
Abstract
Nitrogen heterocycles are part of the structure of natural products and agents with important biological activity, such as antiviral, antibiotic, and antitumor drugs. For this reason, heterocyclic compounds are one of today’s most desirable synthetic targets and the Povarov reaction is a powerful synthetic tool for the construction of highly functionalized heterocyclic systems. This process involves an aromatic amine, a carbonyl compound, and an olefin or acetylene to give rise to the formation of a nitrogen-containing heterocycle. This review illustrates advances in the synthetic aspects of the intramolecular Povarov reaction for the construction of intricate nitrogen-containing polyheterocyclic compounds. This original review presents research done in this field, with references to important works by internationally relevant research groups on this current topic, covering the literature from 1992 to 2022. The intramolecular Povarov reactions are described here according to the key processes involved, using different combinations of aromatic or heteroaromatic amines, and aliphatic, aromatic, or heteroaromatic aldehydes. Some catalytic reactions promoted by transition metals are detailed, as well as the oxidative Povarov reaction and some asymmetric intramolecular Povarov processes.
Similar content being viewed by others


1 Introduction
Nitrogen-containing heterocycles are present in many natural products and agents with important biological activity, such as antiviral, antibiotic, and antitumor drugs [1,2,3,4,5]. For this reason, the synthesis of N-heterocycles and their derivatives has always been an attractive topic in organic synthesis. Food and Drug Administration (FDA) databases reveal the structural importance of nitrogen-based heterocycles in drug design, considering that a large amount of small-molecule drugs contain a nitrogen heterocycle [6, 7]. In addition, N-heterocyclic skeletons are used as building blocks for a number of new drug candidates, due to the ability of the nitrogen atom to readily form hydrogen bonds with biological targets [8]. Hence, in the drug discovery process, the development of practical synthetic routes to access these structural motifs in the simplest possible way is an important goal for synthetic and medicinal chemists.
One of the most straightforward and versatile processes for the preparation of heterocyclic nitrogen compounds is the Povarov reaction. Especially this reaction allows the preparation of tetrahydroquinolines (THQs) and their more aromatized analogs, the quinolines (QUINs), one of the most relevant nitrogen-containing heterocycles (Fig. 1) [9,10,11,12].

A Some marketed synthetic THQs and QUINs and their application. B Some bioactive THQs and QUINs from natural sources
This process was first reported in the 1960s, when Povarov et al. described the formation of two new C–C bonds from N-arylimines 3 with vinyl enolethers 4 under medium-strength Lewis acid catalysis (BF3·OEt2), thus obtaining substituted THQs 5 that were oxidized to the corresponding QUIN 6 (Scheme 1) [13].

First example described by Povarov
Since Povarov’s first reaction, several reports indicated the versatility of this tool for the synthesis of nitrogen heterocycles [14, 15]. In general, in this process an aromatic amine 1, a carbonyl compound 2, and an olefin participate to give rise to the formation of a nitrogen-containing heterocycle. This hetero-Diels–Alder reaction represents a powerful tool for the construction of carbon–carbon and carbon–heteroatom bonds by generating three stereocenters in one step showing high regio- and diastereoselectivity. In cases where mixtures of diastereoisomers are observed, the ratio of endo/exo diastereoisomers formed is modulated and determined by the catalyst and solvent used in the process. Moreover, some developed catalytic enantioselective methods have been reviewed recently [16]. Two types of mechanism for this reaction have been described in the literature. Kobayashi et al. [17] suggested a stepwise mechanism via a cationic intermediate for the reaction catalyzed by rare earth triflates (Ln(OTf)3). On the other hand, Palacios et al. [18] in their combined computational and experimental study using BF3·OEt2 (the same Lewis acid as Povarov), as well as Xu et al. [19] in their work support the hypothesis that the cycloaddition occurs via an asynchronous concerted mechanism.
Apart from the mechanistic aspects, throughout the development and study of the Povarov reaction, other types of variables have been analyzed, such as: (a) the use of different catalysts in the reaction, both Lewis and Brønsted acids; (b) the nature of the dienophiles, olefins, acetylenes, etc.; (c) the design of the step-by-step or multicomponent protocols. These aspects have been extensively covered in excellent general reviews of this methodology in the recent literature [14,15,16, 20, 21].
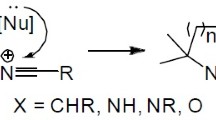
A particular case of a multicomponent reaction is the intramolecular Povarov reaction. For this purpose, aldimines present both, diene and dienophile functionality, in their structure. In general terms, intramolecular Povarov reaction can be explained as a formal intramolecular [4 + 2] cycloaddition reaction of aldimines 9, obtained by condensation between aromatic or heteroaromatic amines 7 and aldehydes 8 functionalized with double or triple bonds (Scheme 2). The initial adduct 10 obtained by the intramolecular cycloaddition of aldimine 9, followed by a double-bond tautomerization would generate the heteroaromatic compound 11 whose subsequent aromatization would give rise to derivatives 12.

Intramolecular Povarov reaction
Therefore, the intramolecular version of the Povarov reaction allows the generation of fused rings of high molecular complexity for the preparation of a wide variety of heterocyclic compounds. Skeletons of different sizes and with various condensed cycles can be obtained depending on the different combinations of aromatic or heteroaromatic amines with aliphatic, aromatic or heteroaromatic aldehydes (Scheme 3).

Scope of intramolecular Povarov reaction
The aim of this review is to illustrate the advances in the synthetic aspects of the intramolecular Povarov reaction for the construction of intricate nitrogen-fused polyheterocyclic compounds. The revision is intended to be a comprehensive, authoritative, critical, and accessible review of general interest to the chemical community, as it contains a broad overview of published data on the intramolecular Povarov reaction covering the literature from 1992 to 2022, described according to the key processes involved, combining different aromatic/heteroaromatic amines and aliphatic/aromatic/heteroaromatic aldehydes. Some catalytic reactions promoted by transition metals are detailed, as well as the oxidative Povarov reaction and some asymmetric intramolecular Povarov processes. In addition, the effect of catalysts and solvents on the preparation of the final products will be examined, reflecting the synthetic potential of this strategy.
2 Aromatic Amines and Aliphatic Aldehydes
2.1 Aliphatic Alkene-Tethered Aldehydes
Laschat’s group developed an intramolecular hetero-Diels–Alder reaction of N-aryl imines derived from aliphatic aldehydes tethered to non-activated olefins to afford 1,2,3,4,4a,9,9a,10-octahydroacridine (OHA) derivatives in high yields [22, 23]. The mechanism of this reaction could be explained either by concerted [4 + 2]-cycloaddition or by a multistep reaction via ionic intermediates. However, the authors suggested the concerted cycloaddition mechanism that would explain better the stereochemistry of the products. Then, treatment of imine 15, obtained from 2-methylaniline 13 and 3,3-dimethylcitronellal 14, with SnCl4 as Lewis acid afforded the trans-1,2,3,4,4a,9,9a,10-octahydroacridine 17 with a good yield (91%) and 1/99 cis/trans-diastereoselectivity. The formation of reaction compounds may be rationalized by cycloaddition through transition states TSI and TSII, which would lead to the formation of the trans- and cis-derivatives 17, respectively (Scheme 4).

Mechanistic approaches to form trans- and cis-adducts 17
Experimentally, both the reactivity and the cis/trans-selectivity of acridine derivatives 21 depended mainly on the substitution pattern at position 3 of the cyclization precursor 19, where steric bulk at C-3 favored the formation of the trans-product (Scheme 5). The successive addition of the Lewis acid and the aldehydes 19 to a precooled solution (−78 °C) of the amines 18 was also investigated with yields and cis/trans-ratios quite similar to the cyclization when isolated imines 20 were used. A broad range of Lewis (LA) or Brønsted (BA) acids can catalyze the formation of octahydroacridines 21, and the selectivity was found to be more dependent on the substrate structure than on the type of catalyst used.

Synthesis of octahydroacridines 21 by using Lewis or Brønsted acids
This process has been extended to chromium complexes. When starting from chromiun tricarbonylamine 22, the highly trans-selective preparation of (octahydroacridine)chromium complexes 24 was accomplished through the intramolecular [4 + 2]-cycloaddition of the (imino-arene)chromium complexes 23 (Scheme 6) [24]. Different conditions were used. For example, the reaction of the imino complexes 23 with catalytic amounts of SnCl4 (10 mol%) gave (1,2,3,4,4a,9,9a,l0-octahydroacridine)chromium tricarbonyl complexes 24 in high yields and stereoselectivity toward the trans-products, which seems to agree with a concerted hetero-Diels–Alder type mechanism. These (octahydrocridine)chromium complexes 24 could be also prepared by direct complexation of octahydroacridines 26 with chromium tricarbonyl 25 [25].

Synthesis of (octahydroacridine)chromium tricarbonyl complexes 25
A major drawback in cyclization and cycloaddition reactions of imines is the necessary activation; therefore, Laschat et al. [26] studied the reaction of anilines with para- and ortho-electron-withdrawing substituents with 3-methylcitronellal 14 in the presence of molecular sieves. The authors observed that the reaction proceeded differently depending largely on the type of molecular sieves used (Scheme 7). When powdered molecular sieves were used, anilines 18 gave very pure imines 27 in almost quantitative yield after 15 min. However, while the 4-nitroaniline 18 (R1 = 4-NO2) could not be converted to the imine 27 with powdered molecular sieves, when molecular sieve beads were used the formation of a mixture of the trans-cyclization products 28 and 29 was observed with other anilines.

Synthesis of octahydroacridines 28 and/or amines 29 with molecular sieves as activating agents
In previous works, a variety of Lewis or Brønsted strong acids has been used for the preparation of octahydroacridine derivatives. Sabitha et al. [27] studied the intramolecular hetero-Diels–Alder reaction of (R)-citronellal 30 and anilines 18 when bismuth (III) chloride was used (Scheme 8). As authors stated, the temperature of the reaction medium seems to play a key role in determining the cis/trans ratio formation, and in this case, the reaction with a catalytic amount of BiCl3 at 0 °C in acetonitrile proceeds in a highly stereoselective fashion giving trans-products 31 diastereoselectively. These trans-adducts 31 were obtained exclusively when non-substituted amines 18 (R1 = H) were used.

Synthesis of octahydroacridines 31 using bismuth(III) chloride as catalyst
A solid-supported catalyst (SiO2/ZnCl2), under MW irradiation and without any solvent, has also been used for the synthesis of octahydroacridines (Scheme 9) [28]. Thus, an environmentally friendly and efficient method consisting on a facile imino-Diels–Alder reaction from N-aryl amines 18 and (R)-citronellal 30 has been developed and corresponding octahydroacridine derivatives 32 have been obtained in good yields.

Synthesis of octahydroacridines 32 using a solid-supported catalyst
Six years later, the same group reported the synthesis of several acridine derivatives with sulfur substituents by using the same catalyst, (R)-citronellal 30 and thio-functionalized anilines 33. In this case, octahydroacridines 34 were obtained in moderate yields and poor diastereoselectivity, obtaining almost stoichiometric mixtures of cis- and trans-diastereoisomers (Scheme 10) [29].

Synthesis of octahydroacridine derivatives 34 using a solid-supported catalyst and thio-functionalized anilines
The intramolecular Povarov reaction mediated by solid-supported catalyst (SiO2/ZnCl2) at room temperature was achieved using 3-(arylthio)citronellal 35 and anilines 18 for the preparation of octahydroacridines 36 (Scheme 11) [29]. The best results were obtained when a mixture of aromatic amines 18 and 3-(phenylthio)citronellal 35 was stirred in the presence of SiO2/ZnCl2 (10 mol%) at room temperature, yielding the corresponding acridine derivatives 36 (Scheme 11). Regarding the stereochemistry of the ring fusion, the formation of a mixture of trans- and cis-adducts was observed, with good selectivity for the trans-fused 3-(phenylthio)octahydroacridines 36 in most of the cases. However, o-toluidine and 1-naphthylamine reacted with aldehyde 35 to afford mainly adducts with cis-selectivity (53:47–63:37 cis/trans).

Synthesis of octahydroacridine derivatives 36 with a solid-supported catalyst and thio-functionalized aldehydes
The synthesis of octahydroacridines 39 has been performed through intramolecular imino-Diels–Alder reaction starting from aniline 37 and citronellal 38 catalyzed by solid acid catalyst (Scheme 12) [30]. Several catalytic materials were studied, firstly, in the presence of pre-reduced Cu catalyst with molecular hydrogen, giving the corresponding octahydroacridines 39 as the only product in high yield. The use of the unreduced CuO/SiO2 (CuO/Si) catalyst at room temperature in the presence of air gave comparable results. Other solid acids can be used in the synthesis of 39, such as silica-alumina cracking catalysts with a 13% content of Al2O3 (SiAl 13) and a 0.6% alumina on silica (SiAl 0.6). Furthermore, two commercial acid-treated clays were also tested, namely Montmorillonite K10 and KSF. However moving from aniline 37 (R1 = H) to electron-rich amines 37 (R1 ≠ H), the selectivity of heterocycles 39 increased. In addition, differences in stereochemistry are evident. Pure Lewis solids (both CuO/silica and SiAl 0.6) promote 40/60 mixtures with a slight excess of trans-isomers. Conversely, the use of Brønsted acids, as Montmorillonite KSF, favor selectively the formation of the cis-isomer.

Synthesis of octahydroacridines 39 catalyzed by different solid acid catalysts
Based on the activation with molecular sieves, Laschat et al. studied the bis-cyclization of N-aryl diimines for the preparation of polycyclic ring systems [26]. In diamines with “separated” aromatic systems the reaction proceeded with molecular sieve beads. In this sense, when aryldiamines 40–42 were treated with 3-methylcitronellal 14 in the presence of 4 Å molecular sieve beads (Scheme 13), the biscyclization was complete after 24 h as determined by nuclear magnetic resonance (NMR) spectroscopy and corresponding compounds 46–48 were isolated. However, treatment of compound 40 (X = CH2) with aldehyde 14 in the presence of powdered molecular sieves gave, as expected, complete formation of the diimine 43 (X = CH2) after 15 min with no further cyclization. On the other hand, treatment of the isolated imine 43 with MeAlCl2 yielded the biscyclization product 46 (X = CH2). When the aromatic diamine has both functionalities in the same aromatic system (diamines 49–52, Scheme 13), the biscyclization reaction was not possible neither with powered molecular sieves nor with 4 Å MS beads. This means that the presence of a second imino function in the same aromatic ring decreases the reactivity of the first one, so that a stronger activation, e.g., by Lewis acids, is required to obtain the biscyclization products. Moreover, only when diamines 49 and 52 are used are the corresponding biscycloadducts obtained, probably because the two amino functions should be separated from each other, avoiding unfavorable steric interactions. Therefore, after formation of corresponding bisimines from amines 49 and 52, the treatment with 2.5 equivalents of MeAlC12 gave the corresponding biscyclization products.

Synthesis of bis-octahydroacridines 46–48
Exploiting the advantages of the solid-phase synthesis methodology, the preparation of octahydroacridines by concomitant formation of the imine and subsequent intramolecular capture of the alkene has been reported [31]. For example, when aniline resin 53 was reacted with (R)-citronellal 30 in the presence of Yb(OTf)3, the only product isolated after cleavage of the resin with TFA was octahydroacridine 55 in excellent yield as a single diastereoisomer (Scheme 14).

Solid-phase synthesis of octahydroacridine 55
Fluorous phase synthesis was also applied for the preparation of octahydroacridines [32]. Intramolecular aza-Diels–Alder reaction of functionalized N-aryl imines 56, produced in situ from aryl amines 18 and citronellal 38, was carried out at room temperature in the presence of trifluoroethanol (TFE), without any additional catalyst. In parallel, same reactions were studied in the presence of 10 mol% TiCl3 (Scheme 15). Regarding the yields of the reactions, a greater amount of product 57 was obtained with the use of the fluorinated solvent; however, worse results were obtained in terms of the stereochemistry of the reaction, while TiCl3 afforded a diastereomeric excess of trans-derivatives 57 the use of TFE gave to equimolecular mixtures of cis- and trans-derivatives.

Synthesis of octahydroacridines 57 by fluorous phase synthesis
The use of Lewis acids for the preparation of octahydroacridines presents some drawbacks, such as long reaction times, the use of low temperatures (−78 °C), and organic solvents. As previously reported, molecular sieves and BiCl3 have been introduced, among other variations, as alternatives for cyclization promoters solving some of these limitations. Moreover, with the use of ionic liquids (IL), easily recoverable after the reaction, no polluting solvents or complementary catalysts are needed, as the ionic liquids perform this function. In this regard, selenium- and tellurium-based ionic liquids have been used as solvents and/or catalysts in the synthesis of octahydroacridines via the hetero-Diels–Alder reaction involving (R)-citronellal 30 and aryl amines 37 (Scheme 16) [33]. When using 5 mol% of ionic liquid 61 or 62 the expected products 63 are obtained in good yield. Moreover, to reduce the reaction times, microwave irradiation was used, the consumption of the starting products was observed in 6 min and the expected products 63 were also obtained in good yields. A cis/trans-mixture of diastereoisomers was obtained as determined by NMR spectroscopy.

Synthesis of octahydroacridines 63 by using selenium- and tellurium-based ionic liquids
In the same way, other ionic liquids, such as 1-butyl-3-methylimidazolium tetrafluoroborate [bmim]BF4, 1-hexyl-3-methylimidazolium tetrafluoroborate [hmim]BF4, and 1-octyl-3-methylimidazolium tetrafluoroborate [octmim]BF4, have resulted suitable solvents to obtain octahydroacridine derivatives 21 (Scheme 17) [34]. The cycloaddition of aryl imines 20, formed in situ from a wide range of anilines 18 and (R)-citronellal 19 (R2 = Me, R3 = H) or 3-methyl citronellal 19 (R2 = R3 = Me), exhibited improved reactivity in the ionic liquid, thus reducing reaction time and significantly improving the yield. For example, the treatment of aniline 18 (R1 = H) with (R)-citronellal 19 (R2 = Me, R3 = H) at room temperature, without the need for any additional catalyst, resulted in the formation of 3,9,9-trimethyl-1,2,3,4,4a,9,9a,10-octahydroacridine 21 in 95% yield as a mixture of cis- and trans-isomers.

Synthesis of octahydroacridines 21 with ionic liquids
Not only citronellal was used as a suitable aldehyde for intramolecular Povarov reaction with anilines. The synthesis of cyclopenta[b]quinoline 70 and 71, as a part of isoschizozygane alkaloids, was accomplished by an intramolecular formal hetero-Diels–Alder Brønsted acid-catalyzed reaction from imine 66 obtained from unsaturated aldehyde 65 and aniline 64 (Scheme 18) [35]. In this case, the acid-catalyzed condensation of aromatic amine 64 with conjugated unsaturated aldehyde 65, followed by the cycloaddition reaction is described as a good route for the preparation of cyclopenta[b]quinoline derivative 70 and 71. The optimized conditions involved catalysis with 5 mol% of TsOH and provided 89% yield of an 86:14 mixture of 70 and 71 that could be readily separated by crystallization or column chromatography. The reaction is highly diastereoselective and produces adducts 70 and 71 with four contiguous stereocenters. The authors suggest in this case that the diastereoselectivity in adduct formation is determined by the nucleophilic attack of the diene on the iminium ion and directed by the C-3 stereocenter. Semiempirical calculations suggest that the closure of the cyclopentane ring would occur via a more stable intermediate 68 and give rise to 69 with a trans arrangement of the allylic cation and an amine. An alternative intermediate 67 is destabilized by the torsional interaction of the dienyl and imino moieties. The electrophilic aromatic substitution reaction between the allylic cation and the aniline in 69 will give rise to the more stable 1,2,4-trisubstituted arenes of structure 70 and 71.

Synthesis of cyclopenta[b]quinolines 70 and 71
Fused acridines have been prepared also by intramolecular Povarov reaction. 1,2,4-Trisubstituted cyclohexadienal 72, obtained by self-condensation of citral in the presence of NaH, is a suitable carbonyl substrate in the Povarov reaction providing molecular complexity and structural diversity. In this way, octahydrobenzo[c]acridines 73 have been prepared by intramolecular Povarov reaction of substituted anilines 18 and aldehyde 72 catalyzed by InCl3 (Scheme 19) [36]. Methylene chloride was selected as the best solvent for this transformation, since ethyl ether, THF, or hexanes were found to be less efficient. The scope of substituted anilines 18 with a varied array of functional groups was studied, showing a strong reaction efficiency effect by steric and electronic factors. For instance, electron-withdrawing groups and sterically crowded anilines lead to acridine derivatives 73 in very low yield.

Preparation of octahydrobenzo[c]acridines 73
Condensation of aldehyde 74 with o-toluidine 13 afforded compound 75 whose subsequent intramolecular hetero-Diels–Alder reaction catalyzed by a Lewis or Brønsted acid produced diastereoselectively the cyclopenta[c]acridine derivatives 76 (Scheme 20) [37]. The formation of the cis- or trans-isomers was modulated depending on the Lewis or Brønsted acid used.

Synthesis of cyclopenta[c]acridine derivatives 76
See Table 1 for the most representative examples of Sect. 2.1.
2.2 Steroid- and Carbohydrate-Derived Aldehydes
The intramolecular Povarov reaction with aliphatic aldehydes has been applied to the preparation of hybrid derivatives of tetrahydroquinolines condensed to a steroid skeleton by using Lewis or Brønsted acids. For this purpose, the reaction of the estrone derivative 77, with an allyl and a formyl group in suitable positions, with different anilines 18 was studied (Scheme 21) [38, 39]. After the reaction of aldehyde 77 and anilines 18 and subsequent treatment with BF3·OEt2, two different cyclic products 80 and 82 were obtained. Although compound 80 is the formal Diels–Alder adduct, the authors indicated that this compound may be obtained in a two-step mechanism from the initially formed iminium ion 78, which led to the carbocation 79 and then an electrophilic aromatic substitution to give 80. However, the iminium ion in 78 might also react with the alkene moiety to afford the cation 81 which could be further transformed by the addition of a nucleophile into compound 82.

Synthesis of quinolines 80 condensed to a steroid skeleton
In a similar manner, other aryl imino steroids 84 were prepared from the aldehyde steroid fragment 83 and various anilines 37 (Scheme 22) [40]. Afterwards, their intramolecular cyclization was studied via a Lewis acid-catalyzed reaction in the presence of BF3·OEt2. The nature of the substituent on the aniline influenced the reactivity since different tetrahydroquinoline derivatives 85 were formed from unsubstituted (R1 = H) or substituted (R1 = Br, OMe) anilines 37 followed by treatment with acetic acid anhydride/potassium acetate. However, when 4-nitroaniline (R1 = NO2) was used, a fluoro-d-homosteroid derivative was isolated, apparently through an intramolecular Prins reaction.

Synthesis of heterocycles 85 from steroid-derived aldehydes
Not only steroid-derived aldehydes have been used in intramolecular Povarov type cycloaddition reactions, as previously indicated, but also carbohydrate-derived aldehydes. Because natural carbohydrates, readily available and affordable, present a certain absolute stereochemistry, they are interesting substrates as chiral auxiliaries or chiral building blocks. Intramolecular hetero-Diels–Alder reactions of carbohydrate-derived aldehydes have been performed by Sabitha et al. [41]. In this work, pyrano[4,3-b]quinolines 88 have been prepared in a highly efficient and stereoselective way (Scheme 23). Aldimines 87 generated in situ from aromatic amines 18 and the O-allyl derivative of the d-glucose aldehyde 86 were treated in acetonitrile in the presence of a catalytic amount of BiCl3. This Lewis acid is the most suitable, since it can be used in substoichiometric amount (10 mol%), while other Lewis acids such as ZnCl2, FeCl3, ZrCl4, AlCl3, and BF3·OEt2 are needed in at least stoichiometric amounts. Cycloadducts 88 were obtained with high selectivity and good to excellent yields. In general, the reactions led to the formation of trans-isomers as major products, although small amounts of cis-isomers were observed. However, when a bulky group, such as tert-butyl (R1 = tBu), was present in the ortho position of the amine 18, only the trans-adduct 88 was exclusively obtained.

Synthesis of pyrano[4,3-b]quinolines 88 from O-allyl carbohydrate-derived aldehydes
On the basis of this protocol, the same group has performed the preparation of tetra- or pentacyclic furo[3,2-h][1,6]naphthyridine derivatives from anilines or 1-naphthylamine and a simple sugar derivative [42]. In this case, an N-prenylated sugar aldehyde 89 and different aromatic amines 18 were used in the condensation reaction to give the imines 90 (Scheme 24). Afterwards, intramolecular hetero-Diels–Alder reaction in the presence of bismuth(III) chloride as catalyst, under very mild conditions, was completed within 30 min to give the corresponding trans-fused products 91 stereoselectively and in good to excellent yields.

Synthesis of furo[3,2-h][1,6]naphthyridines 91 from N-allyl carbohydrate-derived aldehydes
2.3 Nitrogen- or Oxygen-Containing Aliphatic Alkene or Alkyne-Tethered Aldehydes
Some functionalized aldehydes with side chains possessing alkene or alkyne linked to a heteroatom such as oxygen or nitrogen have been described, allowing the preparation of fused heterocycles. For example, aldimines 93 derived from condensation of aromatic amines 37 with glyoxylic acid-derived O-allyl ester 92 in toluene and in the presence of molecular sieves were subjected to Lewis acid-catalyzed intramolecular Povarov reaction (Scheme 25) [43]. Using 1 equivalent of BF3·OEt2 in CH2Cl2 at room temperature, a mixture of quinoline-fused lactones 94 and amines derived from the reduction of aldimines 93 was observed, instead of expected tetrahydroquinoline-fused lactone (Scheme 25). This result suggests that the imine 93 reacts as an oxidant to convert the expected tetrahydroquinoline into quinoline 94, confirming that this oxidation proceeds faster or at the same rate as the intramolecular Povarov cycloaddition. However, the presence of an oxidant such as 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) afforded quinoline 94 in low to moderate yields. Two equivalents of DDQ were necessary to convert aldimines 93 into quinoline-fused-lactone 94 with better yields without any traces of amines derived from the reduction of aldimines 93.

Quinoline-fused lactones 94 obtained by intramolecular Povarov reaction
In a similar way, the glyoxylic acid-derived aldehydes 95 (X = O) react with anilines 18 in the presence of TFA to afford the tetrahydroquinoline lactones 97. A stepwise process in the [4 + 2]-cyclization would provide the trans-diastereoisomer (Scheme 26) [44]. When the reaction was performed with the N-benzylglyoxamides 96 (X = NBn) the corresponding derivatives 98 were obtained. In these cyclizations, the trans-configuration of the major isomer was obtained (Scheme 26).

Intramolecular Povarov reaction with glyoxylic 95 or glyoxamide-derived aldehydes 96
This process has also been extended to α-amino acid-derived aldehydes. Raghunathan et al. [45, 46] described an efficient synthesis of unreported fused pyrroloquinolines through reaction of aldimines 100, resulting from aromatic amines 37 and N-prenylated aliphatic aldehydes 99, in a Lewis acid catalyzed intramolecular Povarov reaction. Aldehydes 99 and anilines 37 were subjected to the intramolecular Povarov reaction using a 20 mol% of InCl3 in MeCN (Scheme 27). Thus, after generation of the corresponding imine, this is trapped by an N-tethered prenyl moiety cyclizing intramolecularly to give the Povarov adducts 101 in excellent chemical yields and trans-selectivity (40:60–23:77 cis/trans). Pyrroloquinolines 101 exhibited good antibacterial activity toward six different bacterial strains with MIC values of 5 mM. Gyrase assays showed the potential of compounds 101 to bind to gyrase, preventing their gene expression [45, 46]. The same group reported the diastereoselective synthesis of trans-fused pyrroloquinolines 101 by the InCl3-promoted intramolecular Povarov reaction of aldimines resulting from the condensation of aromatic amines 37 and alkene-tethered aldehydes derived from (S)-phenylalanine 99 (R2 = Bn) [47]. The cycloaddition reaction resulted to be stereoselective, and trans-pyrroloquinolines 101 were obtained in 86–97% yield.

InCl3-catalyzed intramolecular Povarov reaction for the preparation of pyrroloquinolines 101
Cyclization of the imine formed from cinnamoylaminoaldehyde 102 derived from different amino acids with aniline derivatives 37 using the mild Lewis acid ytterbium triflate yielded the thermodynamically more stable trans-products 103 (Scheme 28). The stereoselectivity was explained by a stepwise mechanism involving the transition states TSI and TSII (Scheme 28). In the electrophilic attack of the ytterbium–imine complex, fewer steric interactions would occur between the R3 substituent and the aromatic ring of the aniline in TSII, maintaining the equatorial orientation of the aryl substituent in the subsequent closure of the second ring. The strategy of the cyclization from aminoaldehydes 102 was also transferred from solution to solid phase [44].

Ytterbium triflate-catalyzed Povarov reaction with N-cinnamoyl-α-aminoaldehydes
Likewise, aldimines 105 prepared from N-(prenylaminomethyl)cinnamaldehydes 104 derived from Morita–Baylis–Hillman adducts of acrylates, and aromatic amines 37, were subjected to an intramolecular cycloaddition reaction to furnish benzonaphthyridine derivatives 106 (Scheme 29) [48]. Several Lewis acids, for instance, Yb(OTf)3, InCl3, Sc(OTf)3, or BiCl3, or even Brønsted acids such as TFA, were used for this transformation; however, only in the presence of BiCl3 did the reaction proceed efficiently to afford azaheterocycles 106 in good to excellent yields, but as a diastereomeric mixture of cis- and trans-adducts.

BiCl3-promoted intramolecular Povarov reaction with N-(prenylaminomethyl)cinnamaldehyde
Fused benzo[b]pyrrolo[1,2-h][1,7]naphthyridine heterocycles 111, without a gem-dimethyl group, were prepared by taking vinyldisilane-terminated N-aryl imine 109 as a precursor obtained from aldehydes derived from l-prolinol 108 (Scheme 30) [49]. This process yielded new benzo[b]pyrrolo[1,2-h][1,7]naphthyridines 111 as a 50:50 mixture of diastereoisomers by Lewis acid-catalyzed cyclization of N-aryl imines 109 as a key step.

Synthesis of fused benzo[b]pyrrolo[1,2-h][1,7]naphthyridines 111
Condensation of the l-proline-derived aldehydes 112 with o-toluidine 13 afforded compounds 113 whose subsequent intramolecular hetero-Diels–Alder reaction catalyzed by a Lewis or Brønsted acid produced diastereoselectively the benzo[b]pyrrolo[1,2-h][1,7]naphthyridine derivatives 114 (Scheme 31) [37, 50, 51]. This cycloaddition reaction, where a second nitrogen atom has been introduced, displayed a remarkable Lewis acid-dependent reversal of the diastereoselectivity. The formation of the cis- or trans-isomers was modulated depending on the Lewis or Brønsted acid used. When using FeCl3, SnCl4, BF3·OEt2, p-TsOH, TFA, AlCl3, and Et2AlCl trans-stereoselectivity was observed, whereas when using EtAlCl2, MeAlCl2 and Me2AlCl2 the diastereoselectivity is favored to the formation of the cis-isomers.

Intramolecular Povarov reaction of l-proline-derived aldehydes
Laschat’s group studied the Povarov reaction of proline-derived aldehydes 115 with aromatic diamines 40 or 116 (Scheme 32) [52]. In this way, bis(benzo[b]pyrrolo[1,2-h][1,7]naphthyridine)methane 118 or 119 were isolated, respectively.

Synthesis of fused bis-benzopyrrolo[1,7]naphthyridine derivatives 118 and 119
The same group studied the Lewis acid-catalyzed cyclization of N-aryl imines 121 obtained from l-phenylalanine derived aldehyde 120, catalyzed by EtAlC12 (Scheme 33) [53]. In this way, the benzo[g]quinolino[2,3-a]quinolidines 122 were obtained. The starting aldehyde 120, prepared from l-phenylalanine, was treated with various aryl amines 18 in the presence of molecular sieves, giving rise to the corresponding imines 121, which were immediately cyclized in the presence of EtAlC12 to the benzo[g]quinolino[2,3-a]quinolidines 122. The formal hetero-Diels–Alder reaction of 121 proceeded with high diastereoselectivity in favor of the cis configured product. The amino-substituent into a rigid pentacyclic system like 122 resulted in a good cytotoxic activity against human brain tumor cell lines.

Synthesis of fused benzo[g]quinolino[2,3-a]quinolidines 122
On the one hand, the corresponding imine intermediate, obtained by condensation of aldehyde 120 with ethyl 4-aminobenzoate 123 in the presence of molecular sieves, directly treated with EtAlCl2, afforded the pentacyclic benzo[b]isoquino[2,3-h][1,7]naphthyridine 125 with high cis-diastereoselectivity (Scheme 34). Whereas, when the same imine was treated with SnCl4 the pentacyclic trans-diastereoisomer 126 was obtained in a dr 0.5/99.5 [54]. The authors highlight the interest of preparing polycyclic all-trans-derivatives because of their more planar shape, which could induce a different interaction mode with DNA.

Synthesis of fused benzo[b]isoquinolino[2,3-h][1,7]naphthyridines 125 and 126
More aromatized fused heterocyclic compounds can be prepared directly by intramolecular Povarov reaction with triple-bond-functionalized aldehydes. When aldimines 129 derived from condensation of aromatic amines 127 with glyoxal-derived alkynes 128 are used, only one equivalent of DDQ is required to carry out the oxidation of dihydroquinoline-fused lactones 130, obtained by intramolecular Povarov cycloaddition using 1 equivalent of BF3·OEt2 in CH2Cl2 at room temperature, affording the corresponding quinoline-fused lactones 131 (Scheme 35) [43].

Synthesis of quinoline-fused lactones 131
Pyrrolo[3,4-b]quinolines 133 can also be synthesized by using propargyl aldehydes derived from α-amino acids 132 and various substituted anilines 18. The intramolecular Povarov reaction requires a strategically positioned aldehyde moiety tethered to an alkynyl group. Hence, the reaction of N-propargyl aldehyde 132 and aromatic amines 18 was carried out in the presence of BF3·OEt2 in dry CH2Cl2. Using these reaction conditions, a series of pyrrolo[3,4-b]quinolines 133 were obtained in excellent yields (Scheme 36) [55].

Synthesis of pyrrolo[3,4-b]quinolines 133
When l-proline-derived aldehyde 134 with tethered triple bond was used, benzo[b]pyrrolo[1,2-h][1,7]naphthyridine 137 was obtained (Scheme 37) [51]. Treatment of imine 135 bearing an internal alkyne moiety with BF3·OEt2 resulted in the clean formation of the indolizino[3,4-b]quinoline 137. Obviously, the initially formed cyclization product 136 undergoes a rapid dehydrogenation to the aromatic compound 137.

Synthesis of benzo[b]pyrrolo[1,2-h][1,7]naphthyridine derivative 137
A small library of A- and D-ring modified luotonin-inspired heterocyclic systems was synthesized in moderate to good yields following a six-step route that starts from phenylalanine. The key step of this total synthesis consists in an intramolecular Povarov reaction of imines obtained from a tetrahydroquinoline-derived alkynyl aldehyde 138 and various aryl amines 18 (Scheme 38). The corresponding N-aryl imines 139 were formed in situ from aldehyde 138 and substituted aryl amines 18 in the presence of 4 Å molecular sieves. Without isolation, subsequent treatment of N-aryl imines 139 with 1.5 equivalents of BF3·OEt2 afforded the target pentacyclic heterocycles 140 in yields that were approximately in the 40–50% range [56].

Synthesis of luotonin A analogs via intramolecular Povarov reaction
When longer side chain with a triple bond is used in aldehyde 141, benzo[b]isoquinolino[2,3-h][1,7]naphthyridine 142 is obtained (Scheme 39) [54]. In this case, aldehyde 141 was treated with ethyl 4-aminobenzoate 123 and the formation of the corresponding imine was observed and used further without purification. Subsequent addition of BF3·OEt2 and aqueous workup resulted in the formation of the quinoline ester 142 in 38% yield. When BF3·OEt2 was replaced by EtAlCl2, a chloro compound was isolated in 7% yield as a minor by-product. The isolation of this latter compound, further supports a cationic cyclization mechanism where in the presence of EtAlCl2 the cyclization of imine should afford carbenium ion 143, which can undergo Friedel–Crafts-type electrophilic aromatic substitution, followed by tautomerization to give 142 after oxidation.

Synthesis of fused benzo[b]isoquinolino[2,3-h][1,7]naphthyridines 142
3 Aromatic Amines and Aromatic Aldehydes
In this section, we disclose the intramolecular Povarov reaction using aromatic amines and aromatic aldehydes which allows the preparation of a diversity of polycyclic nitrogen containing heterocycles. Reactions have been classified considering the structure of the ortho-formylarenes and the way that the dienophile is tethered to the benzene ring.
3.1 C-Alkenyl(Alkynyl) Ortho-Formylarenes
Hexahydrobenzoacridine derivatives can be synthesized by intramolecular Povarov reaction of aldimines 145 derived from aromatic amines 18 and 2-prenylated benzaldehyde 144 (Scheme 40) [57]. The use of amines bearing electron-withdrawing or electron-donating groups in this approach, which was promoted by catalytic amount of bismuth(III) chloride, seems to have no effect on the reaction time or the yield. cis-Annulated hexahydrobenzo[c]acridines 146 were achieved in all cases with selectivities up to 97:3.

2-Prenylated benzaldehyde as carbonyl component in the BiCl3-promoted intramolecular Povarov reaction
Through a tandem allylation/intramolecular Povarov reaction, polycyclic compounds 150 were synthesized by a [4 + 2] cycloaddition process (Scheme 41). The imine group in compound 147 acts as a directing group to enable the introduction of a pendant alkene, thereby enabling a Lewis acid-catalyzed intramolecular Povarov reaction. Specifically, a manganese (I) complex catalyzed the directed C–H allylation with allene 148, producing compound 149 ready for an in situ Povarov cyclization catalyzed by silver (I). Other Lewis acid, including BiCl3, Sc(OTf)2 and Zn(OTf)2, led to significant decomposition of the ketimine 149. The reaction proceeds with high bond-forming efficiency (three C–C bonds), broad substrate scope, high regio- and trans-stereoselectivity, and 100% atom economy (Scheme 41). The polycyclic indenoquinoline, bearing two stereogenic centers, was obtained as a single diastereoisomer. The compatibility of different allenes was also examined, being the symmetric 1,1-dialkyl-substituted allenes the most efficiently coupling partners. The potential synthetic utility was demonstrated by a gram-scale synthesis [58].

Synthesis of polycyclic compounds 150 through previous allylation followed by intramolecular Povarov reaction
Liu et al. reported in 2013 the synthesis of indeno[1,2-b]quinolines 155 by means of reaction of aromatic amines 18 with o-propargylbenzaldehydes 151 (Scheme 42) [59]. Using a water-removing agent such as 4 Å molecular sieves compounds 155 were obtained in 1,2-dichloroethane (DCE) at 80 °C with 2 equivalents of functionalized aniline 18. By reducing the amount of aromatic amine 18 to 1 equivalent, the yield of indenoquinoline 155 was affected by a significant decrease. A widespread diversity of substituted o-propargylbenzaldehydes 151 and aromatic amines 18 were appropriate for this transformation, giving to the formation of indeno[1,2-b]quinoline derivatives 155 in good to high yields. The mechanism of the formation of indenoquinolines 155 can start via initial formation of imine 152 by condensation of aromatic amines 18 with aldehydes 151. The intramolecular Povarov reaction between azadiene moiety and alkyne group of 152 affords intermediates 153. Elimination of OR2 group and subsequent double-bond isomerization furnish indenoquinolines 155 (Scheme 42).

Synthesis of indeno[1,2-b]quinolines through reaction of aromatic amines and o-propargylbenzaldehydes
A modified Povarov reaction involving 2′-alkynylbiaryl-2-carbaldehydes 156 and aryl amines 18 with tandem oxidation was performed using catalytic FeCl3. The outcome was an efficient general synthesis of dibenzo[a,c]acridines 157 with moderate to high yields (Scheme 43). This method offers simplicity in the preparation of substrates, diverse substrate scope, and high atom economy. The optimum reactions conditions for the general synthesis of dibenzo[a,c]acridine derivatives 157 were obtained with a 10 mol% FeCl3 in toluene at 100 °C in open air. The synthesized compounds 157 had significant absorption and emission properties [60].

Synthesis of dibenzo[a,c]acridines using FeCl3
3.2 O-Alkenyl(Alkynyl) Ortho-Formylarenes
The reaction of aniline derivatives 18 with O-allyl derived salicylaldehydes 158 has been widely used for the synthesis of polysubstituted tetrahydrochromeno[4,3-b]quinolines 159 (Scheme 44). The intramolecular [4 + 2] cycloaddition reaction has been catalyzed in the presence of different Brønsted acids, such as trifluoroacetic acid (TFA) [61] and sulfamic acid [62], or in the presence of Lewis acids such as Yb(OTf)3 [61], BiCl3, [63] lithium perchlorate in diethyl ether (LPDE) [64], triphenylphosphonium perchlorate (TPP) [65], and InCl3 [66], and even in the presence of a recyclable ionic liquid as a reaction medium, [bmim]BF4 [67]. The reactions transcurred from good to excellent yields and a mixture of cis/trans-diastereoisomers was obtained in all cases.

Synthesis of polysubstituted tetrahydrochromeno[4,3-b]quinolines 159 using different catalytic conditions
Alternatively, condensation of 2-allyloxynaphthalene-1-carbaldehyde 160 with substituted anilines 37 and subsequent intramolecular cyclization in the presence of BF3·OEt2 yielded benzochromeno[4,3-b]quinolines 161 (Scheme 45) [68]. When the reaction was carried out with TFA, the obtained products were not those expected but their dehydrogenated derivatives 162. In a similar manner, cis-compounds 161 could also be obtained performing the intramolecular aza-Diels–Alder reaction in [bmim]BF4 ionic medium [67], being the last one a green protocol that offers significant advantages over reported methods.

Synthesis of benzochromeno[4,3-b]quinoline derivatives 161 and 162
Substituted chromeno[4,3-b]quinolines 166 were achieved, under mild conditions, by tandem intramolecular aza-Diels–Alder reaction/photooxidation using a strategy of combination of visible-light photoredox and Lewis-acid catalysis. This intramolecular aza-Diels–Alder cycloaddition took place between the in situ generated benzylidene imine 164, derived from aryl amines 18 and salicylaldehydes 163 bearing an alkene-tethered partner, followed by oxidative aromatization to give the products 166 (Scheme 46). The reaction takes place using BF3·OEt2 as Lewis acid and Ru(bpy)3(PF6)2 as photosensitizer in acetonitrile under aerobic condition with the irradiation of visible light [69].

Preparation of chromeno[4,3-b]quinolines via intramolecular aza-Diels–Alder reaction
The reaction between nitrobenzenes 167 and ω-unsaturated aldehydes, that is, 2-(cinnamyloxy)benzaldehydes 168, in the presence of iron as reductant and catalytic amounts of montmorillonite K10 in aqueous citric acid at 80 °C produced trans-fused tetrahydrochromeno[4,3-b]quinolines 170 exclusively with yields ranging from 69% to 87% (Scheme 47) [70]. It is assumed that the sequence of reactions starts with an iron-mediated reduction of the nitrobenzene 167. The resulting aniline reacts with the ω-unsaturated aldehyde 168 to give the corresponding imine 169, which in turn undergoes an intramolecular aza-Diels–Alder reaction. This Povarov-type reaction is catalyzed by montmorillonite K10, proceeds via an exo-transition state structure, and delivers the trans-fused tetrahydrochromeno[4,3-b]quinolines 170 in diastereomerically pure form. The method enables the replacement of anilines with nitrobenzenes as substrates for intramolecular aza-Diels–Alder reactions. The domino reaction can be performed with numerous functionalized nitrobenzenes and a number of 2-(cinnamyloxy)benzaldehydes.

Synthesis of trans-fused tetrahydrochromeno[4,3-b]quinolines from nitrobenzenes 167
Other configurational and functionally diverse heterocyclic compounds have also been prepared through an intramolecular formal aza-Diels–Alder cyclization. The substrates included substituted functionalized salicylaldehydes with a variety of anilines to yield different tetrahydroquinoline products [44]. Thus, cyclization of cinnamyl salicylaldehyde ethers 171 with substituted anilines 18 and treatment with trifluoroacetic acid in acetonitrile at 55 °C for 30 min afford the tetrahydroquinoline cycloadducts 173 in good yield (Scheme 48). Modest variations are well tolerated on the aniline ring, and the process can be extended to an electron-rich cinnamate ester 172 as well, although products 174 were obtained in modest yield. The major products isolated as single isomers after chromatographic or crystallographic purification possess the thermodynamically favored trans-configuration.

Intramolecular aza-Diels–Alder cyclization using O-cinnamyl- and O-cinnamoylsalicylaldehydes
This methodology has also been extended to solid-phase synthesis with acid-sensitive methoxy benzaldehyde polystyrene (AMEBA) resin 177 (Scheme 49). The reaction of immobilized anilines 175 with salicylic aldehyde derivatives 176 containing an electron-rich olefin substituent, catalyzed by both TFA and Yb(OTf)3 yielded polysubstituted tetrahydrochromeno[4,3-b]quinolines 178 as 50:50 mixtures of diastereoisomers, which were subsequently separated by preparative HPLC [71].

Solid-phase preparation of polysubstituted tetrahydrochromeno[4,3-b]quinolines 178 extended to solid phase
In addition, when using diamines and TPP or alternatively [bmim]BF4 as catalysts bis-tetrahydrochromeno[4,3-b]quinolines 181 or 182 could also be obtained (Scheme 50). The reaction of imines 179 or 180 derived from O-allyl salicylaldehydes 158 and 4,4′-methylenedianiline 40 or 4,4′-oxadianiline 41 over anhydrous Na2SO4 in acetonitrile and in the presence of 40 mol% of TPP, underwent intramolecular bis-cyclization to give the corresponding bis-4,4′-methylene 181 or 4,4′-oxatetrahydrochromeno[4,3-b]quinolines 182, respectively, in good yields as a mixture of three isomers cis/cis, cis/trans, and trans/trans in a ratio of 1:1:1 (Scheme 50). The product ratio was determined by examination of the 1H-NMR spectrum of the crude product mixture [65]. Similarly, treatment of 4,4′-methylenedianiline 40 (X = CH2) with the O-prenyl derivative of salicylaldehyde 158 in [bmim]BF4 afforded the biscyclization product as a mixture of cis/cis, cis/trans, and trans/trans-isomers. However, in the case of 4,4′-oxadianiline 41 (X = O), the product was obtained exclusively as cis/trans-bis-adduct 182 under similar conditions [67].

Preparation of bis-tetrahydrochromeno[4,3-b]quinolines using different reaction conditions
Very recently, Kouznetsov et al. [72] have described the synthesis of chromeno[4,3-b]quinolines 186 promoted by I2 and DMSO from easily available aryl amines 18 and O-cynnamyl salicyladehydes 183. Iodine acts as a Lewis acid to catalyze the formation and cyclization of the imines 184, using DMSO as the solvent, to generate the respective tetrahydrochromenoquinolines 185 as intermediates (Scheme 51). Finally, the I2/DMSO catalytic system could mediate the aromatization of 185 to the corresponding chromeno[4,3-b]quinolines 186. The scope and general applicability of the reaction has been widely studied, considering both anilines and 2-(cinnamyloxy)benzaldehydes. The reaction proceeds under mild conditions, tolerates a great range of functional groups and features high step economy, since it constitutes a tandem process.

Synthesis of chromeno[4,3-b]quinolines promoted by I2/DMSO system
Other strategies have been used for the synthesis of chromenoquinoline derivatives involving the use of O-propargyl-substituted salicylaldehyde ethers. In this context, activation of a terminal alkyne C–H bond by transition-metal catalysts is one of the major interests in synthetic organic chemistry. Several reports describe this activation by transition metal catalyst such as Ag, Au(I), Au(III), Cu(I), Ru, and Ir [73,74,75,76]. In this way, Nagarajan’s group [77] described a straightforward approach to chromenoquinolines using a mixture of copper(I) iodide and lanthanum triflate as an efficient catalyst. Moreover, copper compounds are readily available, non-air-sensitive, nontoxic catalysts and inexpensive, compared with other transition metal catalysts. 6H-Chromeno[4,3-b]quinolines 189 can be attained in good yields by intramolecular Povarov reaction of the intermediate aldimine 188 derived from the reaction of aromatic amines 18 with O-propargylated salicyladehydes 187 (Scheme 52). The combination of Cu(I) species/Lewis or Brønsted acids resulted in excellent catalytic properties, since the use of only a Lewis acid such as InCl3, BF3·OEt2, or La(OTf)3; or the use of a copper species such as CuI, CuBr or CuCl, afforded chromenoquinolines 189 with worse chemical yields. Aromatic amines 18 with ring-activating groups in ortho-, meta-, or para-positions participated in this reaction, giving the expected products with remarkably comparable yields (Scheme 52). Conversely, aromatic amines 18 with electron-withdrawing groups (R1 = NO2, CO2R, CN) did not afford the expected chromenoquinolines 189. Moreover, substitution at O-propargylated salicyladehyde ring seems not to affect the reaction.

Efficient synthesis of 6H-chromeno[4,3-b]quinolines through CuI/La(OTf)3 promoted intramolecular Povarov reaction
Alternatively, a green and simple intramolecular domino condensation aza-Diels–Alder reaction between anilines 18 and O-propargylated salicylaldehydes 190 in the presence of CuI as catalyst in H2O/EtOH was used to obtain 6H-chromeno[4,3-b]quinolines 192 in 75–83% yield (Scheme 53) [78]. A plausible mechanism assumes the formation of a copper-acetylide imine intermediates 191 that after the sequential intramolecular [4 + 2] cycloaddition, protonation, and oxidation generate the product 192. The best yield was only obtained using highly electron-rich anilines. The simplicity of the starting materials, good yields of the products, and use of green, cheap, and nontoxic solvents are the main advantages of this method.

Copper-catalyzed intramolecular domino synthesis of 6H-chromeno[4,3-b]quinolines in green conditions
Complementarily, a new method was developed to synthesize 7-halogenated chromenoquinolines 195 and 7-halogenated thiochromenoquinolines 196. The products can be directly obtained through Cu-catalyzed cascade reaction, that is, aza-Diels–Alder reaction of Schiff base 193 or 194, followed by halogenation (chlorination or bromination), using chloranil or bromanil as halogen sources. Cu2O worked as both a Lewis acid and transition-metal catalyst in the aza-Diels–Alder reaction and halogenation reaction, respectively. Chloranil and bromanil also performed dual functions, that is, as a halogen source and oxidant. Although the halogenated products were obtained in moderate yields (Scheme 54), the present method is highly useful in organic synthesis because of mild reaction conditions and experimental simplicity [79].

Synthesis of halogenated chromenoquinolines and thiochromenoquinolines via Cu-catalyzed cascade reaction
Very recently, Wang’s group [80] developed an intramolecular Povarov reaction for the construction of chemically stable chromenoquinoline-based covalent organic frameworks (COF). Thus, the synthesis involves the formation of the imine COF 199 by reaction of 2,5-bis-propargyloxy terephthalaldehyde (BPTA) 198 and 1,3,5-tris(4-aminophenyl)benzene (TAPB) 197 in a mixture of o-dichlorobenzene (o-DCB) and n-butanol (n-BuOH) followed by the addition of aqueous acetic acid (Scheme 55). Next, the intramolecular Povarov reaction to integrate the alkyne moieties into the imine COF 199 and to build the chromenoquinoline ring, was carried out using BF3·OEt2 as catalyst in toluene and in the presence of chloranil as an oxidating agent, leading the chromenoquinoline-COFTAPB-BPTA 200 in a 96% yield (Scheme 55). Instead of TAPB 198 other amines, such as 1,3,6,8-tetrakis(4-aminophenyl)benzene 201, 1,3,5-tris-(4-aminophenyl)triazine (TAPT) 202, and Ni-porphyrin 203, were also used to synthesize additional monomers with different symmetries and functional core moieties (Fig. 2). This novel approach achieves a high cyclization degree of 80–90%, which endows the chromenoquinoline-COFs with excellent chemical stability toward strong acid, base, and redox reagents. The absorption and fluorescence intensities of chromenoquinoline-COFs are sensitive to acid, which allows for dual-mode sensing of strongly acidic environments.

Synthesis of chromenoquinoline-COFTAPB-BPTA 200

Other amines used to synthetize chromenoquinoline-based covalent organic frameworks (COF)
See Table 2 for the most representative examples of Sect. 3.2.
3.3 N-Alkenyl(Alkynyl) Ortho-Formylarenes
As in the case of O-allyl derivatives of salicylaldehydes, BiCl3 was used as Lewis acid to catalyze the intramolecular [4 + 2] cycloaddition reaction of in situ generated aldimines derived from aromatic amines and o-aminobenzaldehyde [81]. Therefore, treatment of anilines 18 with the N-allyl derivative of o-aminobenzaldehyde 204 in the presence of 10 mol% BiCl3 in refluxing acetonitrile resulted in the formation of hexahydrodibenzo[b,h][1,6]naphthyridines 205 as trans- and cis-diastereoisomers in a 1:1 ratio in excellent yields (Scheme 56).

Preparation of hexahydrodibenzo[b,h][1,6]naphthyridines catalyzed by BiCl3 as Lewis acid
Alternatively, 1,6-naphthyridines 209 were achieved by tandem intramolecular aza-Diels–Alder reaction/oxidative aromatization using a strategy of combination of visible-light photoredox and Lewis acid catalysis. This intramolecular aza-Diels–Alder cycloaddition of the in situ generated benzylidene imines 207, derived from aryl amines 18 and 2-aminoaryl aldehydes 206 bearing an alkene-tethered partner, took place followed by oxidative aromatization to give products 209 (Scheme 57). The reaction proceeds using BF3·OEt2 as Lewis acid and Ru(bpy)3(PF6)2 as photosensitizer in acetonitrile under aerobic condition with the irradiation of visible light. This method provided a new access to the synthesis of important heterocycles under mild conditions [69].

Preparation of 1,6-naphthyridines via tandem intramolecular aza-Diels–Alder reaction/oxidative aromatization
Another highly efficient synthesis of 5,6-dihydrodibenzo[b,h][1,6]naphthyridines 212 was achieved by reaction between 2-(N-propargylamino)benzaldehydes 210 and aryl amines 18 in the presence of CuBr2 (Scheme 58). First, other copper halides were tested, such as CuCl, CuBr, and CuBr2, with the last one being the most efficient in terms of yield. Meanwhile, CuI and Cu(OAc)2 resulted to be inefficient. The proposed route involves that the in situ generated electron-deficient heterodienes 211 underwent an intramolecular inverse electron-demand hetero-Diels–Alder reaction followed by spontaneous dehydrogenation. This reaction tolerated a large number of substituents to afford diverse products under mild conditions [82].

Synthesis of 5,6-dihydrodibenzo[b,h][1,6]naphthyridine derivatives via copper catalyzed reaction
4 Aromatic Amines and Heteroaromatic Aldehydes
4.1 Five-Membered Nitrogen-Containing Heterocyclic Alkene-Tethered Aldehydes
N-cinnamyl pyrrole-2-carbaldehyde 213 has been used as carbonyl component for the preparation of intermediate imines 214, which readily cyclized in an intramolecular Povarov reaction to afford pyrrolizino-annulated quinoline derivatives 215 in good yields (Scheme 59) [83]. Indium trichloride proved to be an efficient Lewis acid catalyst for this transformation.

Synthesis of pyrrolizino[1,2-b]quinolines 215 by InCl3 promoted-intramolecular Povarov reaction
The synthesis of indolo-annulated pyrroloquinoline via the imino-Diels–Alder reaction has been described by Nagarajan et al. [84] In this case, Lewis acid-catalyzed intramolecular imino-Diels–Alder reaction of N-prenylated-2-formyl-3-chloroindole 216 (R2 = R3 = Me, R4 = Cl) and substituted anilines or naphthylamines 37 produced indolopyrroloquinolines 217 in moderate to excellent chemical yields and high cis-diastereoselectivity (Scheme 60). An array of Lewis acid catalyst has been tested in this approach and among them, La(OTf)3, Sc(OTf)3, and Yb(OTf)3 gave better diastereoselectivities. Only the cis-isomer was observed in the presence of La(OTf)3, when the reaction was performed at 130–140 °C. Similary, when indole-2-carbaldehydes containing an internal dienophile were used, indolo[2,1-a]pyrrolo[4′,3′:2,3]-7a,8,13,13b-tetrahydroquinolines 217 have been prepared from several substituted aromatic amines 37 through the intramolecular imino-Diels–Alder reaction (Scheme 60) [85]. N-alkenyl indole-2-carbaldehydes 216 (R4 = H) reacted with various p-substituted anilines 1 in the presence of different Lewis acid catalysts, namely AlCl3, BF3·OEt2, ZnCl2, and InCl3. However, the best overall yields were obtained when 20 mol% of InCl3 was used, and under these reaction conditions, the corresponding cycloadducts 217 were obtained in good overall yield and cis-diasteroselectivities ranging from 80:20 to 96:4.

Lewis acid-catalyzed intramolecular Povarov reaction for the synthesis of indolo-annulated pyrroloquinolines
N-alkenyl pyrrolopyrimidine-6-carbaldehydes 218 have also been used as carbonyl component for the preparation of intermediate imines, which readily cyclized in an intramolecular Povarov reaction to afford uracil-annulated quinoline derivatives 223 in good yields and good to excellent stereoselectivities (Scheme 61) [83, 86]. Different Lewis acid catalysts were studied in this transformation, namely BF3·OEt2, Yb(OTf)3, Sc(OTf)3, and InCl3. Indium trichloride proved to be the most efficient with overall yields higher compared with other tested Lewis acid catalysts [86]. These results indicate that the cyclization pathway proceeds by a stepwise mechanism as outlined in Scheme 61. Tetrahydroquinoline-annulated heterocycles 223 (R1 = Cl) were evaluated for their antibacterial activity against six different bacterial strains, being as active as the antibiotic ciprofloxacin and presenting a MIC value of 2.5 mg/mL against Escherichia coli [83].

Synthesis of quinoline-annulated heterocycles by InCl3-assisted intramolecular Povarov reaction
By using N-aryl imines 225, generated in situ from anilines 18 and S-allyl-1H-pyrazole-4-carbaldehyde derivatives 224, through an intramolecular imino-Diels–Alder reaction hexahydropyrazolo[4′,3′:5,6]thiopyrano[4,3-b]quinolines 226 have been prepared in good yields (Scheme 62) [87]. In this case, the reaction has been catalyzed by 5 mol% of BiCl3 and the process is highly diastereoselective by the exclusive isolation of the cis-cycloadduct. Some years later, Raghunathan et al. [88] reported similar synthesis of hexahydropyrazolo[4′,3′:5,6]thiopyrano[4,3-b]quinolines 226 by InCl3-promoted intramolecular Povarov reaction of S-allyl-1H-pyrazole-4-carbaldehyde derivatives 224 with substituted anilines 18 (Scheme 62). Cycloadducts 226 were obtained with 85–96% chemical yields and diastereoselectivities higher than 94:6 in favor of the cis-quinoline derivative.

Pyrazole-annulated sulfur heterocycles by intramolecular Povarov reaction catalyzed by BiCl3 or InCl3
Reaction of aldehydes 224 and bis-aniline derivatives 40 or 41 affords intermediate aldimines 227 or 228, which in the presence of 40 mol% of InCl3 undewent bis-intramolecular Povarov reaction to yield bis-tetrahydropyrazolo-thiopyrano[4,3-b]quinoline derivatives 229 or 230, respectively, as a mixture of three inseparable isomers cis/cis, cis/trans, and trans/trans in favor of the cis/cis-isomer (Scheme 63) [88].

Pyrazole-annulated sulfur heterocycles by bis-intramolecular Povarov reaction catalyzed by InCl3
See Table 3 for the most representative examples of Sect. 4.1.
4.2 Six-Membered Nitrogen- or Oxygen-Containing Heterocyclic Alkene-Tethered Aldehydes
Recently, Zhang’s group [89] reported a one-step construction of substituted indolizino[1,2-b]quinolin-9(11H)-ones by combination of visible-light-photoredox and Brønsted acid catalysis through an intramolecular Povarov cycloaddition reaction under mild conditions. Thus, reaction of pyridine derivative-2-carbaldehyde 231 with anilines 18 in the presence of a photocatalyst and a Brønsted acid catalyst (TsOH) afforded indolizino[1,2-b]quinolin-9(11H)-ones 233 (Scheme 64). Both Ru(bpy)3Cl2·6H2O and Ru(bpy)3(PF6)2 were used as the photocatalyst, giving to the formation of tetracyclic compound 233 in more than 95% yield. Likewise, other acids, such as zinc trifluoromethanesulfonate (Zn(OTf)2), displayed a similarly high catalytic effectiveness. In this catalytic process, the visible-light-promoted dehydrogenation protocol of tetrahydroquinolines 232 constitutes the key procedure. The aniline substitution plays a crucial role in the success of the tetrahydroquinoline dehydrogenation step. Both weakly and strongly electron-donating groups (Me, OMe, OBn) at para-position of the aniline ring undergo excellent yields of compound 233. Conversely, electron-withdrawing groups (Cl, CN) at this position of the aniline ring showed a negative effect on the reaction yield.

Combination of visible-light-photoredox and Brønsted acid-catalyzed intramolecular Povarov reaction for the preparation of indolizino[1,2‑b]quinolin-9(11H)‑ones
A synthetic strategy developed in 2010 by Bai’s group [90] affords an efficient access to a series of libraries of the tetracyclic pyrimidine-fused heterocycles. The key step in this synthetic methodology entails the intramolecular Povarov reaction of imine intermediate formed in situ from the reaction of aromatic amines 18 and allylaminopyrimidine-5-carbaldehydes 234. Trifluoroacetic acid was selected as Brønsted acid catalyst to accomplish this transformation, affording exclusively cis-benzopyrimido[4,5-h][1,6]naphthyridines 235 in good to excellent yields (Scheme 65). Although the use of 10 mol% TFA in acetonitrile yielded the desired product in good yields, increasing the catalyst loading to 2 equivalents led to shorter reaction rates and higher yields.

Intramolecular Povarov reaction in the preparation of benzopyrimido[4,5-h][1,6]naphthyridine libraries
The combination of visible-light-photoredox and acid catalysis has also been applied to the formal synthesis of the precursor of 10-hydroxycamptothecin and irinotecan. The intramolecular Povarov cycloaddition/dehydrogenation aromatization cascade of pyridone carbaldehyde 237 and 4-aminophenol 236 in the presence of a photocatalyst (Ru(bpy)3Cl2·6H2O) and a Brønsted acid catalyst p-toluenesulfonic acid (p-TsOH) yielded pentacyclic derivative 238 in 92% yield (Scheme 66) [89].

Tandem acid-catalyzed intramolecular Povarov reaction/visible-light photoredox for the synthesis of the precursor of 10-hydroxycamptothecin and irinotecan
In 2010 Subba Reddy et al. [91] described the first synthesis of pentacyclic polyaromatic chromenoacridine derivatives in a single-pot operation. This protocol involves the formation of the intermediate imine between para- and ortho-substituted aromatic amines 18 and alkene-tethered chromene-3-carbaldehyde 239, followed by the BF3·OEt2-induced intramolecular Povarov reaction. Under these reaction conditions a set of 18 chromeno[2,3-c]acridines 240 were obtained in 66–88% yield with high trans-steroselectivity (Scheme 67). Other Lewis acids such as AlCl3, FeCl3, ZnCl2, SnCl4, Sc(OTf)3, InCl3, InBr3, In(OTf)3, LiClO4, and Brønsted acid TFA, were ineffective for this transformation in terms of both yield and selectivity. All attempts to extend this protocol to diamines such as 1,5-diaminonaphthalene did not furnish the desired product.

Synthesis of chromenoacridine derivatives through intramolecular Povarov reaction
Alkene-tethered aminochromene-3-carbaldehyde 241 has been employed for the intramolecular inverse electron demand [4 + 2] cycloaddition reaction [92]. 2-(N-Alkenyl-N-aryl)aminochromene-3-carbaldehyde 241 also underwent intramolecular Povarov reaction with aromatic amines 37 in the presence of Lewis acids to furnish chromenonaphthyridines 242 (Scheme 68) [93]. Thus, reaction of para-substituted aromatic amines 37 with 241 in the presence of 40 mol% of PPh3·HClO4 (TPP)[65], afforded cis- or trans-chromenonaphthyridines 242. cis-Adduct 242 is favored when R4 = Me, while trans-chromenonaphthyridines 242 was observed when R4 = Ph.

Synthesis of chromenonaphthyridines via TPP-induced intramolecular Povarov reaction
Raghunathan et al. [94] reported in 2008 a simple procedure for the synthesis of pyrano and thiopyranoquinoline derivatives using indium trichloride supported in silica gel. O-alkenyl 243 and S-alkenylquinoline-3-carbaldehyde 244 are suitable starting materials to undergo intramolecular Povarov reaction with a variety of aromatic amines 37. Therefore, reaction of aromatic amines 37 with 243 or 244 in the presence of InCl3 in acetonitrile furnished a mixture of cis- and trans-pyrano 245 and thiopyranoquinolines 246, respectively, with diastereoselectivities ranging from 65:35 to 84:16 by intramolecular Povarov reaction of the intermediate imine generated in the one-pot reaction (Scheme 69). As a further extension of this work, the same reaction was carried out using InCl3 impregnated in silica gel as Lewis acid catalyst under microwave irradiation. These reaction conditions dramatically increase the overall yields from 55–82% to 75–97%, retaining nearly the same diastereoselectivity ratios. This ecofriendly protocol avoids the use of organic solvents, has general applicability, and notably enhances reaction rates and chemical yields.

Synthesis of pyrano and thiopyranoquinolines through InCl3/silica gel supported catalyzed intramolecular Povarov reaction
In 2008, Zhang’s group developed a new intramolecular Povarov reaction for the preparation of luotonin A analogs [95]. This approach entails the in situ formation of imidates through activation of corresponding chemically stable amides. Thus, bis(triphenyl)oxodiphosphonium trifluoromethanesulfonate, formed in situ from Ph3PO and Tf2O used for the total synthesis of camptothecin [96] and luotonin A [97], works as an amide-activating reagent to convert the amide moiety to its corresponding imidate under mild reaction conditions, and also to promote the subsequent intramolecular Povarov reaction in the desired direction. Using these catalytic conditions, cyclization of N-allyl naphthyridones 247 to afford luotonin A analogs 249 through the corresponding imidates 248 was attained in 64–78% yield (Scheme 70). The formation of compound 249 may be rationalized by the stability of aromatic system, driven by the catalytic system acidity.

Cyclization of N-allyl naphthyridones into luotonin A analogs using amide-activating reagents
Nagaiah et al. [98] reported the diastereoselective synthesis of tetrahydropyrano-chromeno[4,3-b]quinolines 251 by intramolecular Povarov reaction of formal 2-azadienes obtained in situ from aromatic amines 37 and O-prenyladed compounds 250 derived from 8-formyl chromenones (Scheme 71). Several Lewis and Brønsted acid catalysts were tested in this reaction and, among them, Yb(OTf)3 and Sc(OTf)3 were found to be almost equally efficient according to reaction yields, times, and diastereoselectivities. Independent of the nature of the catalyst, in all cases studied exclusive formation of cis-tetrahydrochromenoquinolines 251 was obtained, which may be due to the steric effect of the chromenone moiety. This method allows the use of aromatic amines 37 with electron-withdrawing or electron-donating groups, giving compounds 251 in very good yields. Some of these synthesized tetrahydrochromeno[4,3-b]quinolines 251 exhibited significant antiproliferative activity against MCF-7 breast cancer cell line and low inhibitory activity against MDA-MB-231 breast cancer cell line.

Povarov reaction in the preparation of antiproliferative tetrahydropyranochromeno[4,3-b]quinolines
Very recently, Zhang’s group [99], continuing their work on heterocyclic compound synthesis under visible light, has developed an intramolecular Povarov cycloaddition reaction to construct substituted luotonin A via visible-light-promoted dehydrogenation of pentacyclic pyrroloquinazolines 254 (Scheme 72). The optimized reaction conditions consisted in using eosin Y as the photocatalyst and TsOH·H2O as the co-catalyst in MeOH under the irradiation using a normal 23 W household lamp at room temperature. Thus, when 3-cynnamyl-4-oxo-3,4-dihydroquinazoline-2-carbaldehydes 252 reacted with anilines 18, the pentacyclic pyrroloquinazolines 254 were obtained with up to 97% yield. Control experiments confirmed the necessity of both visible light and oxygen. In the absence of photocatalyst or acid the yield significantly decreased. At the same time, the reaction could not proceed in the dark or under a N2 atmosphere.
See Table 4 for the most representative examples of Sect. 4.2.

Tandem intramolecular Povarov reaction/visible light-promoted dehydrogenation for the construction of substituted luotonin A derivatives
4.3 Oxygen- or Nitrogen-Containing Heterocyclic Alkyne-Tethered Aldehydes
O-Propargylated compounds derived from 8-formyl chromenones 255 have been used as carbonyl compounds in the preparation of pyranochromeno[4,3-b]quinolines 256 [100]. Authors analyze the activity of several copper catalyst for the activation of the terminal alkyne C–H bond in 255 by the use of CuFe2O4 nanoparticles, CuI, Cu(OTf)2, CuCl, and CuBr. CuFe2O4 nanoparticles were found to be the best catalyst for this transformation, which due to their magnetic properties can be easily separated from the reaction mixture and reused without loss of activity. In addition, the choice of DMSO as the solvent among others (i.e. MeCN, toluene, DMF, H2O) was done in terms of reaction efficacy. Electron-donating groups at ortho or para-positions in aromatic amines 18 gave good yields of pyranochromenoquinolines 256 (Scheme 73). However, anilines with electron-withdrawing groups did not afford the desired adducts 256 when reacted with O-propargylated-8-formyl chromenones 255.

O-Propargylated 8-formyl chromenones as carbonyl components in the CuFe2O4-promoted intramolecular Povarov reaction
The intramolecular Povarov reaction using N-containing heterocyclic N-alkyne-tethered aldehydes has been applied for the preparation of alkaloids with fused heterocycles. Menéndez’s group [101] reported in 2017 a small library of benzimidazole-fused pyrrolo[3,4-b]quinolines 259 synthesized from readily available benzimidazole 2-carbaldehyde 257 and various substituted aryl amines 18. Under catalytic-free conditions or in the presence of InCl3, Yb(OTf)3, InBr3, or ammonium cerium(IV) nitrate (CAN), in different solvents such as MeCN, CH2Cl2, and 1,2-dichloroethane (DCE); the corresponding cycloadducts 259 derived from the intramolecular Povarov reaction were not obtained and only the corresponding aldimines 258 were attained instead (Scheme 74). However, treatment of aldimines 258 with 20 mol% of BF3·OEt2 in DCE at 80 °C afforded pyrrolo[3,4-b]quinolines 259 in good yields. In addition, cycloadducts 259 were achieved in 65–80% yield via one-pot intramolecular Povarov reaction when substituted anilines 18 reacted with benzimidazole 2-carbaldehyde 257 in the presence of BF3·OEt2. Compounds, thus synthesized, can be considered as decarbonyl analogs of the anticancer alkaloid luotonin A and were evaluated in a DNA relaxation assay for their ability to inhibit human topoisomerase I.

Synthesis of decarbonyl analogs of the anticancer alkaloid luotonin A by BF3·OEt2-assisted intramolecular Povarov reaction
Batey’s group [102] reported the intramolecular Povarov reaction employing N-propargylic-substituted aldehydes 260 derived from pyridine for the synthesis of the pyrrolo[3,4-b]quinoline nucleus of camptothecin (Scheme 75). When aldehyde 260 and aniline reacted in the presence of 10 mol% of Dy(OTf)3 at room temperature, the corresponding imine 261 was isolated, whereas when the reaction was carried out at 50 °C quinoline 262 was directly obtained. The formation of quinoline derivative 262 (R1 = H) constitutes a formal synthesis of camptothecin, while the obtained compound 262 derived from p-anisidine 37 (R1 = MeO) can be used as precursor in the preparation of topotecan.

Intramolecular Povarov reaction in the formal synthesis of camptothecin
Similarly, this methodology allowed the synthesis of luotonin A 264 in 51% yield from the intramolecular Povarov reaction of N-propargylic-substituted aldehyde derived from quinazoline 263 and aniline 1 in the presence of 10 mol% Dy(OTf)3 in acetonitrile (Scheme 76) [102].

Intramolecular Povarov reaction in the total synthesis of luotonin A
Luotonin A analogs [95] have also been prepared by intramolecular Povarov reaction through in situ formation of imidates by activation of corresponding chemically stable amides. Bis(triphenyl)oxodiphosphonium trifluoromethanesulfonate, an amide-activating reagent, catalyzes the cyclization of N-propargyl naphthyridones 265 yielding luotonin A analogs 266 in moderate yield (Scheme 77).

Cyclization of N-propargyl naphthyridones into luotonin A analogs using amide-activating reagents
5 Heteroaromatic Amines and Aliphatic Aldehydes
Amino-heterocycles have scarcely been exploited as building blocks for either inter- or intramolecular imino-Diels–Alder reaction, even though they can condense with aldehydes to form imine derivatives. As far as we know, only two examples have been reported in the intramolecular Povarov reaction of imines generated from heteroaromatic amines and aliphatic alkene-tethered aldehydes. For instance, a catalyst-free intramolecular imino-Diels–Alder protocol for the synthesis of annulated tetrahydropyridines has been developed by Vilches-Herrera et al. [103]. The corresponding octahydro-1H-pyrrolo[2,3-b]quinoline 269 was obtained in 65% yield via cycloaddition of aldimine 268 obtained by condensation reaction between 2-aminopyrrole 267 and a non-aromatic aldehyde such as citronellal 38 (Scheme 78). The reaction is carried out in water under microwave irradiation at 200 °C and with no catalyst, which fulfills all the requirements for sustainable chemistry.

Intramolecular Povarov reaction using 2-aminopyrrole under microwave conditions
The Nagarajan’s group [104] reported in 2008 the synthesis of indoloacridine 271 in the intramolecular Povarov reaction of imine derived from aliphatic citronellal 38 as dienophile and a heteroaromatic amine such as 3-aminocarbazole 270 (Scheme 79). The reaction proceeded very smoothly in the presence of Lewis acid La(OTf)3. Although the diastereoselectivity is highly temperature dependent, the trans-isomer is the major product isolated in this reaction, and better diastereoselectivities (10:90) were attained at low reaction temperatures.

Povarov reaction of citronellal and 3-aminocarbazole
6 Heteroaromatic Amines and Aromatic Aldehydes
The first example was reported by Tietze et. al. in 1992. They worked out the intramolecular Diels–Alder reaction between benzaldehydes 273 and aminoisoxazole 272 under thermal conditions (Scheme 80) [105]. In this case, the condensation of both reagents generated the corresponding imines 274, which can be isolated, and selectively cyclized to form the cis- or trans-fused tetrahydropyridines 275 (Scheme 80). The selectivity of these reactions could be explained by electronic effects. In the reaction of 273 (R1 = R2 = R3 = R4 = H) with 272 only the trans-annulated tetrahydropyridine 275 was obtained. In addition, during the reaction of 272 with 273 (R1 = R2 = R3 = H, R4 = CO2Me) two diastereoisomers could be formed, but only the trans-annulated tetrahydropyridine 275 was observed. Surprisingly, the reaction of 273 (R1 = R2 = Cl, R3 = R4 = Me) with 272 yielded only the cis-fused compound 275.

Hetero-Diels–Alder reaction of 5-amino-3-methylisoxazole in the synthesis of annulated tetrahydropyridines
A highly diastereoselective methodology for the aza-Diels–Alder cycloaddition using 2-aminopyrrole derivatives 276 (X = CH) or 2-aminopyrazole derivatives 277 (X = N) to construct chiral tetracyclic hexahydrochromenopyrrolo-pyridines 281 and hexahydrochromenopyrazolo-pyridines 282 was developed (Scheme 81) [103]. In a first step, imine derivatives 279 or 280 were previously synthesized through condensation of commercially available 2-hydroxybenzaldehyde derivatives 278 with 276 or 277 and subsequent alkylation reaction. The [4 + 2] cycloaddition reaction was conducted in water under microwave irradiation and with no catalyst (Scheme 81). Moreover, in most of the cases the products precipitate in the reaction media, avoiding the use of solvents for extraction and column chromatography for purification. The reaction is solvent dependent with regard to its stereoselectivity. Only the trans-isomer is obtained if the reaction is performed in water, whereas in a nonpolar solvent such as p-xylene the cis-isomer can also be isolated.

Intramolecular Povarov reaction using 2-aminopyrrole and 2-aminopyrazole under microwave conditions
As an extension of the methodology developed by Nagarajan’s group, which used 3-aminocarbazol 270 as heteroaromatic amine and La(OTf)3 as Lewis acid (vide supra Scheme 79), the reaction was also performed with imines 283 derived from aromatic aldehydes (O-prenylated salicylaldehydes) 176 [104]. Isomeric ellipticine derivatives 284 were obtained in 85–92% yield and very good diastereoselectivities (95:5–98:2) in favor of cis-isomer (Scheme 82).

Intramolecular Povarov reaction of aromatic aldehydes and 3-aminocarbazole
Palacios et al. have described the synthesis of 1,5-naphthyridine derivatives fused with other oxygen-containing heterocycles such as chromenes or chromen-2-ones [106]. The synthetic route involves an intramolecular [4 + 2] cycloaddition reaction using BF3·OEt2 as Lewis acid of functionalized aldimines 288 or 289 obtained by the condensation of 3-aminopyridine derivatives 285 with aldehydes containing a carbon–carbon double bond in ortho position 286 or 287 followed by prototropic tautomerization. The reaction transcurred in a selective manner allowing the generation of three stereogenic centers in a short fashion, and the trans-isomers 290 and 291 were obtained. The subsequent dehydrogenation of the fused tetrahydrochromeno[4,3-b][1,5]naphthyridines 290 and tetrahydrochromeno[4,3-b][1,5]naphthyridin-6-ones 291, using DDQ as oxidant, leads to the formation of the corresponding tetracyclic chromeno[4,3-b][1,5]naphthyridine derivatives 292 and chromeno[4,3-b][1,5]naphthyridin-6-ones 293 in excellent yields (Scheme 83). The use of 2-aminopyridine derivatives, as amine component, afforded the corresponding 1,8-naphthyridine regioisomers [107]. The behavior as topoisomerase I inhibitors of the synthesized 1,5- and 1,8-naphthyridine derivatives was also studied.

Synthesis of chromeno[4,3-b][1,5]naphthyridines and chromeno[4,3-b][1,5]naphthyridin-6-ones
A BF3·OEt2-catalyzed intramolecular Povarov reaction, followed by oxidation with DDQ, was used to synthesize chromenopyridine-fused thiazolino-2-pyridone peptidomimetics with the ability to bind α-synuclein and amyloid-β fibrils in vitro [108]. The reaction works with several O-alkylated salicylaldehydes 295 and amino functionalized thiazolino-2-pyridones 294, to generate polyheterocycles 296 with diverse substitution in moderate to excellent yields (Scheme 84). On the contrary, attempts to synthesize C-7 unsubstituted molecules 296 (R2 = H) through intramolecular Povarov reaction, using O-allylsalicylaldehyde 295 (R2 = H) took place in very low yields, but the use of a vinyl ester moiety as electron-donating auxiliary 296 (R2 = OCOPh), allowed the obtainment of the C-7 unsubstituted compounds 297 in reasonable reaction times and moderate yields after removal of benzoate functionality during the oxidation process.

Synthesis of thiazolino-2-pyridone-based polyheterocycles capable of modulating and binding to α-synuclein and amyloid-β fibrils
Following the intramolecular strategy, hybrid substituted quinolino[4,3-b][1,5]naphthyridines 304 and quinolino[4,3-b][1,5]naphthyridin-6(5H)-ones 305 were synthesized [109]. The derivatives were achieved by an intramolecular Povarov [4 + 2]-cycloaddition reaction using BF3·OEt2 as Lewis acid (Scheme 85). First, the corresponding 5-tosyl functionalized aldehydes 206 (X = CH2) or 299 (X = CO), which tailored a double bond in their structure, condensed with 3-aminopyridines 298 to afford imines 300 or 301, respectively. Subsequent regio- and stereospecific intramolecular cyclization in refluxing chloroform and in the presence of a Lewis acid such as BF3·OEt2 and prototropic tautomerization, gave the corresponding tetrahydro 1,5-naphthyridines 302 or 1,5-naphthyridin-6(5H)-ones 303 respectively, as trans-diastereoisomers. Their dehydrogenation reaction was performed using MnO2 in toluene-yielding compounds 304 or 305. The corresponding deprotection of the tosyl group could be accomplished with magnesium under acidic conditions. The corresponding 1,8-naphthyridine regioisomers could also be prepared when 2-aminopyridine derivatives are used as the amine component in this intramolecular Povarov reaction [107].

Synthesis of quinolino[4,3-b][1,5]naphthyridines and quinolino[4,3-b][1,5]naphthyridin-6(5H)-ones
The previously reported methodology using 2-aminopyrrole 276 and 2-aminopyrazole 277 to construct chiral tetracyclic hexahydrochromenopyrrolo- and hexahydrochromenopyrazolo-pyridines (vide supra Scheme 81) was extended to alkyne bridged aldehydes derived from alkylation of aldehydes 306. Thus, when propargyl bromide was used as the dienophile, the aromatic annulated compounds 309 or 310 were obtained in good yields via spontaneous aromatization of the corresponding cycloadducts (Scheme 86) [103].

Intramolecular Povarov reaction using 2-aminopyrrole or 2-aminopyrazole and alkyne-tethered aldehydes
Similarly, aldimines 314 or 315, derived from the condensation of substituted 2-propargyloxybenzaldehydes 313 and 3-aminopyridine 311 or 2-aminopyridine 312, respectively, afforded the corresponding chromeno[1,5]naphthyridine derivatives 318 or chromeno[1,8]naphthyridine compounds 319 after BF3·OEt2-catalyzed intramolecular Povarov reaction (Scheme 87). It is noteworthy that, with this strategy, from a preparative point of view, the aromatic 1,5- and 1,8-naphthyridine core may be directly obtained [106, 107].

Synthesis of chromeno[4,3-b][1,5] and [1,8]naphthyridines using alkyne-tethered aldehydes
5-Amino-1,3-dimethyl uracil 320 has been used as the amino component in the intramolecular Povarov reaction. Thus, Majumdar et al. [110] reported in 2010 the Lewis acid catalytic intramolecular Povarov reaction between O-propargylated salicylaldehydes 321 and 5-amino-1,3-dimethyl uracil 320 (Scheme 88). Several Lewis acids (BF3·OEt2, Yb(OTf)3, CuBr, and CuI), Brønsted acids (TFA), and solvents (MeCN, THF, DMF, DMSO, EtOH, and toluene) were screened. All these variations of the catalyst and solvent showed that running the reaction in toluene using 10 mol% of BF3·OEt2 as the catalyst provides the best results for the synthesis of chromene-fused pyrido[3,2-d]pyrimidines 322 in good chemical yields (Scheme 88).

Intramolecular Povarov reaction involving 5-amino-1,3-dimethyl uracil for the preparation of chromenopyrido[3,2-d]pyrimidines
Nagarajan et al. [111] have reported the synthesis of isomeric isoellipticine derivatives through a straightforward CuI/La(OTf)3-catalyzed tandem reaction in ionic liquid [bmim][BF4]. Thus, the reaction of bromo-, fluor-, chloro-, methyl-, or methoxy-substituted O-propargylated salicylaldehyde 187 with carbazole-derived amine 270 in the presence of CuI/La(OTf)3 and in ionic liquid afforded isoellipticine fused with dihydro chromene derivatives 323 in 80–96% chemical yield (Scheme 89). After careful analysis, authors identified the intramolecular Povarov occurred through C–4 of the carbazole ring.

Intramolecular Povarov reactions in the synthesis of isoellipticine fused with dihydrochromene derivatives
The synthesis of hybrid substituted quinolino[1,5]naphthyridines 330 and quinolino[1,8]naphthyridines 331 may also be obtained in good yields by BF3·OEt2-catalyzed intramolecular cycloaddition of aldimines 326 or 327, derived from N-propargyl substituted aldehyde 325 and the corresponding aminopyridines 298 or 324 (Scheme 90) [107, 109]. The deprotection of the tosyl group at the nitrogen atom was carried out with Mg under acidic conditions.

Synthesis of quinolino[1,5]naphthyridines and quinolino[1,8]naphthyridines
See Table 5 for the most representative examples of Sect. 6.
7 Heteroaromatic Amines and Heteroaromatic Aldehydes
Only one report has disclosed in the intramolecular Povarov reaction of imine intermediates derived from heteroaromatic aldehydes and heteroaromatic amines. In this way, Nagarajan et al. [104] demonstrated the utility of imines, resulting from the condensation of N-prenylated indole-2-carbaldehydes 333 with aminocarbazoles 332, in the intramolecular Povarov reaction. This protocol efficiently proceeds in the presence of Lewis or Brønsted acids, but diastereoselectivity is influenced by the nature of the catalyst. The best catalytic conditions were found for La(OTf)3 (10 mol%) in 1,4-dioxane at 150–160 °C, yielding isomeric ellipticine ring system derivatives 335 (R3 = R4 = H) in good yields and excellent diastereoselectivities (Scheme 91). The intramolecular Povarov reaction occurred through C–4 of the carbazole ring, and the six-membered piperidine and five-membered pyrrolidine rings were cis-fused. When amine 332 with substitution at C–1 and C–4 (R3 = R4 = Me) reacted with aldehyde 333, the intramolecular cyclization occurred through the C–2 position of the carbazole ring, affording the corresponding product 337 in only 51% yield (Scheme 91).

Synthesis of isomeric ellipticine derivatives by means of intramolecular Povarov reaction of heteroaromatic aldehydes and heteroaromatic amines
8 Intramolecular Povarov Reaction with Secondary Amines
In 1996 Beifuss’s group [112] developed the first intramolecular cationic Povarov cyclization. The condensation of N-substituted anilines 338 and chiral ω-unsaturated aldehydes 339 in situ affords cationic iminium ions 340 (Scheme 92). Subsequent intramolecular [4 + 2]-cycloaddition of 340 leads to the highly trans-diastereoselective formation of octahydroacridines 341 with five stereogenic centers. The best yields were obtained when the transformation was performed with BF3·OEt2 (30 mol%) as the Lewis acid in CH2Cl2.

Intramolecular Povarov reaction of iminium ions for the synthesis of octahydroacridines
A one-pot diastereoselective synthesis of new N-substituted octahydroacridines was successfully achieved by Kouznetsov et al. [113, 114] via BiCl3-catalyzed intramolecular cationic imino-Diels–Alder reaction. The intermediate iminium ions were prepared in situ through condensation of N-protected anilines 342 and (±)-citronellal 38 under mild reaction conditions (Scheme 93). It was observed that bulky N-substituent groups play a key role in the cis/trans ratio of the corresponding octahydroacridines 344. For instance, when R1 = Me, a mixture of 50:50 cis/trans-octahydroacridine derivatives 344 were observed. However, increasing bulkiness (R1 = allyl, propargyl, and benzyl substituents) allows preferential formation of the trans-fused heterocycles in ratios ranging from 22:78 to 3:97 cis/trans-isomers. It was found that use of the N-benzyl group resulted in a highly diastereoselective process that gives easily separable trans-fused N-substituted octahydroacridines 344. The developed protocol was extended to involve the use of citronella essential oil from Cymbopogon nardus as a renewable source of these biologically important heterocyclic molecules. The results obtained from C. nardus essential oil are comparable to those observed from pure (±)-citronellal 38, without changes in the diastereoselectivity. Furthermore, this methodology can be extended to other N-substituted anilines with reactive groups as starting materials to prepare different trans-N-substituted hybrid octahydroacridines 344 as potential bioassay substrates.

BiCl3-catalyzed intramolecular cationic Povarov reaction for the construction of octahydroacridines
Spaller et al. [115] described the application of the intramolecular aza-Diels–Alder transformation to generate a diverse range of quinoline-fused structures with multiple stereogenic centers, many of which resemble lignan and arylnaphthalene-type natural products. In this work, they combined several secondary (N-alkylated) anilines 342 and aldehyde-alkene bridge 345, with various Brønsted and Lewis acid catalysts (Scheme 94). For instances, several Brønsted acids such as TFA, trifluoromethanesulfonic acid (TFMSA), acetic acid, p-TsOH, or l-tartaric acid and Lewis acids such as Yb(OTf)3 or BiCl3 were used for this transformation. The results demonstrated that both Brønsted and Lewis acids have a slight effect on both selectivity and overall yields. The use of 3 equivalents of TFA in acetonitrile at room temperature afforded hexahydropyrrolo[3,4-b]quinolin-1-ones 346 in moderate to excellent yields, and diastereoselectivities ranging from 34:66 to 6:94 (Scheme 94).

Intramolecular cationic Povarov reaction catalyzed by TFA in the preparation of pyrroloquinolinones
N-alkyl substitution in the N-aromatic amine component was rigidified by incorporating a saturated ring system. Thus, trifluoroacetic acid-catalyzed intramolecular Povarov reaction of the corresponding iminium ion intermediate, generated in the condensation between 1,2,3,4-tetrahydroquinoline 347 and aldehyde 348, bearing an alkene-tethered partner, produced cycloadduct 349 in 93% yield and 15:85 in favor of trans-selectivity (Scheme 95) [115].

Synthesis of diazacyclopenta[a]phenalenone by intramolecular cationic Povarov approach
Bai’s group [90] developed an efficient synthesis of tetracyclic pyrimidine-fused heterocycles through the intramolecular Povarov reaction of iminium salts 351 formed in situ from the reaction of secondary aryl amines 338 and allylaminopyrimidine-5-carbaldehyde 350. p-Toluenesulfonic acid was selected as Brønsted acid catalyst, yielding solely cis-benzopyrimido[4,5-h][1,6]naphthyridines 352 in moderate to good yields (Scheme 96).

Intramolecular Povarov reaction in the preparation of benzopyrimido[4,5-h][1,6]naphthyridine libraries
See Table 6 for the most representative examples of Sect. 8.
9 Oxidative Intramolecular Povarov Reaction
In 2011, Mancheño’s group described the oxidative Povarov reaction between glycine derivatives with olefins [116]. In this method, a crucial oxidant was required for both the in situ generation of the iminium intermediate (by a Csp3–H bond oxidation of N-aryl amine) and the final dehydrogenation of the tetrahydroquinoline to form the corresponding heteroaromatic compound. The strategy represents a milestone in organic synthesis and specifically in the Povarov reaction as no prefunctionalization is required in the reaction partners and CH bonds are ubiquitous in organic molecules. Since then, the oxidative Povarov reaction to form quinoline derivatives, also in its intramolecular version, has been extensively studied as the process involves the use of simpler starting materials and less waste generation.
Csp3–H bond oxidation of N-aryl glycine esters and amides can be carried out under catalytic radical cation salt-induced conditions. The peroxyl radical cation, which is generated in the coupling between tris(4-bromophenyl)ammoniumyl hexachloroantimonate (TBPA+·) and oxygen, might be involved to initiate the catalytic oxidation. In this way, Jia’s group [117, 118] reported a direct construction of quinoline-fused lactones accomplished by Csp3–H bond oxidation under catalytic radical salt-induced conditions of starting N-aryl glycine esters. Radical cation salt TBPA+, stable in the solid state but decomposed after 3 h in MeCN in the presence of oxygen, promotes the cyclization of glycine derivatives 353 to yield Povarov adducts. Both electron-withdrawing and electron-donating substituted N-aryl glycine cinnamyl esters 353 were converted to quinoline-fused lactones 358 in moderate to excellent yields (Scheme 97). The lack of substituents or the presence of a substituent at the ortho position of the aniline dramatically diminished the reaction yield; for instance, when R1 = H compound 358 was obtained in 49% and only 20% yield of 358 was observed when R1 = 2-Me. The key step in this transformation comprises the formation of a glycine imine (or iminium ion) that can efficiently add to the styrene moiety to afford Povarov adducts. In this way, authors suggest a plausible mechanism where the sp3 C–H bond adjacent to the aniline group is oxidized by TBPA+· in the presence of oxygen, giving radical intermediate 354, which can be further oxidized to the corresponding glycine imine 355 (Scheme 97, route a). The formation of quinoline-fused lactones 358 may arise from a radical cation salt-induced Povarov reaction. Nevertheless, radical intermediate 354 may also add to the double bond of styrene, and subsequent radical addition to the phenyl group would yield 357. Further oxidation and aromatization would afford 358 (Scheme 97, route b) [118].

Catalytic radical cation salt induced Csp3–H oxidation for the construction of quinoline-fused lactones
The generality of this protocol was determined by the use of glycine amides toward the construction of quinoline-fused lactams 360 under the same catalytic radical salt-induced conditions [118]. All of the tested N-aryl glycine cinnamyl amides 359 displayed good reactivity, yielding the corresponding quinoline-fused lactams 360 in good to excellent yields (Scheme 98). Bulky amide N-protecting groups gave better results, probably due to the closeness of the cinnamyl group to the reactive radical in those derivatives, which favors the intramolecular annulation. In addition, the effect of the substituents on the cinnamyl group was studied, showing that electron-donating groups increase the annulation yields.

Catalytic radical cation salt induced Csp3–H oxidation for the construction of quinoline-fused lactams
More recently, Muthukrishnan’s group [119] reported an intramolecular dehydrogenation promoted by oxone followed by imino-Diels–Alder reaction (Povarov cyclization) of alkyne-tethered N-aryl glycine esters and amides for the preparation of quinoline-fused lactones and lactams. Hence, the dehydrogenative Povarov reaction of N-aryl glycine ester 361 (X = O) was proved by using 5 mol% BF3·OEt2 as a Lewis acid in the presence of 2-iodoxybenzoic acid (IBX) as an oxidant at room temperature. Under these conditions, quinoline fused lactones 363 were obtained in 58% yield (Scheme 99). Other Lewis acids, such as Cu(OTf)2 or Sn(OTf)3, and different peroxide-based oxidants such as PhI(OAc)2, PhI(IOCOCF3)2, Na2S2O8, benzoyl peroxide (BPO) or Oxone (2KHSO5-KHSO4-K2SO4) were screened. The combination of Cu(OTf)2 as Lewis acid and Oxone as oxidant works well, affording cycloadducts 363 in good yields, except for methyl-substituted alkyne 361 (R2 = Me), which was obtained in 17% yield (Scheme 99). Remarkably, Oxone would be a favorable oxidant as it is easy to handle, cheap, and nontoxic. The scope and generality of this approach indicated that different electron-donating and electron-withdrawing groups on the aniline ring as well as the aryl alkyne moiety were well tolerated (Scheme 99). This method was further extended to the preparation of quinoline-fused lactams 364 (X = NR3) (Scheme 99). The intramolecular dehydrogenative Povarov cyclization of N-aryl glycine amide 362 (X = NR3) using the optimized reaction conditions, yielded the cycloadducts 364 in moderate to good yield, although higher temperatures were required for reaction completion [119]. This protocol is very general since electron-donating and electron-withdrawing groups on the aniline ring as well as the aryl alkyne moiety of NR3 protected glycine amide furnished the corresponding products 364 (Scheme 99). The method was further used for the preparation of biologically important quinoline core of uncialamycin and luotonin A analogs.

Oxone promoted intramolecular dehydrogenation followed by Povarov cyclization for the construction of quinoline-fused lactones and lactams
10 Asymmetric Intramolecular Povarov Reaction
Masson’s group first described the asymmetric version of the intramolecular Povarov reaction in 2017 [120]. They developed an efficient asymmetric organocatalytic intramolecular Povarov reaction for the preparation of optically active chromeno-fused quinoline derivatives as well as dibenzo-fused naphthyridine derivatives. A chiral phosphoric acid, (R)-3,3′-bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diyl hydrogen phosphate (369, TRIP) catalyzes the enantioselective intramolecular Povarov-type reaction of alkene-tethered aldehydes 366–368 and primary 2-hydroxy anilines 365. The corresponding tetrahydrochromeno[4,3-b]quinolin-6-ones 370 (X = O) as well as tetrahydrodibenzo[1,6]naphthyridin-6-ones 371 (X = NH) were obtained in excellent yields, with high diastereo- and enantioselectivities ranging from 81% to 98% ees (Scheme 100). The intramolecular cycloaddition proceeds at room temperature with complete diastereoselectivity in favor of the trans,trans-tetrahydrochromeno[4,3-b]quinolin-6-one derivative 370 when the dienophile possesses a benzene ring with a hydroxy group at para-position, while ortho- and meta-analogs were completely unreactive in this cycloaddition. It appears to be reasonable that hydrogen bonding of the phosphoric acid 369 to p-phenol seems to be critical for effective cycloaddition, but not for enantioselectivity, since 35% ee has been observed for 370 when 4-methoxy aniline was used. The use of 2-aminophenol 365 (R1 = H) as starting material might form an additional hydrogen bond with the chiral phosphoric acid 369 to increase facial discrimination of the N-2-hydroxy aldimine in the Povarov cycloaddition. This is observed by a dramatic increase in enantioselectivity. Furthermore, the catalyst loading could be lowered to 1 mol%, and the obtained azacycles were formed in high yield and high purity, after precipitation in the reaction vessel and isolation by filtration without purification by column chromatography. As an extension of a previous paper, Masson et al. [121] reported the enantioselective intramolecular Povarov synthesis of tetrahydrothiochromeno[4,3-b]quinolin-6-ones 372 (X = S, Scheme 100). These new fused nitrogen-containing tetraheterocycles were attained in excellent diastereo- and enantioselectivity (93–96% ee), although the reaction conversion is not completed. However, a slight increase in the catalyst loading to 2 mol% gave the corresponding cycloadducts in excellent yields (91–97%). If the intermediate imine coordinates in a bidentate manner to the chiral phosphoric acid (R)-369 through a nine-membered cyclic transition state, the cyclization pathway may proceed by a stepwise mechanism (Scheme 100).

TRIP-catalyzed enantioselective organocatalytic intramolecular Povarov reaction
To further demonstrate the efficiency and scope of the present method, it was next applied to linear precursors with ether (374, X = O) or amine groups (375–377, X = NR2) as the linker between the aromatic aldehyde ring and the styrene group [121]. Precursors with an ether group as a linker were smoothly converted into the corresponding tetrahydro-6H-chromeno[4,3-b]quinolines 378 in excellent yields, enantioselectivities up to 99% ee, and trans,trans-diastereoselectivities (Scheme 101). In the same way, linear precursors with amine linker provided access to hexahydrodibenzo[b,h][1,6]naphthyridines 379–381, which were obtained with excellent yields, diastereo- and enantioselectivities ranging from 87% to 98% ee (Scheme 101). Diversely substituted 2-aminophenols were suitable partners to afford the corresponding cycloadducts in high yields and enantioselectivities. Modification of the protecting group on nitrogen afforded the desired products with similar yields although with slightly lower enantioselectivity.

Asymmetric intramolecular Povarov reaction for the preparation of enantiomerically enriched tetrahydrochromeno[4,3-b]quinolines and hexahydrodibenzo[b,h][1,6]naphthyridines
Catalytic enantioselective reactions with secondary amine substrates that involve intermediate iminium ions remain far less studied than the corresponding transformations with primary amines or preformed imines. Seidel et al. [122], for the purpose of developing the such transformation, synthesized polycyclic amines containing three contiguous stereogenic centers with excellent stereocontrol in a single step from N-methyl aryl amines 382 and aldehyde 383 possessing a pendant dienophile. Thus, N-methyl aryl amines 382 and O-allylsalicylaldehyde derivative 383 reacted using chiral Brønsted acid catalyst 384, afforded tetrahydrochromeno[4,3-b]pyrrolo[3,2,1-ij]quinolines 385 in moderate to excellent yields and high levels of diastereo- and enantioselectivity (Scheme 102). Catalyst 384, which involves a carboxylic acid group and a thiourea moiety as a covalently connected anion-recognition site, was previously applied to a catalytic enantioselective three-component Povarov reaction [123].

Enantioselective intramolecular Povarov reaction with secondary anilines
Likewise, indoline derivatives 386 and O-allylsalicylaldehyde derivatives 387 with different substituents on the aldehyde phenyl ring, reacted using chiral Brønsted acid catalyst 384, affording tetrahydrochromeno[4,3-b]pyrrolo[3,2,1-ij]quinolines 390 with excellent yields and high levels of diastereo- and enantioselectivity (Scheme 103) [123]. Similarly, the thiosalicylaldehyde-derivative 388 (X = S) was fruitfully converted into the corresponding tetrahydropyrrolo[3,2,1-ij]thiochromeno[4,3-b]quinoline 391 with a slight decrease of diastereomeric ratio; however, excellent yield and enantioselectivity was preserved (Scheme 103). The starting material in which the oxygen linker is replaced with a methylene bridge 389 (X = CH2) was similarly reactive but provides products in racemic form (dr = 2:1). Regarding to the amine component, tetrahydroquinoline reacted to afford the corresponding cycloadduct in 83% chemical yield after 2 days, but with reduced diastereoselectivity (dr = 7:1) and enantioselectivity (81% ee).

Enantioselective intramolecular Povarov reaction with indoline derivatives
As an extension of Seidel’s methodology, the same group introduced a new approach for the rapid synthesis of polycyclic amines through aza-Diels–Alder (Povarov) reaction, applying the kinetic resolution of two-substituted indolines to enhance the stereochemical complexity of the products [124]. Under control of a chiral Brønsted acid catalyst, racemic 2-phenylindoline 392 (R1 = Ph) undergoes intramolecular Povarov reaction with achiral aromatic aldehydes 387 bearing a pendant dienophile. One enantiomer of the indoline reacts preferentially, resulting in the highly enantio- and diastereoselective formation of polycyclic heterocycles, tetrahydrochromeno[4,3-b]pyrrolo[3,2,1-ij]quinolines 393 with four stereogenic centers (Scheme 104). This kinetic resolution approach exploits the differential formation/reactivity of diastereomeric ion pairs. The use of (S)-TRIP 369 provided good results, which were increased lowering the temperature to –10 ºC, improving de enantioselectivity process. The scope of this transformation was evaluated with a range of O-allylsalicylaldehyde derivatives 387. A range of substituents on the aldehyde phenyl ring and the styrene component were readily tolerated in reactions with 2-phenylindoline 392 (R1 = Ph), producing polycyclic heterocycles in excellent yields (82–99%), diastereo- (up to 20:1) and enantioselectivities (80–96% ee). Variation of the indoline cycloaddition partner was evaluated next. A number of two-substituted indolines 392 with diverse substituents (R1 = Ar, Alk) performed well. Even ethyl ester substituent or tert-butyldimethylsilyl (TBS)-protected alcohol were accommodated (Scheme 104).

Enantioselective intramolecular Povarov reaction with two-substituted indoline derivatives
Jørgensen’s group [125] developed an efficient asymmetric organocatalytic one-pot domino Michael addition/intramolecular Povarov reaction for the synthesis of optically active octahydroacridines having four stereocenters. Thus, malononitriles 394 react with a series of aliphatic α,β-unsaturated aldehydes 395 and p-substituted anilines 37, in the presence of diarylprolinol 396 and benzoic acid as additive, to produce octahydroacridines 398 with high yields and excellent enantio- and diastereomeric control (Scheme 105). The conjugate addition of malononitriles 394 to α,β-unsaturated aldehydes 395 using aminocatalysis leads to the formation of proper intermediates 397, which could be trapped in an amine condensation/intramolecular Povarov cascade to afford products 398. This asymmetric organocatalytic one-pot domino protocol displays great tolerance toward different aliphatic α,β-unsaturated aldehydes, malononitriles, and p-substituted anilines.

Enantioselective organocatalytic one-pot domino Michael/intramolecular Povarov reaction with malononitriles
A similar strategy, settled by Wang’s group [126], entails an effective organocatalytic one-pot domino Michael/intramolecular Povarov reaction using substituted indolinones 399, α,β-unsaturated aldehydes 395 and aromatic amines 18 (Scheme 106). Different commercially available chiral secondary amines were scrutinized, due to their recognized abilities to activate α,β-unsaturated aldehydes toward asymmetric transformation. The diarylprolinol 400 with bulky ether groups such as O-triethylsilyl (O-TES) led to enantiomerically enriched spirooctahydroacridine-3,3′-oxindoles 402 with the generation of five sterogenic centers at the same time with higher diasteroselectivities and enantioselectivities. Next, authors examined the addition of additives, reveling that the combination of diarylprolinol 400 with chiral Brønsted acid 401 gave products 402 in higher yield and excellent stereoselectivities. Under optimal conditions, the scope to probe the generality of this procedure was examined, exhibiting an excellent acceptance toward a variety of different substrates furnishing enantiomerically enriched spirooctahydroacridine-3,3′-oxindole 402 in good yields, diastereoselectivities up to 20:1, and enantioselectivities ranging from 84% to > 99% (Scheme 106).

Enantioselective organocatalytic one-pot domino Michael/intramolecular Povarov reaction with indolinones
11 Conclusions
The Povarov reaction allowed the preparation of heterocyclic compounds, tetrahydroquinoline skeleton, in a chemo-, regio- and stereoselective way by [4 + 2] cycloaddition between aromatic aldimines and dienophiles, in the presence of Lewis or Brønsted acids and under mild reaction conditions. A particular type of Povarov reaction is its intramolecular version when both, the aromatic imine (diene) and the dienophile system, are present in the same initial chemical structure, so that in the presence of Lewis or Brønsted acids this strategy allows the synthesis of a wide variety of fused heterocyclic compounds. These present important applications in medicinal, biological, and materials chemistry.
The intramolecular Povarov reaction provides a simple way to modulate the structural variety of the heterocyclic compounds to be prepared. Thus, the review began with a description of the heterocycles formed from aromatic amines with aliphatic, aromatic, and heteroaromatic aldehydes, followed by an analysis of the heterocycles obtained from heteroaromatic amines with different aldehydes. Likewise, special sections have been devoted to the intramolecular Povarov reaction with secondary amines and to the oxidative Povarov reaction demonstrating the applicability of this tool. Finally, the enantioselective preparation of fused heterocycles has been also addressed. As an improvement of the methods described so far, further exploration remains to be done in this field to discover new synthetic strategies for the preparation of new molecules and the development of new efficient asymmetric catalytic protocols to obtain compounds in an enantioselective way.
Data availability
The data reported in this review article is available in the internet, as in the references stated below. The authors also confirm that the data supporting the findings of this study are available within the article.
Abbreviations
- AMEBA:
-
Acid-sensitive methoxy benzaldehyde polystyrene
- BA:
-
Brønsted acid
- bmim:
-
1-Butyl-3-methylimidazolium
- BPO:
-
Benzoyl peroxide
- BPTA:
-
2,5-Bis-propargyloxy terephthalaldehyde
- CAN:
-
Ammonium cerium (IV) nitrate
- COF:
-
Covalent organic framework
- o-DCB:
-
o-Dichlorobenzene
- DCE:
-
1,2-Dichloroethane
- DCM:
-
Dichloromethane
- DDQ:
-
2,3-Dichloro-5,6-dicyano-p-benzoquinone
- DMB:
-
2,4-Dimethoxybenzyl
- DME:
-
Dimethoxyethane
- DMF:
-
Dimethylformamide
- DMSO:
-
Dimethylsulfoxide
- eq:
-
Equivalents
- FDA:
-
Food and Drug Administration
- hmim:
-
1-Hexyl-3-methylimidazolium
- IBX:
-
2-Iodoxybenzoic acid
- IL:
-
Ionic liquids
- LA:
-
Lewis acid
- LPDE:
-
Lithium perchlorate in diethyl ether
- MCF-7:
-
Breast cancer cell line
- MCR:
-
Multicomponent reaction
- MDA-MB-231:
-
Breast cancer cell line
- MIC:
-
Minimum inhibitory concentration
- Ms:
-
Mesyl
- MS:
-
Molecular sieves
- MW:
-
Microwave
- NiP:
-
Ni-porphyrin
- NMR:
-
Nuclear magnetic resonance
- Ns:
-
Nosyl
- octmim:
-
1-Octyl-3-methylimidazolium
- Piv:
-
Pivaloyl
- QUIN:
-
Quinoline
- rt:
-
Room temperature
- TAPB:
-
1,3,5-Tris(4-aminophenyl)benzene
- TAPT:
-
1,3,5-Tris(4-aminophenyl)triazine
- TBAF:
-
Tetra-n-butylammonium fluoride
- TBPA:
-
Tris(4-bromophenyl)ammoniumyl hexachloroantimonate
- TBS:
-
tert-Butyldimethylsilyl
- O-TES:
-
O-Triethylsilyl
- TFA:
-
Trifluoroacetic acid
- TFE:
-
Trifluoroethanol
- TFMSA:
-
Trifluoromethanesulfonic acid
- THF:
-
Tetrahydrofurane
- THQ:
-
Tetrahydroquinoline
- O-TMS:
-
O-Trimethylsilyl
- TRIP:
-
3,3′-Bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diyl hydrogen phosphate
- TPP:
-
Triphenylphosphonium perchlorate
- Ts:
-
Tosyl
- TS:
-
Transition state
References
Aatif M, Raza M, Javed K, Nashre-ul-Islam SM, Farhan M, Alam M (2022) Potential nitrogen-based heterocyclic compounds for treating infectious diseases: a literature review. Antibiotics 11:1750
Tran TN, Henary M (2022) Synthesis and applications of nitrogen-containing heterocycles as antiviral agents. Molecules 27:2700
Mermer A, Keles T, Sirin Y (2021) Recent studies of nitrogen containing heterocyclic compounds as novel antiviral agents: a review. Bioorg Chem 114:105076
Liu J, Jiang J, Zheng L, Liu ZQ (2020) Recent advances in the synthesis of nitrogen heterocycles using arenediazonium salts as nitrogen sources. Adv Synth Catal 362:4876–4895
Heravi MM, Zadsirjan V (2020) Prescribed drugs containing nitrogen heterocycles: an overview. RSC Adv 10:44247–44311
Bhutani P, Joshi G, Raja N, Bachhav N, Rajanna PK, Bhutani H, Paul AT, Kumar RUS (2021) FDA approved drugs from 2015–June 2020: a perspective. J Med Chem 64:2339–2381
Vitaku E, Smith DT, Njardarson JT (2014) Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among US FDA approved pharmaceuticals. J Med Chem 57:10257–10274
Kerru N, Gummidi L, Maddila S, Gangu KK, Jonnalagadda SB (1909) A Review on recent advances in nitrogen-containing molecules and their biological applications. Molecules 2020:25
Cabrele C, Reiser O (2016) The modern face of synthetic heterocyclic chemistry. J Org Chem 81:10109–10125
Sridharan V, Suryavanshi PA, Menendez JC (2011) Advances in the chemistry of tetrahydroquinolines. Chem Rev 111:7157–7259
Carey JS, Laffan D, Thomson C, Williams MT (2006) Analysis of the reactions used for the preparation of drug candidate molecules. Org Biomol Chem 4:2337–2347
Schreiber SL (2000) Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 287:1964–1969
Povarov LS, Mikhailov BM (1963) A new type of Diels–Alder reaction. Izv Akad Nauk SSR 1963:955–956
Ferreira de Paiva W, de Freitas Rego Y, de Fátima A, Fernandes SA (2022) The Povarov reaction: a versatile method to synthesize tetrahydroquinolines, quinolines and julolidines. Synthesis 54:3162–3179
Ghashghaei O, Masdeu C, Alonso C, Palacios F, Lavilla R (2018) Recent advances of the Povarov reaction in medicinal chemistry. Drug Discov Today Technol 29:71–79
Clerigue J, Ramos MT, Menendez JC (2022) Enantioselective catalytic Povarov reactions. Org Biomol Chem 20:1550–1581
Kobayashi S, Ishitani H, Nagayama S (1995) Lanthanide triflate catalyzed imino Diels–Alder reactions; convenient syntheses of pyridine and quinoline derivatives. Synthesis 20:1195–1202
Palacios F, Alonso C, Arrieta A, Cossío FP, Ezpeleta JM, Fuertes M, Rubiales G (2010) Lewis acid activated aza Diels−Alder reaction of N-(3-pyridyl)aldimines: an experimental and computational study. Eur J Org Chem 25:2091–2099
Xu H, Zuend SJ, Woll MG, Tao Y, Jacobsen EN (2010) Science 327:986–990
Lemos BC, Venturini Filho E, Fiorot RG, Medici F, Greco SJ, Benaglia M (2022) Enantioselective Povarov reactions: an update of a powerful catalytic synthetic methodology. Eur J Org Chem 25:e202101171
Ajani OO, Iyaye KT, Ademosun OT (2022) Recent advances in chemistry and therapeutic potential of functionalized quinoline motifs—a review. RSC Adv 12:18594–18614
Laschat S, Lauterwein J (1993) Intramolecular hetero-Diels–Alder reaction of N-arylimines. Applications to the synthesis of octahydroacridine derivatives. J Org Chem 58:2856–2861
Temme O, Froehlich R, Laschat S (1998) Convenient preparation of aromatic aldehydes via DDQ oxidation. Application in the synthesis of a tripodand containing octahydroacridine moieties. J Prakt Chem 25(340):2–5
Laschat S, Noe R, Riedel M, Krueger C (1993) Novel (imino-η6-arene)chromium complexes and their diastereoselective intramolecular hetero-Diels–Alder reactions. Organometallics 129:3738–3742
Schulte JL, Laschat S, Kotila S, Hecht J, Fröhlich R, Wibbeling BT (1996) Synthesis of η6-(octahydroacrydine)chromium tricarbonyl complexes with non-polar tails via molecular sieves-catalyzed cyclization of N-arylimines and subsequent diastereoselective complexation. Heterocycles 43:2713–2724
Temme O, Laschat S (1995) Effect of molecular sieves on the formation and acid-catalyzed mono- and bis-cyclization of N-arylimines: easy entry to polycyclic ring systems by a novel cascade reaction. J Chem Soc Perkin Trans 1:125–131
Sabitha G, Reddy EV, Yadav JSY (2002) Bismuth(III) chloride: an efficient catalyst for the one-pot stereoselective synthesis of octahydroacridines. Synthesis 20:409–412
Jacob RG, Perin G, Botteselle GV, Lenardao EJ (2003) Clean and atom-economic synthesis of octahydroacridines: application to essential oil of citronella. Tetrahedron Lett 44:6809–6812
Jacob RG, Silva MS, Mendes SR, Borges EL, Lenardao EJ, Perin G (2009) Atom-economic synthesis of functionalized octahydroacridines from citronellal or 3-(phenylthio)-citronellal. Synth Commun 39:2747–2762
Zaccheria F, Santoro F, Dhiaul Iftitah E, Ravasio N (2018) Brønsted and Lewis solid acid catalysts in the valorization of citronellal. Catalysts 8:410
Kiselyov AS, Smith L, Armstrong RW (1998) Solid support synthesis of polysubstituted tetrahydroquinolines via three-component condensation catalyzed by Yb(OTf)3. Tetrahedron 54:5089–5096
Mayekar NV, Nayak SK, Chattopadhyay S (2004) Two convenient one-pot strategies for the synthesis of octahydroacridines. Synth Commun 34:3111–3119
Lenardao EJ, Mendes SR, Ferreira PC, Perin G, Silveira CC, Jacob RG (2006) Selenium- and tellurium-based ionic liquids and their use in the synthesis of octahydroacridines. Tetrahedron Lett 47:7439–7442
Yadav JS, Reddy BVS, Chetia L, Srinivasulu G, Kunwar AC (2005) Ionic liquid accelerated intramolecular hetero-Diels–Alder reactions: a protocol for the synthesis of octahydroacridines. Tetrahedron Lett 46:1039–1044
Magomedov NA (2003) Efficient Construction of Cyclopenta[b]quinoline Core of isoschizozygane alkaloids via intramolecular formal hetero-Diels−Alder reaction. Org Lett 5:2509–2512
Chen Y, Ramanathan M, Liu S-T (2015) Intramolecular aza-Diels–Alder cyclization of a dimerized citral with anilines catalyzed by InCl3. J Chin Chem Soc 62:761–765
Linkert F, Laschat S, Knickneier M (1995) Chelation control by a second nitrogen atom in formal hetero-Diels–Alder reactions of N-arylimines. Liebigs Annal 25:985–993
Wölfling J, Frank E, Schneider G, Bes MT, Tietze LF (1998) Synthesis of azasteroids and D-homosteroids by intramolecular cyclization reactions of steroid arylimines. Synlett 20:1205–1206
Wölfling J, Frank E, Schneider G, Tietze LF (1999) Synthesis of novel steroid alkaloids by cyclization of arylimines from estrone. Eur J Org Chem 25:3013–3020
Magyar A, Wölfling J, Kubas M, Cuesta Seijo JA, Sevvana M, Herbst-Irmer R, Forgó P, Schneider G (2004) Synthesis of novel steroid-tetrahydroquinoline hybrid molecules and D-homosteroids by intramolecular cyclization reactions. Steroids 69:301–312
Sabitha G, Reddy EV, Yadav JS, Rama Krishna KVS, Ravi Sankar A (2002) Stereoselective synthesis of octahydro-3bH-[1,3]dioxolo[4",5":4’,5’]furo[2’,3’:5,6]pyrano[4,3-b]quinolines via intramolecular hetero-Diels–Alder reactions catalyzed by bismuth(III) chloride. Tetrahedron Lett 43:4029–4032
Sabitha G, Maruthi C, Reddy EV, Srinivas C, Yadav JS, Dutta SK, Kunwar AC (2006) Intramolecular hetero-Diels–Alder reactions catalyzed by BiCl3: stereoselective synthesis of benzoannelated decahydrofuro[3,2-h][1,6]-naphthyridine derivatives. Helv Chim Acta 89:2728–2731
Desrat S, van de Weghe P (2009) Intramolecular imino-Diels–Alder reaction: progress toward the synthesis of uncialamycin. J Org Chem 74:6728–6734
Spaller MR, Thielemann WT, Brennan PE, Bartlett PA (2002) Combinatorial synthetic design. Solution and polymer-supported synthesis of heterocycles via intramolecular aza Diels–Alder and imino alcohol cyclizations. J Comb Chem 4:516–522
Jayagobi M, Poornachandran M, Raghunathan R (2009) A novel heterotricyclic assembly through intramolecular imino-Diels–Alder reaction: synthesis of pyrrolo[3,4-b]quinolones. Tetrahedron Lett 50:648–650
Jayagobi M, Raghunathan R, Sainath S, Raghunathan M (2011) Synthesis and antibacterial property of pyrrolopyrano quinolinones and pyrroloquinolines. Eur J Med Chem 46:2075–2082
Jayagobi M, Raghunathan R (2012) Novel diastereoselective synthesis of trans-fused pyrrolo[3,4-b]quinolines through intramolecular imino-Diels–Alder reaction. Synth Commun 42:2917–2930
Mishra A, Rastogi N, Batra S (2012) 2-(N-Allylaminomethyl)cinnamaldehydes as substrates for syntheses of aza-polycycles via intramolecular cycloaddition reactions. Tetrahedron 68:2146–2154
Dickner T, Laschat S (2000) Stereoselective synthesis and binding properties of novel concave-shaped indolizino[3,4-b]quinolines. J Prakt Chem 342:804–811
Linkert F, Laschat S (1994) Intramolecular hetero-Diels–Alder reaction of prolinal-derived N-arylimines. Lewis acid-dependent reversal of the diastereoselectivity. Synlett 25:125–126
Linkert F, Laschat S, Kotila S, Fox T (1996) Evidence for a stepwise mechanism in formal hetero-Diels–Alder reactions of N-arylimines. Tetrahedron 52:955–970
Temme O, Dickner T, Laschat S, Froehlich R, Kotila S, Bergander K (1998) Synthesis of aza polycyclic systems based on the indolizino[3,4-b]quinoline skeleton. A diastereoselective entry to potential oligodentate artificial receptors. Eur J Org Chem 25:651–659
Monsees A, Laschat S, Hotfilder M, Jones PG (1998) Diastereoselective synthesis of octahydro-14H-benzo[g]quinolino-[2,3-a]quinolidines. Improved cytotoxic activity against human brain tumor cell lines as a result of the increased rigidity of the molecular backbone. Bioorg Med Chem Lett 8:2881–2884
Koepler O, Mazzini S, Bellucci MC, Mondelli R, Baro A, Laschat S, Hotfilder M, Viseur C, Frey W (2005) Synthesis and DNA binding properties of novel benzo[b]isoquino[2,3-h]naphthyridines. Org Biomol Chem 3:2848–2858
Almansour AI, Arumugam N, Kumar RS, Menéndez JC, Ghabbour HA, Fun H-K, Kumar RR (2015) Straightforward synthesis of pyrrolo[3,4-b]quinolines through intramolecular Povarov reactions. Tetrahedron Lett 56:6900–6903
Almansour AI, Kumar RS, Arumugam N, Bianchini G, Menéndez JC, Al-thamili DM, Periyasami G, Altaf M (2019) Design and synthesis of A- and D ring-modified analogues of luotonin A with reduced planarity. Tetrahedron Lett 60:1514–1517
Sabitha G, Shankaraiah K, Sindhu K, Latha BM (2015) Bismuth(III) chloride catalyzed intramolecular hetero-Diels–Alder reaction: access to cis-fused angular hexahydrobenzo[c]acridines. Synthesis 47:124–128
Chen S-H, Li Q, Liu X-G, Wu J-K, Zhang S-S, Wang H (2017) Polycyclization enabled by relay catalysis: one-pot manganese-catalyzed C–H allylation and silver-catalyzed Povarov reaction. Chemsuschem 10:2360–2364
Chen M, Sun N, Liu Y (2013) Environmentally benign synthesis of indeno[1,2-b]quinolines via an intramolecular Povarov reaction. Org Lett 15:5574–5577
Chakraborty B, Kar A, Chanda R, Umasishn J (2020) Application of the Povarov reaction in biaryls under iron catalysis for the general synthesis of dibenzo[a, c]acridines. J Org Chem 85:9281–9289
Jones W, Kiselyov AS (2000) Intramolecular cyclization of aromatic imines: an approach to tetrahydrochromeno[4,3-b]quinolines. Tetrahedron Lett 41:2309–2312
Rajagopal N, Magesh CJ, Perumal PT (2004) Inter- and intramolecular imino-Diels–Alder reactions catalyzed by sulfamic acid: a mild and efficient catalyst for a one-pot synthesis of tetrahydroquinolines. Synthesis 1:69–74
Sabitha G, Reddy EV, Yadav JS (2001) Bismuth(III) chloride-catalyzed intramolecular hetero-Diels–Alder reaction: application to the synthesis of tetrahydrochromano[4,3-b]quinoline derivatives. Synthesis 13:1979–1984
Yadav JS, Reddy BVS, Rao CV, Srinivas R (2002) LPDE-catalyzed intramolecular cyclization of arylimines: a facile synthesis of tetrahydrochromanoquinolines. Synlett 6:993–995
Anniyappan M, Muralidharan D, Perumal PT (2003) Triphenylphosphonium perchlorate as an efficient catalyst for mono- and bis-intramolecular imino-Diels–Alder reactions: synthesis of tetrahydrochromenoquinolines. Tetrahedron Lett 44:3653–3657
Elamparuthi E, Anniyappan M, Muralidharan D, Perumal PT (2005) InCl3 as an efficient catalyst for intramolecular imino-Diels–Alder reactions: synthesis of tetrahydrochromenoquinolines. ARKIVOC 11:6–16
Yadav JS, Reddy BVS, Kondaji G, Sowjanya S, Nagaiah K (2006) Intramolecular imino-Diels–Alder reactions in [bmim]BF4 ionic medium: Green protocol for the synthesis of tetrahydrochromenoquinolines. J Mol Cat A Chem 258:361–366
Tomashevskaya MM, Tomashenko OA, Tomashevskii AA, Sokolov VV, Potekhin AA (2007) New one-step procedure for the synthesis of 6H-chromeno[4,3-b]quinolines and 8a,9,14,14atetrahydro-8H-benzo[5,6]chromeno[4,3-b]quinolines. Rus J Org Chem 43:77–82
Wuheng D, Yao Y, Xiaoshuang G, Bei H, Xiaomin X, Zhaoguo Z (2018) Merging visible-light photoredox and Lewis acid catalysis for the intramolecular aza-Diels–Alder reaction: synthesis of substituted chromeno[4,3-b]quinolines and [1,6]naphthyridines. ChemCatChem 10:2878–2886
Imrich H-G, Conrad J, Beifuss U (2016) The first domino reduction/imine formation/intramolecular aza-Diels–Alder reaction for the diastereoselective preparation of tetrahydrochromano[4,3-b]quinolones. Eur J Org Chem 25:5706–5715
Zhang D, Kiselyov AS (2001) A solid-phase approach to tetrahydrochromano[4,3-b]quinolines. Synlett 7:1173–1175
Peñaranda A, Puerto CE, Macías MA, Ochoa-Puentes C, Kouznetsov VV (2021) I2/DMSO-Promoted synthesis of chromeno[4,3-b]quinolines through an imine formation/aza-Diels–Alder/aromatization tandem reaction under metal-catalyst- and photosensitizer-free conditions. Synthesis 53:1857–1869
Foley BJ, Bhuvanesh N, Zhou J, Ozerov OV (2020) Combined experimental and computational studies of the mechanism of dehydrogenative borylation of terminal alkynes catalyzed by PNP complexes of iridium. ACS Catal 10:9824–9836
Qiao C, Cao Y, He L-N (2018) Transition metal-catalyzed carboxylation of terminal alkynes with CO2. Mini-Rev Org Chem 15:283–290
Liu Y, Yang Y, Shi Y, Wang X, Zhang L, Cheng Y, You J (2016) Rhodium-catalyzed oxidative coupling of benzoic acids with terminal alkynes: an efficient access to 3-ylidenephthalides. Organometallics 35:1350–1353
Zhang Y-Q, Kepcija N, Kleinschrodt M, Diller K, Fischer S, Papageorgiou AC, Allegretti F, Bjork J, Klyatskaya S, Klappenberger F, Ruben M, Barth JV (2012) Homo-coupling of terminal alkynes on a noble metal surface. Nat Commun 3:1286
Ramesh S, Gaddam V, Nagarajan R (2010) A flexible approach to the chromenoquinolines under copper/Lewis acid catalysis. Synlett 25:757–760
Rahimzadeh G, Soheilizad M, Kianmehr E, Larijani B, Mahdavi M (2018) Copper-catalyzed intramolecular domino synthesis of 6H-chromeno[4,3-b]quinolines in green condition. ARKIVOC v:20–28
Yu X, Wang J, Xu Z, Yamamoto Y, Bao M (2016) Copper-catalyzed aza-Diels−Alder reaction and halogenation: an approach to synthesize 7-halogenated chromenoquinolines. Org Lett 18:2491–2494
Ren X-R, Bai B, Zhang Q, Hao Q, Guo Y, Wan L-J, Wang D (2022) Constructing stable chromenoquinoline-based covalent organic frameworks via intramolecular Povarov reaction. J Am Chem Soc 144:2488–2494
Sabitha G, Reddy EV, Maruthi Ch, Yadav JS (2002) Bismuth(III) chloride-catalyzed intramolecular hetero-Diels–Alder reactions: a novel synthesis of hexahydrodibenzo[b, h][1,6]naphthyridines. Tetrahedron Lett 43:1573–1575
Muthukrishnan I, Vinoth P, Vivekanand T, Nagarajan S, Maheswari CM, Menéndez JC, Sridharan V (2016) Synthesis of 5,6-dihydrodibenzo[b, h][1,6]naphthyridines via copper bromide catalyzed intramolecular [4+2] hetero-Diels−Alder reactions. J Org Chem 81:1116–1124
Ramesh E, Manian RDRS, Raghunathan R, Sainath S, Raghunathan M (2009) Synthesis and antibacterial property of quinolines with potent DNA gyrase activity. Bioorg Med Chem 17:660–666
Gaddam V, Nagarajan R (2007) Highly diastereoselective synthesis of new indolopyrroloquinolines through intramolecular imino-Diels–Alder reactions. Tetrahedron Lett 48:7335–7338
Manian RDRS, Jayashankaran J, Raghunathan R (2007) Indium trichloride catalyzed one-pot synthesis of indolo[2,1-a]pyrrolo[4’,3’:2,3]-7a,8,13,13b-tetrahydroquinolines through intramolecular imino-Diels–Alder reactions. Tetrahedron Lett 48:4139–4142
Ramesh E, Raghunathan R (2008) Indium chloride catalyzed intramolecular cyclization of N-aryl imines: synthesis of pyrrolo[2,3-d]pyrimidine annulated tetrahydroquinoline derivatives. Tetrahedron Lett 49:2583–2587
Sabitha G, Reddy ChS, Maruthi Ch, Reddy EV, Yadav JS (2003) BiCl3-catalyzed diastereoselective intramolecular [4+2] cycloaddition reactions leading to pyrazole annulated new sulfur heterocycles. Synth Commun 33:3063–3070
Manian RDRS, Jayashankaran J, Ramesh R, Raghunathan R (2006) Rapid synthesis of tetrahydroquinolines by indium trichloride catalyzed mono- and bis-intramolecular imino-Diels–Alder reactions. Tetrahedron Lett 47:7571–7574
Dong W, Yuan Y, Hu B, Gao X, Gao H, Xie X, Zhang Z (2018) Combining visible-light-photoredox and Lewis acid catalysis for the synthesis of indolizino[1,2-b]quinolin-9(11H)-ones and irinotecan precursor. Org Lett 20:80–83
Yang F, Zheng L, Xiang J, Dang Q, Bai X (2010) Synthesis of hexahydrobenzo[b]pyrimido[4,5-h][1,6]naphthyridines via an intramolecular hetero-Diels–Alder reaction. J Comb Chem 12:476–481
Subba Reddy BV, Antony A, Yadav JS (2010) Novel intramolecular aza-Diels–Alder reaction: a facile synthesis of trans-fused 5H-chromeno[2,3-c]acridine derivatives. Tetrahedron Lett 51:3071–3074
Maiti S, Panja SK, Bandyopadhyay C (2010) Substituent-controlled domino-Knoevenagel-hetero-Diels–Alder reaction-a one-pot synthesis of polycyclic heterocycles. Tetrahedron 66:7625–7632
Maiti S, Panja SK, Sadhukhan K, Ghosh J, Bandyopadhyay C (2012) Effects of substituent and catalyst on the intramolecular Povarov reaction–synthesis of chromenonaphthyridines. Tetrahedron Lett 53:694–696
Ramesh E, Sree Vidhya TK, Raghunathan R (2008) Indium chloride/silica gel supported synthesis of pyrano/thiopyranoquinolines through intramolecular imino-Diels–Alder reaction using microwave irradiation. Tetrahedron Lett 49:2810–2814
Dai X, Cheng C, Ding C, Yao Q, Zhang A (2008) Synthesis of 2,7-naphthyridine-containing analogues of luotonin A. Synlett 25:2989–2992
Zhou H-B, Liu G-S, Yao Z-J (2007) Highly efficient and mild cascade reactions triggered by bis(triphenyl)oxodiphosphonium trifluoromethanesulfonate and a concise total synthesis of camptothecin. Org Lett 9:2003–2006
Zhou H-B, Liu G-S, Yao Z-J (2007) Short and efficient total synthesis of luotonin A and 22-hydroxyacuminatine using a common cascade strategy. J Org Chem 72:6270–6272
Nagaiah K, Venkatesham A, Srinivasa Rao R, Saddanapu V, Yadav JS, Basha SJ, Sarma AVS, Sridhar B, Addlagatta A (2010) Synthesis of new cis-fused tetrahydrochromeno[4,3-b]quinolines and their antiproliferative activity studies against MDA-MB-231 and MCF-7 breast cancer cell lines. Bioorg Med Chem Lett 20:3259–3264
Wu F, Dong W, Fan S, Yuan Y, Liang C, Chen A, Yin Z, Zhang Z (2022) Rapid synthesis of Luotonin A derivatives via synergistic visible-light photoredox and acid catalysis. J Org Chem 87:1302–1312
Kumara BN, Venkatesham A, Nagaiah K, Babu NJ (2015) Synthesis of new pyrano[2’,3’:5,6]chromeno[4,3-b]quinolin-4-ones via aza-Diels–Alder reaction. Helv Chim Acta 98:417–426
Almansour AI, Arumugam N, Kumar RS, Mahalingam SM, Sau S, Bianchini G, Menéndez JC, Altaf M, Ghabbour HA (2017) Design, synthesis and antiproliferative activity of decarbonyl luotonin analogues. Eur J Med Chem 138:932–941
Twin H, Batey RA (2004) Intramolecular hetero-Diels–Alder (Povarov) approach to the synthesis of the alkaloids luotonin A and Camptothecin. Org Lett 6:4913–4916
Lezana N, Matus-Pérez M, Galdámez A, Lührb S, Vilches-Herrera M (2016) Highly stereoselective and catalyst-free synthesis of annulated tetrahydropyridines by intramolecular imino-Diels–Alder reaction under microwave irradiation in water. Green Chem 18:3712–3717
Gaddam V, Nagarajan R (2008) An efficient, one-pot synthesis of isomeric ellipticine derivatives through intramolecular imio-Diels–Alder reaction. Org Lett 10:1975–1978
Tietze LF, Utecht J (1992) Inter- and intramolecular hetero-Diels–Alder reactions Unusual stereocontrol in intramolecular hetero-Diels–Alder reactions of 2-aza-1,3-butadienes. A stereoselective sequential synthesis of annulated tetrahydropyridines. Chem Ber 125:2259–2263
Martín-Encinas E, Rubiales G, Knudssen BR, Palacios F, Alonso C (2019) Straightforward synthesis and biological evaluation as topoisomerase I inhibitors and antiproliferative agents of hybrid chromeno[4,3-b][1,5]naphthyridines and chromeno[4,3-b][1,5]naphthyridin-6-ones. Eur J Med Chem 178:752–766
Martín-Encinas E, Rubiales G, Knudsen BR, Palacios F, Alonso C (2021) Fused chromeno and quinolino[1,8]naphthyridines: synthesis and biological evaluation as topoisomerase I inhibitors and antiproliferative agents. Bioorg Med Chem 40:116177
Adolfsson DE, Tyagi M, Singh P, Deuschmann A, Ådén J, Gharibyan AL, Jayaweera SW, Lindgren AEG, Olofsson A, Almqvist F (2020) Intramolecular Povarov reactions for the synthesis of chromenopyridine fused 2-pyridone polyheterocycles binding to α-synuclein and amyloid-β fibrils. J Org Chem 85:14174–14189
Martín-Encinas E, Selas A, Tesauro C, Rubiales G, Knudsen BR, Palacios F, Alonso C (2020) Synthesis of novel hybrid quinolino[4,3-b][1,5]naphthyridines and quinolino[4,3-b][1,5]naphthyridin-6(5H)-one derivatives and biological evaluation as topoisomerase I inhibitors and antiproliferatives. Eur J Med Chem 195:112292
Majumdar KC, Ponra S, Ganai S (2010) Copper(I)-catalyst-free approach for the synthesis of chromene-fused pyrido[3,2-d]pyrimidines by Lewis acid catalyzed aza-Diels–Alder reaction. Synlett 25:2575–2578
Gaddam V, Ramesh S, Nagarajan R (2010) CuI/La(OTf)3 catalyzed, one-pot synthesis of isomeric ellipticine derivatives in ionic liquid. Tetrahedron 66:4218–4222
Beifuss U, Herde A, Ledderhose S (1996) Highly diastereoselective synthesis of octahydroacridines by domino imine condensation-intramolecular polar [4π++2π]-cycloaddition of anilines and ω-unsaturated aldehydes. Chem Commun 25:1213–1214
Acelas M, Romero Bohórquez AR, Kouznetsov VV (2017) Highly diastereoselective synthesis of new trans-fused octahydroacridines via intramolecular cationic imino-Diels–Alder reaction of N-protected anilines and citronellal or citronella essential oil. Synthesis 49:2153–2162
Acelas M, Camargo HA, Henao JA, Kouznetsov VV, RomeroBohórquez AR, Dugarte-Dugarte A, Delgado JM, Díaz de Delgado G (2020) Synthesis, characterization and crystal structure of two polymorphs of trans N-benzyl-3,9,9-trimethyl-1,2,3,4,4a,9,9a,10-octahydroacridine. J Mol Struct 1215:128222
Muhuhi J, Spaller MR (2006) Expanding the synthetic method and structural diversity potential for the intramolecular aza-Diels–Alder cyclization. J Org Chem 71:5515–5526
Richter H, García Mancheño O (2011) TEMPO Oxoammonium salt-mediated dehydrogenative Povarov/oxidation tandem reaction of N-alkyl anilines. Org Lett 13:6066–6069
Jia X, Peng F, Qing C, Huo C, Wang X (2012) Catalytic radical cation salt induced Csp3–H functionalization of glycine derivatives: synthesis of substituted quinolines. Org Lett 14:4030–4033
Wang Y, Peng F, Liu J, Huo C, Wang X, Jia X (2015) Radical cation salt-promoted catalytic aerobic sp3 C−H oxidation: construction of quinoline-fused lactones and lactams. J Org Chem 80:609–614
More DA, Shinde GH, Shaikh AC, Muthukrishnan M (2019) Oxone promoted dehydrogenative Povarov cyclization of N-aryl glycine derivatives: an approach towards quinoline fused lactones and lactams. RSC Adv 9:30277–30291
Jarrige L, Blanchard F, Masson G (2017) Enantioselective organocatalytic intramolecular aza-Diels–Alder reaction. Angew Chem Int Ed 56:10573–10576
Jarrige L, Gandon V, Masson G (2020) Enantioselective synthesis of complex fused heterocycles through chiral phosphoric acid catalyzed intramolecular inverse-electron-demand aza-Diels–Alder reactions. Chem Eur J 26:1406–1413
Min C, Lin C-T, Seidel D (2015) Catalytic enantioselective intramolecular aza-Diels–Alder reactions. Angew Chem Int Ed 127:6608–6612
Min C, Mittal N, Sun DX, Seitel D (2013) Conjugate-base-stabilized Brønsted acids as asymmetric catalysts: enantioselective Povarov reactions with secondary aromatic amines. Angew Chem Int Ed 52:14084–14088
Min C, Seidel D (2016) Stereochemically rich polycyclic amines from the kinetic resolution of indolines through intramolecular Povarov reactions. Chem Eur J 22:10817–10820
Dickmeiss G, Jensen KL, Worgull D, Franke PT, Jørgensen KA (2011) An asymmetric organocatalytic one-pot strategy to octahydroacridines. Angew Chem Int Ed 50:1580–1583
Wu H, Wang Y-M (2014) One-pot organocatalytic enantioselective Michael/Povarov domino strategy for the construction of spirooctahydroacridine-3,3′-oxindole scaffolds. Chem Eur J 20:5899–5904
Acknowledgements
Financial support from the Ministerio de Ciencia, Innovación y Universidades (MCIU) (PID2021-122558OB-I00, UE), by Gobierno Vasco, Universidad del País Vasco (GV, IT1701-22; UPV) and by Fundación Vital (VITAL21/01) is gratefully acknowledged.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors do not have any conflicts of interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Masdeu, C., de los Santos, J.M., Palacios, F. et al. The Intramolecular Povarov Tool in the Construction of Fused Nitrogen-Containing Heterocycles. Top Curr Chem (Z) 381, 20 (2023). https://doi.org/10.1007/s41061-023-00428-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41061-023-00428-7