0.5 mL of water in a stainless-steel mold. The mold had an internal diameter of 10 mm and applied 10 kg/cm2 pressure to manufacture the 10 mm high cylindrical tablet using a manual hydraulic press. After the compression stress was stable, the compressed tablet was placed in a specific environment at 37 °C with 100% of saturated water vapor pressure for different days. MTS (858 Mini Bionix II, USA) with a loading ratio of 0.5 mm/min was used to measure the compressive strength until it was broken, then we could get the maximum load (kgf) of the material. Dividing the maximum load by the material cross-sectional area could get the compressive strength (MPa).
Biocompatibility test
To evaluate the biocompatibility of the material, the cellular viability was evaluated using a murine fibroblast cell line (L929) which was cultured at the 37 °C and 5% of CO2 environment. Different weight of set material was immersed in the cell culture medium for 24 h. And 1 mL of the soaking solution was co-cultured with the cell for 24 h. Then, the MTT test was carried out using an Elisa reader (Sunrise remote F039300, Austria) to measure the optical density. The value of the control group with normal culture conditions was set as 100% cellular viability.
Results and discussion
Characterization of SrC 3 S + HAp ceramics
The crystal structure of synthesized 75SrC3S-25HAp powder has been studied by XRD as shown in Fig. 1. The characteristic peaks of SrC3S fall at 2θ = 32°、34° and 46.7°, while the HAp peaks are located at 2θ = 25.7°, 32.4°, 46.7°. Meanwhile, the FTIR result is shown in Fig. 2. The functional groups SiO44−, PO43−, OH− and can be found at 873 ~ 997 cm−1, 500 ~ 700 cm−1 also 1040 cm−1, and 3640 cm−1, respectively. These groups are commonly used to identify as HAp groups. Also, the functional group of CO32− can be found at 1450 cm−1. The results of XRD and FTIR show that the 75SrC3S-25HAp composite has been successfully synthesized using a two-stage sol–gel process.

XRD patterns of 75SrC3S-25HAp powders

FTIR spectra of 75SrC3S-25HAp powders
Working time and setting time
The working time and setting time of SrC3S and 75SrC3S-25HAp are listed in Table 1. The working time of SrC3S is 8.8 min and the setting time is 69.0 min, while the working time of 75SrC3S-25HAp is 15.2 min, and the setting time is 43.6 min. The 75SrC3S-25HAp composite material provides a longer working time and shorter setting time than SrC3S. This improvement can bring benefits while practical application.
XRD patterns
The 75SrC3S-25HAp samples have been preserved at 37 °C with saturation vapor pressure for 1–14 days. The XRD results are shown in Fig. 3. We can find that the characteristic peaks of HAp are at 2θ = 25.7° and 39.3°. The characteristic peak of SrC3S is at 2θ = 32°. The characteristic peak of Ca(OH)2 is at 2θ = 34°. During the 14 days, the intensity of the characteristic peak at 2θ = 32° has gradually weakened due to the hydration reaction of SrC3S. Thus, the strength of the characteristic peak is decreased as the preservation time increases. Due to the hydration reaction, the SrC3S crystal structure will transfer from the low Ca/Si ratio of Tobermorite to the high Ca/Si ratio of Jennite. Therefore, the characteristic peak of the crystal surface of 2θ = 29.2° is Jennite which indicates composite material of 75SrC3S-25HAp has a certain level of hydration after 7 days of the experiment.

XRD patterns of the 75SrC3S-25HAp paste after setting at 37 °C, 100% humidity for 1, 3, 7, and 14 days
The 75SrC3S-25HAp has been soaked in the simulated body fluid for 1–14 days. The XRD technique has been used to evaluate crystal structure, as shown in Fig. 4. The result indicates that the characteristic peaks of HAp are at 2θ = 25.7°, 39.3°. The characteristic peak of SrC3S is 2θ = 32°. The characteristic peak of Ca(OH)2 is 2θ = 32°. The intensity of the characteristic peak at 2θ = 32° does not weaken over time due to 75SrC3S-25HAp being covered by a layer of HAp precipitation. The mass transfer in the water is slower than placed under a saturated steam environment. Therefore, the slower hydration does not transfer the crystal structure from the low Ca/Si ratio of the Tobermorite phase to the high Ca/Si ratio of the Jennite phase. The results suggested an incomplete hydration reaction of SrC3S and showed that hydration speed is slower than in the saturated steam environment.

XRD patterns of the 75SrC3S-25HAp paste after soaking in SBF at 37 °C for 1, 3, 7, and 14 days
SEM image
The surface morphologies of 75SrC3S-25HAp under saturated steam and simulated body fluid for days are shown in Fig. 5A-D and E–H, respectively. Figure 5A-D demonstrate particles aggregated during the initial experiment. The porous structure gets denser as the time increases in saturated steam. The porous structure is hardly found on the surface after 14 days of the investigation. The hydration reaction happened on 75SrC3S-25HAp and the C-S–H began to form a larger piece of hydration structure. Consequently, the reaction leads the pore to become smaller and fewer. The hydraulic ability of 75SrC3S-25HAp also demonstrates the phenomenon. Figure 5E-H suggest a similar result as the XRD pattern. The hydration reaction is lower, resulting in an incompletely densification porous structure, as shown in Fig. 5H. Although there are some HAp precipitations on the surface.

SEM micrographs of the 75SrC3S-25HAp paste after setting at 37 °C, 100% humidity for A 1, B 3, C 7, and D 14 days and soaking in SBF at 37 °C for E 1, F 3, G 7, and H 14 days
Mechanical property
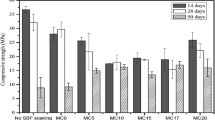
The compressive strength of SrC3S and 75SrC3S-25HAp have been evaluated after soaking in SBF for 7 and 14 days, respectively. The compressive strength results have been shown in Fig. 6. The compressive strength results are 18.8 ± 0.8, and 22.1 ± 2.6 MPa for the 7 and 14 days experiment, respectively. By comparing to SrC3S results, the compressive strengths of SrC3S are 17.9 ± 4.7 and 42.9 ± 1.3 MPa for 7 and 14 days, respectively. The compressive strength of 75SrC3S-25HAp was increased by increasing hydration time; the hydration process gradually changed the crystal structure of 75SrC3S-25HAp from a low Ca/Si ratio of Tobermorite to a high Ca/Si ratio of hexahydrate phase (Jennite) as shown in Fig. 3 XRD result. Meanwhile, the SEM images in Fig. 5 suggested the densification of 75SrC3S-25HAp after 14 days of hydration experiment. This result indicates that the 75SrC3S-25HAp composite has weakened the strength of SrC3S.

Compressive strength of the SrC3S and 75SrC3S-25HAp pastes after setting at 37 °C, 100% humidity for 7 and 14 days
Ion release in SBF
From the results of Fig. 7, the amount of ion changed of 75SrC3S-25HAp for 14 days in the SBF. The ion release results suggest that the concentration of Ca2+, Sr2 and PO43− at day 14 are 501.4, 49.5, and 107.4 ppm, respectively. The concentration of PO43− at day 1 is 79.2 ppm, which is higher than the concentration of PO43− in the SBF about 40.6 ppm. On day 3, the concentration of PO43− slightly decreased to 74.5 ppm and then rose again after day 14 of the experiment. The concentration of PO43− is up to 107.4 ppm. The ion release results demonstrate that the concentration of PO43− on day 1 is higher than the initial concentration of PO43− in the SBF. The higher PO43− concentration is based on 75SrC3S-25HAp released PO43− in the SBF. Furthermore, the concentration of PO43 decreased on day 3; because of the apatite precipitation on the surface of 75SrC3S-25HAp. For the day 3 to day 14 experiment, the concentration of PO43− rises again, explaining the ion-exchanged reaction on the surface of 75SrC3S-25HAp, which leads to the concentration of PO43−increased.

Changes in Ca, Sr, and P concentrations of the SBF solution after soaking with the 75SrC3S-25HAp paste with a liquid/powder ratio of 0.5 g/mL for various time
The pH value of the SBF solution
The pH value of the 75SrC3S-25HAp composite soaked in the simulated body fluid for 14 days is shown in Fig. 8. The pH value of 75SrC3S-25HAp composite is lower than 9 during the experiment. By comparing the pH of SrC3S, the values suggest that the pH of different amounts of SrC3S at the range from 11 and 12 on day 14. However, the 75SrC3S-25HAp composite changed the SrC3S from a highly alkaline material to a low-alkaline composite material. The reason for this change is that the initial pH value of SBF is 7.4. When the pH value of tricalcium phosphate is pH > 4.2, the most stable phase is HAp. So the OH− will react with lots of Ca2+ and PO43−, gradually depositing on the surface of the 75SrC3S-25HAp. Thus, the HAp precipitation on the surface of the composite consequently leads to the concentration of PO43− getting higher while 75SrC3S-25HAp is soaked in 37 °C SBF. Therefore, the pH value of the 75SrC3S-25HAp composite was lower than 9 in this experiment. Finally, the overall composite pH value can be reduced by doping strontium and HAp in tricalcium silicate, which is a great advantage of this material.

Changes the pH value of the SBF solution after soaking with the 75SrC3S-25HAp paste with a Liquid/Powder ratio of 0.5 g/mL for various time
Biocompatibility
The cellular viability of set material co-culture with L929 cell for a day is shown in Fig. 9. The cell survival rates of both samples are higher than 100% at 5 mg/mL concentration. The cell viability remained more than 90% at a 10 mg/mL concentration. However, the cell viability started to decrease while the concentration was above 25 mg/mL in the SrC3S sample. The hydrolysis reaction in the SrC3S sample contributes to a highly alkaline environment with a pH value of up to 11, which is cytotoxic. The 75SrC3S-25HAp sample’s cell viability results suggest higher survival rates, accounting for up to 100% within 25 mg/mL concentration; the survival rate 75SrC3S-25HAp sample remained for more than 70% after the concentration of the material is up to 50 mg/mL. Note that the survival rate of 75SrC3S-25HAp is higher than SrC3S. The reason is that 75SrC3S-25HAp composite is less alkaline than high alkaline SrC3S. This lower pH value can provide the most suitable environment for cells (6 < pH < 9), so the cells have a better survival rate.

The effect of SrC3S and 75SrC3S-25HAp pastes setting at 37 °C, 100% humidity for 1-day extracts with different extract concentrations on L929 cell proliferation
Conclusions
The 75SrC3S-25HAp composite has been successfully synthesized through a sol–gel process. The Hap-doped Sr0.0525Ca2.9475SiO5 can increase working from 8.8 to 15.2 min and reduce the setting time from 69 to 43.7 min, providing more working time and fast setting during the operation. Although, the compressive strength of 75SrC3S-25HAp under a saturated steam environment is lower than SrC3S. The compressive strength of 75SrC3S-25HAp remained at 22.1 MPa for 14 days experiment. The 75SrC3S-25HAp demonstrates good biocompatibility in the cell viability test. The cell viability stays more than 100% at the concentration of 75SrC3S-25HAp composite up to 25 mg/mL. This high survival rate is based on low-alkaline composite providing a proper environment for cells. Thus, the 75SrC3S-25HAp composite has great potential for bone cement application.
References
Charnley, J.: Anchorage of the femoral head prosthesis to the shaft of the femur, The Journal of bone and joint surgery. British 42(1), 28–30 (1960)
Hench, L.L., Splinter, R.J., Allen, W., Greenlee, T.: Bonding mechanisms at the interface of ceramic prosthetic materials. J. Biomed. Mater. Res. 5(6), 117–141 (1971)
Andersson, Ö., Karlsson, K., Hero, H., Vedel, E., Yli-Urpo, A., Pajamäki, K., Lindholm, T.: Bioactive double glass coatings for Co-Cr-Mo alloy. J. Mater. Sci. - Mater. Med. 6(4), 242–247 (1995)
Raksujarit, A., Pengpat, K., Rujijanagul, G., Tunkasiri, T.: Processing and properties of nanoporous hydroxyapatite ceramics. Mater. Des. 31(4), 1658–1660 (2010)
Yamaguchi, I., Tokuchi, K., Fukuzaki, H., Koyama, Y., Takakuda, K., Monma, H., Tanaka, J.: Preparation and microstructure analysis of chitosan/hydroxyapatite nanocomposites, Journal of Biomedical Materials Research: An Official Journal of The Society for Biomaterials, The Japanese Society for Biomaterials, and The Australian Society for Biomaterials and the Korean Society for. Biomaterials 55(1), 20–27 (2001)
Younesi, M., Bahrololoom, M.: Optimizations of wear resistance and toughness of hydroxyapatite nickel free stainless steel new bio-composites for using in total joint replacement. Mater. Des. 31(1), 234–243 (2010)
Constantz, B.R., Ison, I.C., Fulmer, M.T., Poser, R.D., Smith, S.T., VanWagoner, M., Ross, J., Goldstein, S.A., Jupiter, J.B., Rosenthal, D.I.: Skeletal repair by in situ formation of the mineral phase of bone. Science 267(5205), 1796–1799 (1995)
Schildhauer, T., Bennett, A., Wright, T., Lane, J., O’Leary, P.: Intravertebral body reconstruction with an injectable in situ-setting carbonated apatite: biomechanical evaluation of a minimally invasive technique. J. Orthop. Res. 17(1), 67–72 (1999)
Li, Y., Leong, J., Lu, W., Luk, K., Cheung, K., Chiu, K., Chow, S.: A novel injectable bioactive bone cement for spinal surgery: a developmental and preclinical study. J. Biomed. Mater. Res. 52(1), 164–170 (2000)
Ocampo, J.I.G., de Paula, M.M.M., Bassous, N.J., Lobo, A.O., Orozco, C.P.O., Webster, T.J.: Osteoblast responses to injectable bone substitutes of kappa-carrageenan and nano hydroxyapatite. Acta Biomater. 83, 425–434 (2019)
Zhao, W., Chang, J.: Two-step precipitation preparation and self-setting properties of tricalcium silicate. Mater. Sci. Eng., C 28(2), 289–293 (2008)
Zhao, W., Chang, J.: Sol–gel synthesis and in vitro bioactivity of tricalcium silicate powders. Mater. Lett. 58(19), 2350–2353 (2004)
Zhao, W., Wang, J., Zhai, W., Wang, Z., Chang, J.: The self-setting properties and in vitro bioactivity of tricalcium silicate. Biomaterials 26(31), 6113–6121 (2005)
Leu, T.H., Wei, Y., Hwua, Y.S., Huang, X.J., Huang, J.T., Chung, R.J.: Fabrication of PLLA/C3S composite membrane for the prevention of bone cement leakage. Polymers 11(12), 17 (2019)
Chung, R.J., Wang, A.N., Wang, H.Y., Wu, Y.C.: Investigation of collagen/tricalcium silicate/carboxymethyl cellulose composite bone glue for drug controlled release. J. Aust. Ceram. Soc. 53(2), 329–342 (2017)
Phan, P.V., Grzanna, M., Chu, J., Polotsky, A., El-Ghannam, A., Van Heerden, D., Hungerford, D.S., Frondoza, C.G.: The effect of silica-containing calcium-phosphate particles on human osteoblasts in vitro. J. Biomed. Mater. Res. Part A 67(3), 1001–1008 (2003)
Roberts, H.W., Toth, J.M., Berzins, D.W., Charlton, D.G.: Mineral trioxide aggregate material use in endodontic treatment: a review of the literature. Dent. Mater. 24(2), 149–164 (2008)
Zhang, W., Shen, Y., Pan, H., Lin, K., Liu, X., Darvell, B.W., Lu, W.W., Chang, J., Deng, L., Wang, D.: Effects of strontium in modified biomaterials. Acta Biomater. 7(2), 800–808 (2011)
Buehler, J., Chappuis, P., Saffar, J., Tsouderos, Y., Vignery, A.: Strontium ranelate inhibits bone resorption while maintaining bone formation in alveolar bone in monkeys (Macaca fascicularis). Bone 29(2), 176–179 (2001)
Looney, M., O’Shea, H., Redington, W., Kelly, G., Boyd, D.: High-temperature X-ray analysis of phase evolution in Sr-doped zinc-silicate glasses. J. Non-Cryst. Solids 357(10), 2097–2102 (2011)
Liu, W.-C., Hu, C.-C., Tseng, Y.-Y., Sakthivel, R., Fan, K.-S., Wang, A.-N., Wang, Y.-M., Chung, R.-J.: Study on strontium doped tricalcium silicate synthesized through sol-gel process. Mater. Sci. Eng., C 108, 110431 (2020)
Daglilar, S., Erkan, M.E., Gunduz, O., Ozyegin, L.S., Salman, S., Agathopoulos, S., Oktar, F.N.: Water resistance of bone-cements reinforced with bioceramics. Mater. Lett. 61(11–12), 2295–2298 (2007)
Kokubo, T., Takadama, H.: How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 27(15), 2907–2915 (2006)
Acknowledgements
Technical assistance from the Precision Analysis and Material Research Center of the National Taipei University of Technology (Taipei Tech) is appreciated.
Funding
This research is financially supported by the Ministry of Science and Technology of Taiwan (MOST 109–2622-E-027–003-CC3).
Author information
Authors and Affiliations
Contributions
Senthilkumar Thirumurugan: writing—original draft, writing—review and editing, methodology, formal analysis. Yu-Chien Lin: writing—review and editing, methodology, data curation, visualization. Guan-Yi Lin: writing—original draft, writing—review and editing, methodology, formal analysis. Kuei-Sheng Fan: investigation, formal analysis, data curation, validation. Yung-He Liang: writing—review and editing, methodology, project administration, funding acquisition. Yi-Jie Kuo: conceptualization, methodology, supervision, writing—review and editing. Ren-Jei Chung: conceptualization, methodology, supervision, project administration, funding acquisition, writing—review and editing.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thirumurugan, S., Lin, YC., Lin, GY. et al. Study of strontium-doped tricalcium silicate/hydroxyapatite composite cement prepared through sol–gel process. J Aust Ceram Soc 58, 1357–1366 (2022). https://doi.org/10.1007/s41779-022-00783-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41779-022-00783-6