
Abstract
Tubulin posttranslational modifications (PTMs) add “tubulin code” to generate functional diversities of microtubules. Several types of tubulin PTMs accumulate on axonemes and basal bodies of cilia, including acetylation, glutamylation, glycylation and detyrosination. Among them, glutamylation, glycylation and detyrosination are mostly enriched in the B-tubules, whereas acetylation occurs on both A- and B-tubule of the microtubule doublets in a similar level. Recent studies indicate that tubulin PTMs are critical for the fine tuning of assembly/disassembly, maintenance, motility, and signaling of cilia. Dysregulated tubulin PTMs are strongly implicated in human disorders including ciliopathies and neuron degeneration. Here, we review the current understanding how tubulin PTMs regulate cilia formation and function, and their relevance to human health.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Microtubules, assembled from highly conserved α/β-tubulin heterodimers, are the key cytoskeletal elements for constructing various subcellular organelles. Despite their uniformed structure, microtubules can adapt to a large diversity of functions through spatial–temporal generation of specialized identities. The ‘tubulin code’, which is generated by the expression of different tubulin isotypes and posttranslational modifications (PTMs), confers the dynamic, functional diversity on microtubules (Gadadhar et al. 2017a; Janke 2014; Magiera et al. 2018a, b; Song and Brady 2015). The PTMs not only accumulate on a subset of long-lived microtubules, including those found in the centrosomes, cilia, and axons of neurons, but also on the highly dynamic ones found in mitotic spindle and marginal bands of blood platelets (Gadadhar et al. 2017a; Magiera et al. 2018a, b). In living cells, microtubules can interact with a variety set of microtubule-associated proteins (MAPs), such as microtubule motor proteins, microtubule plus end tracking proteins (+TIPs) and severing enzymes. Mechanistically, PTMs control microtubule functions either by direct alteration of their mechanical properties or by modulating their interactions with other proteins. Many types of PTMs have been discovered on tubulin. Some of them, including acetylation, phosphorylation, and methylation, also occur on non-tubulin substrates. While the others, such as (poly) glutamylation, (poly) glycylation and tyrosination/detyrosination, are mostly abundant in tubulin, thus allowing the generation of locally restricted and specialized functions on microtubules.
The cilium is a hair-like protrusion on cell surface of most eukaryotic cells, which is mainly compose of microtubule-cored axoneme anchored by the basal body transformed from the mother centriole. There are two main types of cilia: motile cilia and the primary cilia. In general, motile cilia are characterized by the 9 + 2 arrangement of microtubule doublets: 9 pairs of outer microtubule doublets and a central pair. The dynein arms and radial spokes connect with the outer doublets to generate force for cilia beating. With this kinetic capability, motile cilia tightly control cell locomotion, sexual reproduction and fluid flow generation. The primary cilia lack of central pair and motility and present a 9 + 0 arrangement. All cilia are built and maintained by a microtubule-based intraflagellar transport (IFT) (Rosenbaum and Witman 2002). The IFT particle is composed of two multimeric subcomplexes (IFT-A and IFT-B). In a simple model, anterograde transport is regulated by kinesin-2, whereas dynein regulates retrograde transport. IFT machinery mediates the bidirectional movement of IFT cargos that are required for the biogenesis, maintenance, and signaling of all cilia (Berbari et al. 2009; Pazour and Rosenbaum 2002; Pedersen and Rosenbaum 2008; Rosenbaum and Witman 2002; Scholey 2008; Scholey and Anderson 2006).
Sensory transduction capabilities of cilia are highly conserved across species. Polarized cells utilize primary cilia to receive environmental stimuli that are converted into physiological responses (Nauli et al. 2003; Praetorius and Spring 2001, 2003a, b). Once overlooked as an evolutionary vestige, it have recently been identified as the ubiquitous sensory antenna of many pivotal signaling pathways, such as Hedgehog, Polycystin, GPCR, platelet-derived growth factor receptor (PDGFR), planar cell polarity (PCP) and TGF-β signaling (Goetz and Anderson 2010; Nishimura et al. 2019). With rapid advancements in the positional cloning of human disease genes, 35 disorders (~187 causal loci), such as polycystic kidney disease (ADPKD and ARPKD), Bardet-Biedl syndrome (BBS), Joubert syndrome (JBTS), nephronophthisis (NPHP), and Meckel–Gruber syndrome (MKS), have been characterized molecularly as cilia-related diseases, or ciliopathies (Adams et al. 2008; Badano et al. 2006). Consistent with the presence of cilia on most cell surfaces in human body, most ciliopathies occur as syndromic disorders that affect many organs during development, including the kidneys, limbs, central nervous system (CNS), liver, eyes, and fat storage tissue. Despite the physiological and clinical relevance of cilia, the molecular mechanisms that regulate cilia formation and function and the connections between disease gene functions and pathology remain largely elusive.
The microtubule doublets of the axoneme consist of A-tubule and B-tubule that attached to the A-tubule. A-tubule forms a complete microtubule with 13 protofilaments, whereas B-tubule forms an incomplete microtubule structure with 10 protofilaments (Ichikawa et al. 2017; Ma et al. 2019). Unlike the highly dynamic non-axonemal MTs, the axonemal MTs are long-lived and endow cilia with stability, long-range transport, and structural basis for sensory function (Orbach and Howard 2019). Tubulin PTMs, including acetylation, (poly) glutamylation, (poly) glycylation and tyrosination/detyrosination, occur predominantly along the axoneme. Dysregulated tubulin PTMs are closely linked to a variety of human diseases. Here, we review current understanding of tubulin PTMs in cilia and related human disorders (Fig. 1, Table 1).

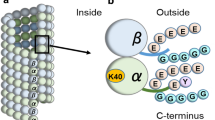
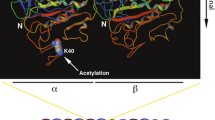
Tubulin posttranscriptional modifications on cilia axoneme. A Basic structure and microtubule doublets arrangement patterns of cilia. Cilium is a membrane-bound structure and composed of microtubule-cored axoneme running from the basal body, diffusion barrier and so on. The ciliary membrane is continuous with the cell membrane, but specialized by distinct lipid compositions and membrane proteins. The motile cilia have 9 + 2 axoneme compositions. The primary cilia have 9 + 0 axoneme compositions and lack of inner and outer dynein arms, radial spokes, and central sheath that compose of motile cilia. Driven by microtubule motors dynein and kinesin, the intraflagellar transport (IFT) machineries bidirectionally transport along the axoneme that is essential for the formation, maintenance of cilia and the proper ciliary translocation of signaling proteins. B Cartoon depicting the cross section of microtubule doublet with PTMs (αK40 acetylation: yellow; polyglycylation chain: black; polyglutamylation chain: red). C Schematic representation of the distribution of PTMs on α/β tubulin heterodimer in cilia and their corresponding involved enzymes
ACETYLATION
Acetylation at residue lysine 40 (K40) of α-tubulin, the predominant form of tubulin acetylation and the only tubulin PTM that occurs inside the microtubule lumen, is associated with long-lived subsets of microtubule structures including cilia axoneme (Gadadhar et al. 2017a; Janke and Bulinski 2011; LeDizet and Piperno 1987; Lhernault and Rosenbaum 1985; Soppina et al. 2012). The level of αK40 acetylation in A-tubule and B-tubule is similar (Orbach and Howard 2019). Of note, recent studies also revealed the existence of a novel acetylation modification of lysine 252 (K252) of β-tubulin (Choudhary et al. 2009; Chu et al. 2011; Liu et al. 2015), but with its distribution and physiological importance awaiting further characterization.
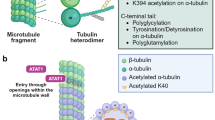
αK40 acetylation is majorly catalyzed by the highly conserved tubulin acetyl transferase αTAT1 and removed by the deacetylases histone deacetylase 6 (HDAC6) and sirtuin 2 (SIRT2) (Akella et al. 2010; Hubbert et al. 2002; Kalebic et al. 2013b; North et al. 2003). HDAC6 and SIRT2 are both enriched in cilia proper and at cilia base (de Diego et al. 2014; Pugacheva et al. 2007; Zhou et al. 2014). Strong biochemical evidences suggest that acetyl-K40 only takes place on microtubule lattice but not the cytosolic tubulin heterodimers (Bulinski et al. 1988; Lhernault and Rosenbaum 1983; Maruta et al. 1986; Piperno et al. 1987). Accordingly, αTAT1 preferentially modifies polymeric tubulin in vitro (Kalebic et al. 2013a). Therefore, αTAT1 must access to the narrow lumen of microtubules to acetylate αK40. Two proposed models attempt to elucidate the entry mechanism of αTAT1 (Coombes et al. 2016). The first model is based on the observation that lattice defects and protofilament switches have been observed in vitro (Chretien et al. 1992; Schaedel et al. 2015). Although it has not been formally visualized in microtubule lumen, αTAT1 may locally and transiently enters into the microtubule lumen through these cracks. Accordingly, αTAT1 preferentially acetylates the highly curved areas of microtubules, the same regions that accumulate lattice openings or cracks, suggesting that αTAT1 can locally modify the mechanical properties of the microtubule to protect it against mechanical stresses. However, in view of the low frequency of microtubule defects, this model may not explain how the acetylation spread over the entire microtubule. αTAT1 has shown a higher affinity for microtubule ends. Alternatively, the high density of exposed luminal sites at the tapered extremities could be captured by αTAT1. This proposed mechanism is supported by studies showing the preferential entry of αTAT1 at open microtubule ends (Coombes et al. 2016; Ly et al. 2016; Szyk et al. 2014). It is conceivable that the entry mechanism of αTAT1 may depend on the way by which microtubules were assembled and the mechanical stresses they were experiencing. In contrast to αTAT1, how deacetylase HDAC6 and SIRT2 act in vivo is less defined. HDAC6 can act on both free tubulin dimers and polymerized microtubules in vitro (Hubbert et al. 2002; Matsuyama et al. 2002; Miyake et al. 2016; Zhao et al. 2010). It also can interact with the microtubule plus end protein EB1, suggesting the end of the microtubules might be the important entry site for HDAC6 (Zilberman et al. 2009).
Although the αK40 acetylation of tubulin has been discovered over 30 years, its biological importance in cilia only emerges recently. Cells lacking of αTAT1 lost acetylation but preserve unaffected cilia morphology (Kalebic et al. 2013b; Shida et al. 2010). Deletion of HDAC6 results in tubulin hyperacetylation but shows no impact on cilia biogenesis and morphology, too (Pugacheva et al. 2007; Ran et al. 2015; Zhang et al. 2008). Loss of αTAT1 appears to delay the assembly rate of primary cilia (Ran et al. 2015) while pharmacological and genetic suppression of HDAC6 protects primary cilia from disassembly (Pugacheva et al. 2007; Ran et al. 2015). Intriguingly, inhibition of SIRT2 not only blocks cilia disassembly but also increase ciliation ratio and cilia length (Zhou et al. 2014). Overexpression of HDAC6 or SIRT2 decreases cilia number and length (Ran et al. 2015; Zhou et al. 2014). The discrepancies of the effects of αTAT1, HDAC6 and SIRT2 on ciliogenesis and cilia length may be attributable to non-tubulin acetylation activities of those enzymes (Drazic et al. 2016; Narita et al. 2019).
There are evidences suggesting that K40 acetylation may be implicated in regulation of motility of molecular motors. αK40 acetylation has been shown to increase axonemal dynein motility (Alper et al. 2014). However, there is controversial views on whether kinesins could be affected acetylation or not, with an in vitro study showing that kinesin-1 shows less binding and motility along the axoneme with α-tubulin K40R mutant (Reed et al. 2006), while the others reported acetylation state of tubulin alone did not affect the motility of kinesin-1 on the microtubule track by using enzymatically generated acetylated or deacetylated microtubules (Kaul et al. 2014; Walter et al. 2012). Given that tubulin acetylation is located in the inaccessible lumen of microtubules, the finding that this modification can control motility of molecular motors indeed came as a surprise. It remains as an open question that acetylation modification might influence other characteristics of the microtubules to indirectly regulate motor motility. For example, tubulin detyrosination is significantly decreased in αTAT1-deleted cells (Xu et al. 2017) and tubulin glycylation deficiency is associated increased levels of acetyl-K40 in Tetrahymena mutants (Wloga et al. 2009). Despite its elusive role in regulating motor motility, acetyl-K40 could be the key protecting mechanism for cilia to cope with mechanical stresses. Cilia are frequently exposed to mechanical forces that can cause microtubules breakage on axoneme, such as the fluid flow in kidney. αK40 acetylation has been shown to soften the microtubules by weakening inter-protofilament interactions, thus enhances its flexibility and confers resilience against mechanical stresses to ensure the persistence of long-lived microtubules (Portran et al. 2017; Xu et al. 2017).
In neurons, tubulin acetylation contributes to axon branching (Dan et al. 2018), cortical neurons migration and morphological development (Li et al. 2012). Links between decreased tubulin acetylation and axonal transport defects have been found in a range of neurodegenerative disorders, such as Huntington’s disease (Dompierre et al. 2007), Charcot-Marie-Tooth disease (Benoy et al. 2017; d'Ydewalle et al. 2011), amyotrophic lateral sclerosis (ALS) (Lazo-Gomez et al. 2013), and Parkinson’s disease (Godena et al. 2014). However, the contribution and mechanism of abnormal tubulin acetylation in neurodegenerative disorders remains to be elucidated, as the mice lacking αTAT1 merely showed defects in touch sensation but did not develop any of the expected degenerative phenotypes (Morley et al. 2016).
Tubulin acetylation also regulates blood clotting. The discoidal shape of blood platelets in the fast bloodstream is maintained by a closed ring of microtubules, called the marginal band (MB) (White and Rao 1998). Platelets are activated after vessel injury and undergo a major shape change known as disc to sphere transition that result from the contraction of MB, which leads to blood clotting (Johnson et al. 2007). MB microtubules in resting platelets are heavily acetylated (Diagouraga et al. 2014; Patel-Hett et al. 2008), and defects in tubulin acetylation of microtubules in marginal band affect the maturation of the precursors of platelets and the following platelet formation (Iancu-Rubin et al. 2012). Tight regulation of acetylation/deacetylation process is critical during platelet spreading after activation (Sadoul et al. 2012). Abnormal tubulin acetylation of sperm flagella has been directly linked to male subfertility, as the mice lacking of αTAT1 show abnormal sperm morphology and motility (Kalebic et al. 2013b). Recently, a study found that mutations in Joubert syndrome genes ARMC9 or TOGARAM1 results in short cilia with decreased axonemal acetylation (Latour et al. 2019).
Interestingly, in the context of ciliopathies, dysfunction of several ciliopathy proteins is correlated with globally aberrant tubulin acetylation, in either the axoneme or cytoplasmic microtubules. Bardet-Biedl syndrome protein BBIP10 is required for cytoplasmic microtubule polymerization and acetylation, which is likely through inhibiting HADC6 (Loktev et al. 2008). Deletion of Joubert syndrome gene KIF7 results in decreased level of acetylated microtubule in the cytoplasm (Dafinger et al. 2011). In addition, deletion of the ADPKD gene Pkd1 increases SIRT2 protein levels and decreases total tubulin acetylation levels, which lead to abnormal centrosome amplification and polyploidy (Zhou et al. 2014). It was shown that cilia ablation by depleting structural gene Ift88 and Kif3a leads to increased αTAT1 activities and hyperacetylation of cytosolic microtubules, with similar impact evident in the kidneys of ARPKD patients (Berbari et al. 2013). These observations raise an interesting perspective that cilia may orchestrate activities of tubulin acetylases/deacetylases to impact global microtubule acetylation.
GLUTAMYLATION
The γ-carboxyl groups of glutamate residues in α/β-tubulin and non-tubulin proteins can be added with single (monoglutamylation) or multiple glutamates (polyglutamylation) (Alexander et al. 1991; Edde et al. 1990; Regnard et al. 2000; Rüdiger et al. 1992; van Dijk et al. 2008). These modifications occur most abundantly on stable microtubule structures such as the ones found in neurons, centrosomes or basal bodies and cilia, whereas it also enriched in the highly dynamic mitotic spindle during mitosis (Audebert et al. 1994; Bobinnec et al. 1998; Fouquet et al. 1994; Mary et al. 1996; Regnard et al. 1999). Specific antibodies are used for detecting glutamylation: GT335 is specific to the branching point of the glutamate side chain and thus recognizes mono- and all forms of polyglutamylated proteins (Wolff et al. 1992); B3 and PolyE antibody recognizes polyglutamylated side chains with a minimum size of two and three glutamate residues, respectively (Gagnon et al. 1996; Kann et al. 2003; van Dijk et al. 2007). The polyglutamylation reaction is initiated by the formation of an isopeptide bond with the γ-carboxyl group of the glutamate acceptor site, and followed by side chain elongation consists of the formation of regular peptide bonds (Janke et al. 2008). In cilia, glutamylation is abundant on the B-tubules of the outer-axoneme doublets (Kubo et al. 2010; Lechtreck and Geimer 2000; Orbach and Howard 2019; Suryavanshi et al. 2010; Wloga et al. 2017). Like other protein modifications, polyglutamylation could generate massive microtubule heterogeneity by varying the density of the modification, choice of the tubulin subunit or isotype, choice of specific glutamate acceptor sites within the tubulin tail, and the length of the added side chain, which licensing the elaborate orchestration of the microtubule-associated physiology.
Microtubule polyglutamylation is a reversible process coordinated by tubulin glutamylases and tubulin deglutamylases in a cooperative manner. Enzymes catalyzing glutamylation in mammals belong to the tubulin tyrosine ligase-like (TTLL) protein family, which is characterized by the conserved core TTL domain with ATPase activity. Each of glutamylases shows intrinsic reaction and substrate specificity (Ikegami et al. 2006; Janke et al. 2005; Regnard et al. 1998; van Dijk et al. 2007): TTLL4, 5 and 7 preferentially catalyze the initiating step, whereas TTLL1, 6, 11 and 13 show more reaction specificity on the elongation step. As to the substrate specificity, TTLL1, 5, 6, 11, and 13 modify mostly on α-tubulin, while TTLL4 and 7 show a preference toward β-tubulin. Structural studies of glutamylases suggest that their catalytic specificities are determined by binding of the enzymes to the entire microtubule lattice (Garnham et al. 2015; Natarajan et al. 2017). However, the tubulin subunit preference of glutamylases could be overwritten by saturation of particular enzymes under certain physiological conditions (van Dijk et al. 2007). Of note, TTLL1 is active only when complexed with other proteins, whereas the others act in an autonomous manner (Janke et al. 2005; van Dijk et al. 2007). The enzymes that catalyze deglutamylation (deglutamylases) belong to the cytosolic carboxypeptidase (CCP) family (Kimura et al. 2010; Rogowski et al. 2010). CCP1, 4 and 6 act as the long-chain deglutamylases which catalyze shortening of glutamate side chains. So far, CCP5 is the only deglutamylase and has been identified that it specifically removes the branching point glutamate. However, CCP5 can also hydrolyze C-terminal glutamate residues from linear peptide chains similar to other members of the CCP family (Berezniuk et al. 2013). Both TTLL glutamylases and CCP deglutamylases have been reported to catalyze non-tubulin proteins such as myosin light chain kinase and telokin (Rogowski et al. 2010).
TTLLs have been reported to localize to the basal body or the axoneme (He et al. 2018; Lee et al. 2012; Suryavanshi et al. 2010; van Dijk et al. 2007; Wloga et al. 2008). In mammalian cells, TTLL4, 5, 6 and 7 are detected in both basal bodies and cilia, whereas TTLL1, 9 and 11 specifically label basal bodies. Intriguingly, overexpressed TTLL5 or TTLL6 shows three distinct ciliary localization patterns: densely punctate labeling surrounding the ciliary base, or exclusive cilia localization, or both, suggesting the ciliary import of TTLL5/6 is a transient and dynamic process (He et al. 2018). Although considered as cytoplasmic glutamylases, evidences hint that TTLL5/6 are transported to the ciliary base via association with specific group of vesicles, which is regulated by the ARL13B-RAB11-FIP5-mediated trafficking pathway (He et al. 2018). Binding of polyglutamylases complex to MTs in cilia, centrioles and neurons require the adaptor PGs1, whose deficiency in the ROSA22 mouse significantly reduces microtubule polyglutamylation (Campbell et al. 2002; Ikegami et al. 2007; Janke et al. 2005; Regnard et al. 2003). In agreement with the gradient pattern of glutamylation modification in cilia, the ciliary level of TTLL5/6 gradually decreases form the proximal to distal end (He et al. 2018; Lee et al. 2012; van Dijk et al. 2007). For deglutamylases, ccp2, ccp5, and ccp6 are expressed in ciliated cells in zebrafish, whereas ccp1 expression is restricted to the nervous system (Pathak et al. 2014). Only ccp5 knockdown increases cilia tubulin glutamylation, suggesting that ccp5 is the principal tubulin deglutamylase that maintains functional levels of tubulin glutamylation in cilia (Pathak et al. 2014). In mammalian cells, CCP5 constitutively and evenly distributes along the whole axoneme (He et al. 2018). In consideration of that the careful balance of TTLLs and CCPs is critical for proper level of microtubule polyglutamylation, the difference in ciliary distribution of CCPs and TTLLs generates the gradually tilted deglutamylation/glutamylation balance toward cilia tip. Surprisingly, in hypoglutamylated axoneme that induced by deletion of TTLL5/6 or overexpression of CCP5, residual axonemal glutamylation is always well preserved at the proximal end of primary cilia corresponding the transition zone (He et al. 2018; Hong et al. 2018), suggesting either unidentified glutamylase/deglutamylases are responsible for TZ polyglutamylation, or this specific region is protected from deglutamylation via a distinct mechanism.
It was proposed that negative charged interfaces generated by polyglutamylation make the microtubule “sticky”, and therefore regulates the interaction of other proteins with microtubules (Mitchell 2010). Polyglutamylation regulates activity of microtubule-severing enzymes spastin and katanin (Dymek et al. 2004; Lacroix et al. 2010; Lu et al. 2004; Sharma et al. 2007; Shin et al. 2019; Valenstein and Roll-Mecak 2016). Interestingly, polyglutamylation biphasically regulates spastin mediated microtubule severing: the spastin microtubule-severing activity increases as the number of glutamates per tubulin rises from one to eight, but decreases beyond this glutamylation threshold (Valenstein and Roll-Mecak 2016). To this end, polyglutamylation may stabilize/assemble or destabilize/disassemble microtubules in different context. The polyglutamylated tubulin in centrioles, spindle, and cilia could be targeted and stabilized by the microtubule stabilizing factor centriole and spindle-associated protein (CASP), which is required for normal brain development and proper left–right asymmetry (Backer et al. 2012; Ohta et al. 2015). Reduced tubulin polyglutamylation suppresses flagellar shortness in Chlamydomonas (Kubo et al. 2015). In Tetrahymena, hyperglutamylation of tubulin can either stabilize or destabilize microtubules in the same cell (Wloga et al. 2010). Although it remains debating that whether CCPP-1 (CCP1 homolog) is a real deglutamylase in C. elegans, the cilia of ccpp-1 worms display a progressive degeneration (Kimura et al. 2010; O'Hagan et al. 2011). In drastic contrast, disruption of axonemal polyglutamylation in mammalian cilia does not affect cilia biogenesis, but only promotes disassembly induced by deciliation signals (He et al. 2018; Hong et al. 2018).
Tubulin polyglutamylation also regulates the activity of inner-arm dynein to control the beating behavior of motile cilia (Kubo et al. 2010; Suryavanshi et al. 2010). Hypoglutamylation induced by mutations in specific TTLLs has been shown to compromise cilia motility in different ciliated organisms (Grau et al. 2012, 2013; Ikegami et al. 2010; Kubo et al. 2010; Pathak et al. 2011; Suryavanshi et al. 2010). However, hyperglutamylation induced by knockdown of ccp5 in zebrafish or overexpressing Ttll6Ap in Tetrahymena also disrupts cilia motility (Janke et al. 2005; Pathak et al. 2014). Whether this discrepancy is caused by non-tubulin glutamylation needs to be further examined. In C. elegans cilia, Chlamydomonas flagella, and mammalian primary cilia, the velocity and processivity of kinesin motors of IFT particles can be promoted by tubulin polyglutamylation (Hong et al. 2018; Ikegami et al. 2007; Kimura et al. 2018; O'Hagan et al. 2011; Sirajuddin et al. 2014). Emerging evidences also highlight the essential role of axoneme polyglutamylation in controlling ciliary localization of signaling molecules (He et al. 2018; Hong et al. 2018). Polycystin 1 (PC1) and polycystin 2 (PC2), two major proteins mutated ADPKD, colocalize to the primary cilium and may form a receptor/channel complex to sense environmental cues (Lee and Somlo 2014; Patel and Honore 2010; Nauli et al. 2003; Torres and Harris 2006). Depletion of CCPP-1 causes excess PKD-2 accumulation both inside cilia and below ciliary base in C. elegans (O'Hagan et al. 2011). Consistently, in mammalian primary cilia, axoneme hypoglutamylation compromises the ciliary localization of PC2, which can be restored by concomitant depletion of CCP5 (He et al. 2018). This suggests axoneme polyglutamylation likely anchors polycystins on the ciliary surface. Since short chain polyglutamylation alone is sufficient for anchoring ciliary polycystin (He et al. 2018), it is very likely that unknown adaptors tether cytoplasmic tails of polycystins with glutamylated tubulins. In addition, transduction of hedgehog (Hh) signaling depends on proper ciliary targeting of signaling molecules such as GLI3 and SMO (Bangs and Anderson 2017). Axonemal hypoglutamylation impairs SAG-induced cilia tip translocation of GLI3 and represses downstream Hh signaling (He et al. 2018; Hong et al. 2018).
Remarkably, defective polyglutamylation is correlated with a variety of typical ciliopathy phenotypes, such as male infertility in mice (Campbell et al. 2002; Giordano et al. 2019; Konno et al. 2016; Mullen et al. 1976; Vogel et al. 2010; Wu et al. 2017), respiratory disorders in mice (Ikegami et al. 2010), dysfunctional ependymal cilia in the brain ventricles in mice ( Grau et al. 2013), and axis curvature, pronephric cysts, and abnormal otolith number in zebrafish (Pathak et al. 2011). Interestingly, mutations in the several human Joubert syndrome genes, including CEP41, ARL13B, ARMC9 and TOGARAM1, cause dramatic reduction in axonemal polyglutamylation (He et al. 2018; Latour et al. 2019; Lee et al. 2012), suggesting axoneme hypoglutamylation could be one of the key pathogenic mechanisms for Joubert syndrome.
Polyglutamylation is enriched during neuronal differentiation and is therefore considered a potential key regulator of neuronal microtubules (Audebert et al. 1994; Kapitein and Hoogenraad 2015). The extensive studies in Purkinje cell degeneration (pcd) mice that carrying Ccp1 inactivating mutation have directly linked tubulin hyperglutamylation to neurodegeneration (Fernandez-Gonzalez et al. 2002; Greer and Shepherd 1982; Mullen et al. 1976; Rogowski et al. 2010; Shashi et al. 2018). Interestingly, for the three CCPs (CCP2, 3, 5) that associated with cilia, Ccp2−/−(Tort et al. 2014), Ccp3−/−(Tort et al. 2014), and Ccp5−/−(Wu et al. 2017; Xia et al. 2016) mice are generally healthy without neural degeneration phenotype. This suggests that neuron degeneration observed in pcd mice is probably caused by upregulated polyglutamylation of axon microtubules but not the axoneme. Although Ccp5−/− mice with presumably hyperglutamylated cilia are generally healthy without ciliopathy phenotypes, hyperglutamylation was correlated with retinal degeneration in either human (Astuti et al. 2016; Branham et al. 2016; Kastner et al. 2015; Sergouniotis et al. 2014) or mouse models (Grau et al. 2017; Marchena et al. 2011). It is argued that the pathogenesis of hyperglutamylation-associated retinal degenerative diseases may be tubulin-independent. A photoreceptor specific ORF15 variant of retinitis pigmentosa GTPase regulator (RPGRORF15), the product of the major causal gene of retinal dystrophy, localizes to the connecting cilium and can be glutamylated by TTLL5 in vivo (Rao et al. 2016). TTLL5 mutations lead to complete loss of RPGR glutamylation and retinal pathology, without marked changes in tubulin glutamylation levels and defects in ultrastructure of microtubule doublets in connecting cilia (Lee et al. 2013; Sergouniotis et al. 2014; Sun et al. 2016).
GLYCYLATION
Tubulin glycylation was initially discovered on Paramecium tetraurelia tubulins that generate side chains of glycine on the γ-carboxyl groups of specific glutamate residues (Redeker et al. 1994), which is mostly enriched in the B-tubule of axoneme (Orbach and Howard 2019). Glycylation has been extensively studied by using specific antibodies that can detect glycylation modification in different length: with the antibody TAP952 detecting monoglycylation and AXO49 detecting chains with three or more glycine residues (polyglycylation) (Bre et al. 1996, 1998; Levilliers et al. 1995), and PolyG detects chains with four or more glycine residues (Tort et al. 2014; Xia et al. 2000). Glycylation modification occurs predominantly in cilia or flagella (Bre et al. 1996; Gadadhar et al. 2017b; Redeker et al. 1994; Ru et al. 1995; Weber et al. 1996; Xia et al. 2000), suggesting its unique roles in regulating cilia/flagella functions. One exception is that the cilia of Kupffer’s vesicle in zebrafish appear to be free of mono- or polyglycylation (Pathak et al. 2011). Whether this is caused by species specificity of anti-glycylation antibodies need to be further examined.
Tubulin glycylation is generated by a subset of enzymes that belong to the same protein family as that of glutamylation, the tubulin tyrosine ligase-like proteins (TTLLs) (Ikegami and Setou 2009; Rogowski et al. 2009). The enzymes catalyzing deglycylation process remain unidentified, though. In mammals, TTLL3 and TTLL8 catalyze the initiation whereas TTLL10 catalyzes the elongation steps of polyglycylation (Ikegami and Setou 2009; Rogowski et al. 2009). In Drosophila melanogaster, no TTLL10 gene has been found (Rogowski et al. 2009). The polyglycylation in Drosophila is generated by bifunctional initiating/elongating glycylases dmTTLL3A and dmTTLL3B (Rogowski et al. 2009). Interestingly, unlike other mammalian species, the axoneme of humans cells only carry monoglycylation, which is caused by a mutation in TTLL10 that inactive its elongating activity (Rogowski et al. 2009). This suggests that the function of microtubule glycylation might be sufficiently fulfilled by monoglycylation.
Tubulin glycylation has been linked to stability and maintenance of motile cilia (Grau et al. 2013; Pathak et al. 2011; Rogowski et al. 2009; Wloga et al. 2009). In Tetrahymena cells, deletion of TTLL3 leads to subtle defect in the tubulin turnover and results in slightly shorter cilia (Wloga et al. 2009). In zebrafish, loss of tubulin polyglycylation causes either shortening or loss of motile cilia in several organs (Wloga et al. 2009). In mammalian cilia, glycylation is redundantly generated by the enzymes TTLL3 and TTLL8 in most tissues. Absence or reduction of glycylation along the axoneme destabilizes motile ependymal cilia (Grau et al. 2013). The existence of glycylation in primary cilia was not confirmed until a novel glycylation antibody (gly-pep) was generated recently (Gadadhar et al. 2017b). The glycylation in primary cilium is unevenly distributed, prominent at the proximal part of the cilia but not confined to the transition zone. Deletion or overexpression of glycylases modulates the length of primary cilia in cultured mammalian cells (Gadadhar et al. 2017b).
In agreement with its importance in motile and primary cilia, loss of glycylation results in cilia-related anomalies. Deletion of in dmTTLL3B in Drosophila causes defects in sperm individualization and axoneme structure (Rogowski et al. 2009). In zebrafish, loss of glycylation contributes to randomization of multicilia orientation in embryos (Pathak et al. 2011). In mice, absence of glycylation leads to ciliary disassembly in ependymal cells (Grau et al. 2013) and shortening of the connecting cilium of photoreceptors and affecting retinal degeneration (Grau et al. 2017). Surprisingly, cilia glycylation is also associated with the development of colorectal cancer. TTLL3 is the only glycylase in colon. Suppression of TTLL3 leads to reduced number of primary cilia and strongly enhanced colon carcinogenesis (Rocha et al. 2014), suggesting the correlation between primary cilia and cell cycle progression.
It is worth to noting that, glycylation and glutamylation occur within the same cluster of glutamates, indicative of a reciprocal or competition pathway of these two PTMs may exist. It is consistent with the observations that loss of tubulin glutamylation or glycylation alone shows mild defects in ultrastructural axonemal structure, while combined loss of glycylation and glutamylation causes near complete loss of cilia motility and induces a variety of dramatic axonemal ultrastructural defects in zebrafish (Pathak et al. 2011). Similarly, Tetrahymena mutants with glutamates on the C-terminal tail of β-tubulin mutated to abolish both glycylation and glutamylation are lethal or possess severe axonemal defects (Redeker et al. 2005; Thazhath et al. 2004; Xia et al. 2000). Loss of glycylation in Tetrahymena, Drosophila and mouse is accompanied by tubulin hyperglutamylation (Gadadhar et al. 2017b; Rogowski et al. 2009; Wloga et al. 2009). Thus, tubulin glycylation and glutamylation are likely regulated together and may coordinate cilia formation and/or function in certain contexts.
DETYROSINATION/∆2-TUBULIN
Encoded in α-tubulin genes, the carboxyl-terminal residue of most nascent α-tubulin has a tyrosine which is removed by detyrosination and re-added by tyrosination (Arce and Barra 1983; Kumar and Flavin 1981). In cilia, detyrosinated tubulins are enriched on the B-tubules of outer doublets (Johnson 1998; Orbach and Howard 2019). Tubulin tyrosine ligase (TTL), the first identified tubulin-modifying enzyme, catalyzes the tyrosination modification (Ersfeld et al. 1993; Murofushi 1980; Prota et al. 2013; Schroder 1985; Szyk et al. 2011). TTL works exclusively and efficiently on detyrosinated tubulin heterodimers and thus the newly assembled microtubules are mostly tyrosinated. The enzymes catalyzing detyrosination have just been identified recently (Aillaud et al. 2017; Nieuwenhuis et al. 2017). When complexed with small vasohibin binding protein (SVBP), vasohibins (VASH1 and VASH2) exhibit robust and specific Tyr/Phe carboxypeptidase activity on polymerized microtubules. Interestingly, pharmacological and genetic suppression of vasohibins didn’t abolish the detyrosinated α-tubulin, and the remaining detyrosinated pools were specifically concentrated in neuron axon, implicating the occurrence of other detyrosinases for detyrosinating axonal microtubules (Aillaud et al. 2017). Although it hasn’t been studied, axonemal microtubules in cilia may be modified by the same detyrosination machinery as mentioned. After detyrosination, the exposed glutamate residues on C-terminal of α-tubulin can further be removed by cytosolic carboxypeptidases (CCPs; CCP1, 2, 3, 4, 6), generating the ∆2- and ∆3-tubulin (Aillaud et al. 2016; Berezniuk et al. 2012; de la Vega et al. 2007; Kalinina et al. 2007; Kimura et al. 2010; Paturle-Lafanechere et al. 1991; Rogowski et al. 2010; Tort et al. 2014). ∆2-tubulin cannot undergo tyrosination and it accumulates in long-lived microtubules in cilia and axonal microtubules.
Abnormal detyrosinated/∆2-tubulin has been linked to defects in brain development. Ttl-knockout mice die perinatally due to massive defects in brain architecture, particularly in the cortico-thalamic loop, which is very likely due to the aberrant timing and extent of neurite outgrowth (Erck et al. 2005). Tubulin detyrosination is also implicated in muscle and cardiac functions. It accumulates at early steps of muscle cell differentiation and affects mechanotransduction (Kerr et al. 2015; Robison et al. 2016). Excess or diminished tubulin detyrosination changes the stiffness of the cardiomyocytes and may lead to cardiac dysfunction. Upregulated tubulin detyrosination was found in patients diagnosed with hypertrophic and dilated cardiomyopathies (Kerr et al. 2015; Robison et al. 2016). Detyrosination has also been detected on microtubules of the mitotic and meiotic spindles, midbody and centrioles, and implicated in controlling the precision of cell division. Detyrosination facilitates the binding of a kinetochore-associated motor protein CENP-E, helps guide all chromosomes toward the metaphase plate in mitosis, and absence of this tubulin modification leads to misaligned chromosomes (Barisic et al. 2015). In female meiosis, detyrosinated microtubules are asymmetrically enriched on one half of the meiotic spindle, which drives non-Mendelian chromosome transmission in mouse oocytes (Akera et al. 2017). In light of its essential role in cell division, deregulated detyrosination is linked to several types of cancer. Differential expression of TTL correlates with poor prognosis in neuroblastoma tumors (Kato et al. 2004), and detyrosination of tubulin was particularly prominent in aggressive subtypes of breast cancer (Mialhe et al. 2001). Moreover, vasohibin is associated with micro-vessel densities, histology grades, invasions, poor clinical features, metastasis, and dissemination in abdominal cavities, as well as EMT (Du et al. 2017). The function of detyrosination/∆2-tubulin in context of cilia remains largely unknown. The anterograde intraflagellar transport (IFT) is driven by kinesin-2 on the B-tubule of axonemal microtubule doublets (Kozminski et al. 1995; Pigino et al, 2009; Stepanek and Pigino 2016). It is worth noting that detyrosinated tubulins are strongly enriched on the B-tubules. In vitro, detyrosination promotes the motility of kinesin-2 (Sirajuddin et al. 2014), suggesting a potential correlation between tubulin detyrosination and selective transportation for anterograde IFT.
CONCLUSION AND PERSPECTIVE
Tubulin PTMs occur on the surface or inside the lumen of microtubules to change their mechanical properties or interactome, by which it fine-tunes the function and confers specialized identities for different microtubule structures. Emerging evidences unveil the physiological importance of tubulin PTMs in both motile and primary cilia. Functional studies in model organisms with mutations in PTMs modifying enzymes and the rapid advances in human genetics strongly suggest the causal role of dysfunctional tubulin PTMs in the pathogenesis of ciliopathies and a wide spectrum of human disorders. However, due to our limited knowledge of tissue specificity, function redundancy, subcellular distribution, and non-tubulin activities for microtubule PTMs modifying enzymes, precisely dissecting the role of tubulin PTMs in the context of cilia and ciliopathies remains challenging. To this end, new tools/reagents/systems are greatly needed to be developed to ensure precise and local manipulation of PTMs modifying enzymes in cilia. Also, as the PTMs modifying enzymes are accessible targets for drug development (Huq and Wei 2010), small-molecule activators/inhibitors of PTM enzymes are desired for basic research and, importantly, may represent promising therapeutic strategies for ciliopathies, neuron degeneration diseases, and other microtubule-associated human disorders.
References
Adams M, Smith UM, Logan CV, Johnson CA (2008) Recent advances in the molecular pathology, cell biology and genetics of ciliopathies. J Med Genet 45:257–267
Aillaud C, Bosc C, Peris L, Bosson A, Heemeryck P, Van Dijk J, Le Friec J, Boulan B, Vossier F, Sanman LE, Syed S, Amara N, Couté Y, Lafanechère L, Denarier E, Delphin C, Pelletier L, Humbert S, Bogyo M, Andrieux A, Rogowski K, Moutin M-J (2017) Vasohibins/SVBP are tubulin carboxypeptidases (TCPs) that regulate neuron differentiation. Science 358:1448–1452
Aillaud C, Bosc C, Saoudi Y, Denarier E, Peris L, Sago L, Taulet N, Cieren A, Tort O, Magiera MM, Janke C, Redeker V, Andrieux A, Moutin M-J (2016) Evidence for new C-terminally truncated variants of alpha- and beta-tubulins. Mol Biol Cell 27:640–653
Akella JS, Wloga D, Kim J, Starostina NG, Lyons-Abbott S, Morrissette NS, Dougan ST, Kipreos ET, Gaertig J (2010) MEC-17 is an alpha-tubulin acetyltransferase. Nature 467:218–222
Akera T, Chmátal L, Trimm E, Yang K, Aonbangkhen C, Chenoweth DM, Janke C, Schultz RM, Lampson MA (2017) Spindle asymmetry drives non-Mendelian chromosome segregation. Science 358:668–672
Alexander JE, Hunt DF, Lee MK, Shabanowitz J, Michel H, Berlin SC, MacDonald TL, Sundberg RJ, Rebhun LI, Frankfurter A (1991) Characterization of posttranslational modifications in neuron-specific class III beta-tubulin by mass spectrometry. Proc Natl Acad Sci USA 88:4685–4689
Alper JD, Decker F, Agana B, Howard J (2014) The motility of axonemal dynein is regulated by the tubulin code. Biophys J 107:2872–2880
Arce CA, Barra HS (1983) Association of tubulinyl-tyrosine carboxypeptidase with microtubules. FEBS Lett 157:75–78
Astuti GD, Arno G, Hull S, Pierrache L, Venselaar H, Carss K, Raymond FL, Collin RW, Faradz SM, van den Born LI, Webster AR, Cremers FPM (2016) Mutations in AGBL5, encoding alpha-tubulin deglutamylase, are associated with autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci 57:6180–6187
Audebert S, Koulakoff A, Berwald-Netter Y, Gros F, Denoulet P, Edde B (1994) Developmental regulation of polyglutamylated alpha- and beta-tubulin in mouse brain neurons. J Cell Sci 107(Pt 8):2313–2322
Backer CB, Gutzman JH, Pearson CG, Cheeseman IM (2012) CSAP localizes to polyglutamylated microtubules and promotes proper cilia function and zebrafish development. Mol Biol Cell 23:2122–2130
Badano JL, Mitsuma N, Beales PL, Katsanis N (2006) The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet 7:125–148
Bangs F, Anderson KV (2017) Primary cilia and mammalian hedgehog signaling. Cold Spring Harb Perspect Biol 9:a028175
Barisic M, e Sousa RS, Tripathy SK, Magiera MM, Zaytsev AV, Pereira AL, Janke C, Grishchuk EL, Maiato H (2015) Microtubule detyrosination guides chromosomes during mitosis. Science 348:799–803
Benoy V, Vanden Berghe P, Jarpe M, Van Damme P, Robberecht W, Van Den Bosch L (2017) Development of improved HDAC6 inhibitors as pharmacological therapy for xonal Charcot-Marie-Tooth disease. Neurotherapeutics 14:417–428
Berbari NF, O'Connor AK, Haycraft CJ, Yoder BK (2009) The primary cilium as a complex signaling center. Curr Biol 19:R526–535
Berbari NF, Sharma N, Malarkey EB, Pieczynski JN, Boddu R, Gaertig J, Guay-Woodford L, Yoder BK (2013) Microtubule modifications and stability are altered by cilia perturbation and in cystic kidney disease. Cytoskeleton (Hoboken) 70:24–31
Berezniuk I, Lyons PJ, Sironi JJ, Xiao H, Setou M, Angeletti RH, Ikegami K, Fricker LD (2013) Cytosolic carboxypeptidase 5 removes alpha- and gamma-linked glutamates from tubulin. J Biol Chem 288:30445–30453
Berezniuk I, Vu HT, Lyons PJ, Sironi JJ, Xiao H, Burd B, Setou M, Angeletti RH, Ikegami K, Fricker LD (2012) Cytosolic carboxypeptidase 1 is involved in processing alpha- and beta-tubulin. J Biol Chem 287:6503–6517
Bobinnec Y, Moudjou M, Fouquet JP, Desbruyeres E, Edde B, Bornens M (1998) Glutamylation of centriole and cytoplasmic tubulin in proliferating non-neuronal cells. Cell Motil Cytoskeleton 39:223–232
Branham K, Matsui H, Biswas P, Guru AA, Hicks M, Suk JJ, Li H, Jakubosky D, Long T, Telenti A, Nariai N, Heckenlively JR, Frazer KA, Sieving PA, Ayyagari R (2016) Establishing the involvement of the novel gene AGBL5 in retinitis pigmentosa by whole genome sequencing. Physiol Genomics 48:922–927
Bre MH, Redeker V, Quibell M, Darmanaden-Delorme J, Bressac C, Cosson J, Huitorel P, Schmitter JM, Rossler J, Johnson T, Adoutte A, Levilliers N (1996) Axonemal tubulin polyglycylation probed with two monoclonal antibodies: widespread evolutionary distribution, appearance during spermatozoan maturation and possible function in motility. J Cell Sci 109(Pt 4):727–738
Bre MH, Redeker V, Vinh J, Rossier J, Levilliers N (1998) Tubulin polyglycylation: differential posttranslational modification of dynamic cytoplasmic and stable axonemal microtubules in paramecium. Mol Biol Cell 9:2655–2665
Bulinski JC, Richards JE, Piperno G (1988) Posttranslational modifications of alpha tubulin: detyrosination and acetylation differentiate populations of interphase microtubules in cultured cells. J Cell Biol 106:1213–1220
Campbell PK, Waymire KG, Heier RL, Sharer C, Day DE, Reimann H, Jaje JM, Friedrich GA, Burmeister M, Bartness TJ, Russell LD, Young LJ, Zimmer M, Jenne DE, MacGregor GR (2002) Mutation of a novel gene results in abnormal development of spermatid flagella, loss of intermale aggression and reduced body fat in mice. Genetics 162:307–320
Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325:834–840
Chretien D, Metoz F, Verde F, Karsenti E, Wade RH (1992) Lattice-defects in microtubules - protofilament numbers vary within individual microtubules. J Cell Biol 117:1031–1040
Chu CW, Hou F, Zhang J, Phu L, Loktev AV, Kirkpatrick DS, Jackson PK, Zhao Y, Zou H (2011) A novel acetylation of beta-tubulin by San modulates microtubule polymerization via down-regulating tubulin incorporation. Mol Biol Cell 22:448–456
Coombes C, Yamamoto A, McClellan M, Reid TA, Plooster M, Luxton GW, Alper J, Howard J, Gardner MK (2016) Mechanism of microtubule lumen entry for the alpha-tubulin acetyltransferase enzyme alphaTAT1. Proc Natl Acad Sci USA 113:E7176–E7184
d'Ydewalle C, Krishnan J, Chiheb DM, Van Damme P, Irobi J, Kozikowski AP, Berghe PV, Timmerman V, Robberecht W, Van Den Bosch L (2011) HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1–induced Charcot-Marie-Tooth disease. Nat Med 17:968–974
Dafinger C, Liebau MC, Elsayed SM, Hellenbroich Y, Boltshauser E, Korenke GC, Fabretti F, Janecke AR, Ebermann I, Nurnberg G, Nürnberg P, Zentgraf H, Koerber F, Addicks K, Elsobky E, Benzing T, Schermer B, Bolz HJ (2011) Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J Clin Invest 121:2662–2667
Dan W, Gao N, Li L, Zhu JX, Diao L, Huang J, Han QJ, Wang S, Xue H, Wang Q, Wu Q-F, Zhang X, Bao L (2018) alpha-tubulin acetylation restricts axon overbranching by dampening microtubule plus-end dynamics in neurons. Cereb Cortex 28:3332–3346
de Diego AS, Alonso Guerrero A, Martínez-A C, van Wely KHM (2014) Dido3-dependent HDAC6 targeting controls cilium size. Nature Commun 5:1–11
de la Vega MR, Sevilla RG, Hermoso A, Lorenzo J, Tanco S, Diez A, Fricker LD, Bautista JM, Avilés FX (2007) Nna1-like proteins are active metallocarboxypeptidases of a new and diverse M14 subfamily. FASEB J 21:851–865
Diagouraga B, Grichine A, Fertin A, Wang J, Khochbin S, Sadoul K (2014) Motor-driven marginal band coiling promotes cell shape change during platelet activation. J Cell Biol 204:177–185
Dompierre JP, Godin JD, Charrin BC, Cordelieres FP, King SJ, Humbert S, Saudou F (2007) Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci 27:3571–3583
Drazic A, Myklebust LM, Ree R, Arnesen T (2016) The world of protein acetylation. Biochim Biophys Acta 1864:1372–1401
Du H, Zhao J, Hai L, Wu J, Yi H, Shi Y (2017) The roles of vasohibin and its family members: beyond angiogenesis modulators. Cancer Biol Ther 18:827–832
Dymek EE, Lefebvre PA, Smith EF (2004) PF15p is the chlamydomonas homologue of the Katanin p80 subunit and is required for assembly of flagellar central microtubules. Eukaryot Cell 3:870–879
Edde B, Rossier J, Le Caer JP, Desbruyeres E, Gros F, Denoulet P (1990) Posttranslational glutamylation of alpha-tubulin. Science 247:83–85
Erck C, Peris L, Andrieux A, Meissirel C, Gruber AD, Vernet M, Schweitzer A, Saoudi Y, Pointu H, Bosc C, Salin PA, Job D, Wehland J (2005) A vital role of tubulin-tyrosine-ligase for neuronal organization. Proc Natl Acad Sci USA 102:7853–7858
Ersfeld K, Wehland J, Plessmann U, Dodemont H, Gerke V, Weber K (1993) Characterization of the tubulin-tyrosine ligase. J Cell Biol 120:725–732
Fernandez-Gonzalez A, La Spada AR, Treadaway J, Higdon JC, Harris BS, Sidman RL, Morgan JI, Zuo J (2002) Purkinje cell degeneration (pcd) phenotypes caused by mutations in the axotomy-induced gene, Nna1. Science 295:1904–1906
Fouquet JP, Edde B, Kann ML, Wolff A, Desbruyeres E, Denoulet P (1994) Differential distribution of glutamylated tubulin during spermatogenesis in mammalian testis. Cell Motil Cytoskeleton 27:49–58
Gadadhar S, Bodakuntla S, Natarajan K, Janke C (2017a) The tubulin code at a glance. J Cell Sci 130:1347–1353
Gadadhar S, Dadi H, Bodakuntla S, Schnitzler A, Bieche I, Rusconi F, Janke C (2017b) Tubulin glycylation controls primary cilia length. J Cell Biol 216:2701–2713
Gagnon C, White D, Cosson J, Huitorel P, Edde B, Desbruyeres E, Paturle-Lafanechere L, Multigner L, Job D, Cibert C (1996) The polyglutamylated lateral chain of alpha-tubulin plays a key role in flagellar motility. J Cell Sci 109(Pt 6):1545–1553
Garnham CP, Vemu A, Wilson-Kubalek EM, Yu I, Szyk A, Lander GC, Milligan RA, Roll-Mecak A (2015) Multivalent microtubule recognition by tubulin tyrosine ligase-like family glutamylases. Cell 161:1112–1123
Giordano T, Gadadhar S, Bodakuntla S, Straub J, Leboucher S, Martinez G, Chemlali W, Bosc C, Andrieux A, Bieche I, Arnoult C, Geimer S, Janke C (2019) Loss of the deglutamylase CCP5 perturbs multiple steps of spermatogenesis and leads to male infertility. J Cell Sci 132:226951
Godena VK, Brookes-Hocking N, Moller A, Shaw G, Oswald M, Sancho RM, Miller CCJ, Whitworth AJ, De Vos KJ (2014) Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat Commun 5:1–11
Goetz SC, Anderson KV (2010) The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 11:331–344
Grau BM, Rochas C, Meunier A, Spassky N, Janke C (2012) Tubulin posttranslational modifications as regulators of microtubule-based functions in cells. Cilia 1:1
Grau BM, Gonzalez Curto G, Rocha C, Magiera MM, Marques Sousa P, Giordano T, Spassky N, Janke C (2013) Tubulin glycylases and glutamylases have distinct functions in stabilization and motility of ependymal cilia. J Cell Biol 202:441–451
Grau MB, Masson C, Gadadhar S, Rocha C, Tort O, Sousa PM, Vacher S, Bieche I, Janke C (2017) Alterations in the balance of tubulin glycylation and glutamylation in photoreceptors leads to retinal degeneration. J Cell Sci 130:938–949
Greer CA, Shepherd GM (1982) Mitral cell degeneration and sensory function in the neurological mutant mouse Purkinje cell degeneration (PCD). Brain Res 235:156–161
He K, Ma X, Xu T, Li Y, Hodge A, Zhang Q, Torline J, Huang Y, Zhao J, Ling K, Hu J (2018) Axoneme polyglutamylation regulated by Joubert syndrome protein ARL13B controls ciliary targeting of signaling molecules. Nat Commun 9:3310
Hong SR, Wang CL, Huang YS, Chang YC, Chang YC, Pusapati GV, Lin CY, Hsu N, Cheng HC, Chiang YC, Huang WE, Shaner NC, Rohatgi R, Inoue T, Lin YC (2018) Spatiotemporal manipulation of ciliary glutamylation reveals its roles in intraciliary trafficking and Hedgehog signaling. Nat Commun 9:1732
Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP (2002) HDAC6 is a microtubule-associated deacetylase. Nature 417:455–458
Huq MDM, Wei L-N (2010) Protein posttranslational modification: a potential target in pharmaceutical development. Pharm Biotechnol. https://doi.org/10.1002/9780470571224.pse302
Iancu-Rubin C, Gajzer D, Mosoyan G, Feller F, Mascarenhas J, Hoffman R (2012) Panobinostat (LBH589)-induced acetylation of tubulin impairs megakaryocyte maturation and platelet formation. Exp Hematol 40:564–574
Ichikawa M, Liu D, Kastritis PL, Basu K, Hsu TC, Yang S, Bui KH (2017) Subnanometre-resolution structure of the doublet microtubule reveals new classes of microtubule-associated proteins. Nat Commun 8:1–22
Ikegami K, Heier RL, Taruishi M, Takagi H, Mukai M, Shimma S, Taira S, Hatanaka K, Morone N, Yao I, Campbell PK, Yuasa S, Janke C, Macgregor GR, Setou M (2007) Loss of alpha-tubulin polyglutamylation in ROSA22 mice is associated with abnormal targeting of KIF1A and modulated synaptic function. Proc Natl Acad Sci USA 104:3213–3218
Ikegami K, Mukai M, Tsuchida J, Heier RL, Macgregor GR, Setou M (2006) TTLL7 is a mammalian beta-tubulin polyglutamylase required for growth of MAP2-positive neurites. J Biol Chem 281:30707–30716
Ikegami K, Sato S, Nakamura K, Ostrowski LE, Setou M (2010) Tubulin polyglutamylation is essential for airway ciliary function through the regulation of beating asymmetry. Proc Natl Acad Sci USA 107:10490–10495
Ikegami K, Setou M (2009) TTLL10 can perform tubulin glycylation when co-expressed with TTLL8. FEBS Lett 583:1957–1963
Janke C (2014) The tubulin code: molecular components, readout mechanisms, and functions. J Cell Biol 206:461–472
Janke C, Bulinski JC (2011) Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol 12:773–786
Janke C, Rogowski K, van Dijk J (2008) Polyglutamylation: a fine-regulator of protein function? 'Protein Modifications: beyond the usual suspects' review series. EMBO Rep 9:636–641
Janke C, Rogowski K, Wloga D, Regnard C, Kajava AV, Strub JM, Temurak N, van Dijk J, Boucher D, van Dorsselaer A, Suryavanshi S, Gaertig J, Eddé B (2005) Tubulin polyglutamylase enzymes are members of the TTL domain protein family. Science 308:1758–1762
Johnson GJ, Leis LA, Krumwiede MD, White JG (2007) The critical role of myosin IIA in platelet internal contraction. J Thromb Haemost 5:1516–1529
Johnson KA (1998) The axonemal microtubules of the Chlamydomonas flagellum differ in tubulin isoform content. J Cell Sci 111(Pt 3):313–320
Kalebic N, Martinez C, Perlas E, Hublitz P, Bilbao-Cortes D, Fiedorczuk K, Andolfo A, Heppenstall PA (2013a) Tubulin acetyltransferase αTAT1 destabilizes microtubules independently of its acetylation activity. Mol Cell Biol 33:1114–1123
Kalebic N, Sorrentino S, Perlas E, Bolasco G, Martinez C, Heppenstall PA (2013b) alphaTAT1 is the major alpha-tubulin acetyltransferase in mice. Nat Commun 4:1962
Kalinina E, Biswas R, Berezniuk I, Hermoso A, Aviles FX, Fricker LD (2007) A novel subfamily of mouse cytosolic carboxypeptidases. FASEB J 21:836–850
Kann ML, Soues S, Levilliers N, Fouquet JP (2003) Glutamylated tubulin: diversity of expression and distribution of isoforms. Cell Motil Cytoskeleton 55:14–25
Kapitein LC, Hoogenraad CC (2015) Building the neuronal microtubule cytoskeleton. Neuron 87:492–506
Kastner S, Thiemann IJ, Dekomien G, Petrasch-Parwez E, Schreiber S, Akkad DA, Gerding WM, Hoffjan S, Günes S, Günes S, Bagci H, Epplen JT (2015) Exome sequencing reveals AGBL5 as novel candidate gene and additional variants for retinitis pigmentosa in five Turkish families. Invest Ophthalmol Vis Sci 56:8045–8053
Kato C, Miyazaki K, Nakagawa A, Ohira M, Nakamura Y, Ozaki T, Imai T, Nakagawara A (2004) Low expression of human tubulin tyrosine ligase and suppressed tubulin tyrosination/detyrosination cycle are associated with impaired neuronal differentiation in neuroblastomas with poor prognosis. Int J Cancer 112:365–375
Kaul N, Soppina V, Verhey KJ (2014) Effects of alpha-tubulin K40 acetylation and detyrosination on kinesin-1 motility in a purified system. Biophys J 106:2636–2643
Kerr JP, Robison P, Shi G, Bogush AI, Kempema AM, Hexum JK, Becerra N, Harki DA, Martin SS, Raiteri R, Prosser BL, Ward CW (2015) Detyrosinated microtubules modulate mechanotransduction in heart and skeletal muscle. Nat Commun 6:8526
Kimura Y, Kurabe N, Ikegami K, Tsutsumi K, Konishi Y, Kaplan OI, Kunitomo H, Iino Y, Blacque OE, Setou M (2010) Identification of tubulin deglutamylase among Caenorhabditis elegans and mammalian cytosolic carboxypeptidases (CCPs). J Biol Chem 285:22936–22941
Kimura Y, Tsutsumi K, Konno A, Ikegami K, Hameed S, Kaneko T, Kaplan OI, Teramoto T, Fujiwara M, Ishihara T, Blacque OE, Setou M (2018) Environmental responsiveness of tubulin glutamylation in sensory cilia is regulated by the p38 MAPK pathway. Sci Rep 8(1):3892
Konno A, Ikegami K, Konishi Y, Yang HJ, Abe M, Yamazaki M, Sakimura K, Yao I, Shiba K, Inaba K, Setou M (2016) Ttll9-/- mice sperm flagella show shortening of doublet 7, reduction of doublet 5 polyglutamylation and a stall in beating. J Cell Sci 129:2757–2766
Kozminski KG, Beech PL, Rosenbaum JL (1995) The Chlamydomonas kinesin-like protein FLA10 is involved in motility associated with the flagellar membrane. J Cell Biol 131:1517–1527
Kubo T, Hirono M, Aikawa T, Kamiya R, Witman GB (2015) Reduced tubulin polyglutamylation suppresses flagellar shortness in Chlamydomonas. Mol Biol Cell 26:2810–2822
Kubo T, Yanagisawa HA, Yagi T, Hirono M, Kamiya R (2010) Tubulin polyglutamylation regulates axonemal motility by modulating activities of inner-arm dyneins. Curr Biol 20:441–445
Kumar N, Flavin M (1981) Preferential action of a brain detyrosinolating carboxypeptidase on polymerized tubulin. J Biol Chem 256:7678–7686
Lacroix B, van Dijk J, Gold ND, Guizetti J, Aldrian-Herrada G, Rogowski K, Gerlich DW, Janke C (2010) Tubulin polyglutamylation stimulates spastin-mediated microtubule severing. J Cell Biol 189:945–954
Latour BL, Van De Weghe JC, Rusterholz TDS, Letteboer SJF, Gomez A, Shaheen R, Gesemann M, Grout ME, van Reeuwijk J, Van Beersum SEC, Miller CV, Dempsey JC, Morsy H, Bamshad MJ, Nickerson DA, Neuhauss SCF, Boldt K, Ueffing M, Alkuraya FS, Bachmann-Gagescu R, Roepman R, Doherty D (2019) ARMC9 and TOGARAM1 define a Joubert syndrome-associated protein module that regulates axonemal post-translational modifications and cilium stability. bioRxiv. doi: 10.1101/817213
Lazo-Gomez R, Ramirez-Jarquin UN, Tovar YRLB, Tapia R (2013) Histone deacetylases and their role in motor neuron degeneration. Front Cell Neurosci 7:243
Lechtreck KF, Geimer S (2000) Distribution of polyglutamylated tubulin in the flagellar apparatus of green flagellates. Cell Motil Cytoskeleton 47:219–235
LeDizet M, Piperno G (1987) Identification of an acetylation site of Chlamydomonas alpha-tubulin. Proc Natl Acad Sci USA 84:5720–5724
Lee GS, He Y, Dougherty EJ, Jimenez-Movilla M, Avella M, Grullon S, Sharlin DS, Guo C, Blackford JA Jr, Awasthi S, Zhang Z, Armstrong SP, London EC, Chen W, Dean J, Simons SS Jr (2013) Disruption of Ttll5/stamp gene (tubulin tyrosine ligase-like protein 5/SRC-1 and TIF2-associated modulatory protein gene) in male mice causes sperm malformation and infertility. J Biol Chem 288:15167–15180
Lee JE, Silhavy JL, Zaki MS, Schroth J, Bielas SL, Marsh SE, Olvera J, Brancati F, Iannicelli M, Ikegami K, Schlossman AM, Merriman B, Attié-Bitach T, Logan CV, Glass IA, Cluckey A, Louie CM, Lee JH, Raynes HR, Rapin I, Castroviejo IP, Setou M, Barbot C, Boltshauser E, Nelson SF, Hildebrandt F, Johnson CA, Doherty DA, Valente EM, Gleeson JG (2012) CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat Genet 44:193–199
Lee SH, Somlo S (2014) Cyst growth, polycystins, and primary cilia in autosomal dominant polycystic kidney disease. Kidney Res Clin Pract 33:73–78
Levilliers N, Fleury A, Hill AM (1995) Monoclonal and polyclonal antibodies detect a new type of post-translational modification of axonemal tubulin. J Cell Sci 108(Pt 9):3013–3028
Lhernault SW, Rosenbaum JL (1983) Chlamydomonas alpha-tubulin Is posttranslationally modified in the Flagella during flagellar assembly. J Cell Biol 97:258–263
Lhernault SW, Rosenbaum JL (1985) Chlamydomonas alpha-tubulin is posttranslationally modified by acetylation on the epsilon-amino group of a lysine. Biochemistry-US 24:473–478
Li L, Wei D, Wang Q, Pan J, Liu R, Zhang X, Bao L (2012) MEC-17 deficiency leads to reduced -tubulin acetylation and impaired migration of cortical neurons. J Neurosci 32:12673–12683
Liu N, Xiong Y, Ren Y, Zhang L, He X, Wang X, Liu M, Li D, Shui W, Zhou J (2015) Proteomic profiling and functional characterization of multiple post-translational modifications of tubulin. J Proteome Res 14:3292–3304
Loktev AV, Zhang Q, Beck JS, Searby CC, Scheetz TE, Bazan JF, Slusarski DC, Sheffield VC, Jackson PK, Nachury MV (2008) A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev Cell 15:854–865
Lu C, Srayko M, Mains PE (2004) The Caenorhabditis elegans microtubule-severing complex MEI-1/MEI-2 katanin interacts differently with two superficially redundant beta-tubulin isotypes. Mol Biol Cell 15:142–150
Ly N, Elkhatib N, Bresteau E, Pietrement O, Khaled M, Magiera MM, Janke C, Le Cam E, Rutenberg AD, Montagnac G (2016) alphaTAT1 controls longitudinal spreading of acetylation marks from open microtubules extremities. Sci Rep 6:35624
Ma M, Stoyanova M, Rademacher G, Dutcher SK, Brown A, Zhang R (2019) Structure of the decorated ciliary doublet microtubule. Cell 179:909–922.e912
Magiera MM, Singh P, Gadadhar S, Janke C (2018a) Tubulin posttranslational modifications and emerging links to human disease. Cell 173:1323–1327
Magiera MM, Singh P, Janke C (2018b) SnapShot: functions of tubulin posttranslational modifications. Cell 173(1552–1552):e1551
Marchena M, Lara J, Aijon J, Germain F, de la Villa P, Velasco A (2011) The retina of the PCD/PCD mouse as a model of photoreceptor degeneration. A structural and functional study. Exp Eye Res 93:607–617
Maruta H, Greer K, Rosenbaum JL (1986) The acetylation of alpha-tubulin and its relationship to the assembly and disassembly of microtubules. J Cell Biol 103:571–579
Mary J, Redeker V, Le Caer JP, Rossier J, Schmitter JM (1996) Posttranslational modifications in the C-terminal tail of axonemal tubulin from sea urchin sperm. J Biol Chem 271:9928–9933
Matsuyama A, Shimazu T, Sumida Y, Saito A, Yoshimatsu Y, Seigneurin-Berny D, Osada H, Komatsu Y, Nishino N, Khochbin S, Horinouchi S, Yoshida M (2002) In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J 21:6820–6831
Mialhe A, Lafanechere L, Treilleux I, Peloux N, Dumontet C, Bremond A, Panh MH, Payan R, Wehland J, Margolis RL, Job D (2001) Tubulin detyrosination is a frequent occurrence in breast cancers of poor prognosis. Cancer Res 61:5024–5027
Mitchell DR (2010) Polyglutamylation: the GLU that makes microtubules sticky. Curr Biol 20:R234–236
Miyake Y, Keusch JJ, Wang L, Saito M, Hess D, Wang X, Melancon BJ, Helquist P, Gut H, Matthias P (2016) Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat Chem Biol 12:748–754
Morley SJ, Qi Y, Iovino L, Andolfi L, Guo D, Kalebic N, Castaldi L, Tischer C, Portulano C, Bolasco G, Shirlekar K, Fusco CM, Asaro A, Fermani F, Sundukova M, Matti U, Reymond L, Ninno AD, Businaro L, Johnsson K, Lazzarino M, Ries J, Schwab Y, Hu J, Heppenstall PA (2016) Acetylated tubulin is essential for touch sensation in mice. Elife 5:e20813. https://doi.org/10.7554/eLife.20813
Mullen RJ, Eicher EM, Sidman RL (1976) Purkinje cell degeneration, a new neurological mutation in the mouse. Proc Natl Acad Sci USA 73:208–212
Murofushi H (1980) Purification and characterization of tubulin-tyrosine ligase from porcine brain1. J Biochem 87:979–984
Narita T, Weinert BT, Choudhary C (2019) Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol 20:156–174
Natarajan K, Gadadhar S, Souphron J, Magiera MM, Janke C (2017) Molecular interactions between tubulin tails and glutamylases reveal determinants of glutamylation patterns. EMBO Rep 18:1013–1026
Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J (2003) Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33:129–137
Nieuwenhuis J, Adamopoulos A, Bleijerveld OB, Mazouzi A, Stickel E, Celie P, Altelaar M, Knipscheer P, Perrakis A, Blomen VA, Brummelkamp TR (2017) Vasohibins encode tubulin detyrosinating activity. Science 358:1453–1456
Nishimura Y, Kasahara K, Shiromizu T, Watanabe M, Inagaki M (2019) Primary cilia as signaling hubs in health and disease. Adv Sci 6:1801138
North BJ, Marshall BL, Borra MT, Denu JM, Verdin E (2003) The human Sir2 ortholog, SIRT2, is an NAD(+)-dependent tubulin deacetylase. Mol Cell 11:437–444
O'Hagan R, Piasecki BP, Silva M, Phirke P, Nguyen KC, Hall DH, Swoboda P, Barr MM (2011) The tubulin deglutamylase CCPP-1 regulates the function and stability of sensory cilia in C. elegans. Curr Biol 21:1685–1694
Ohta S, Hamada M, Sato N, Toramoto I (2015) Polyglutamylated tubulin binding protein C1orf96/CSAP is involved in microtubule stabilization in mitotic spindles. PLoS ONE 10:e0142798
Orbach R, Howard J (2019) The dynamic and structural properties of axonemal tubulins support the high length stability of cilia. Nat Commun 10:1838
Patel-Hett S, Richardson JL, Schulze H, Drabek K, Isaac NA, Hoffmeister K, Shivdasani RA, Bulinski JC, Galjart N, Hartwig JH, Italiano JE Jr (2008) Visualization of microtubule growth in living platelets reveals a dynamic marginal band with multiple microtubules. Blood 111:4605–4616
Patel A, Honore E (2010) Polycystins and renovascular mechanosensory transduction. Nat Rev Nephrol 6:530–538
Pathak N, Austin-Tse CA, Liu Y, Vasilyev A, Drummond IA (2014) Cytoplasmic carboxypeptidase 5 regulates tubulin glutamylation and zebrafish cilia formation and function. Mol Biol Cell 25:1836–1844
Pathak N, Austin CA, Drummond IA (2011) Tubulin tyrosine ligase-like genes ttll3 and ttll6 maintain zebrafish cilia structure and motility. J Biol Chem 286:11685–11695
Paturle-Lafanechere L, Edde B, Denoulet P, Van Dorsselaer A, Mazarguil H, Le Caer JP, Wehland J, Job D (1991) Characterization of a major brain tubulin variant which cannot be tyrosinated. Biochemistry-US 30:10523–10528
Pazour GJ, Rosenbaum JL (2002) Intraflagellar transport and cilia-dependent diseases. Trends Cell Biol 12:551–555
Pedersen LB, Rosenbaum JL (2008) Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol 85:23–61
Pigino G, Geimer S, Lanzavecchia S, Paccagnini E, Cantele F, Diener DR, Rosenbaum JL, Lupetti P (2009) Electron-tomographic analysis of intraflagellar transport particle trains in situ. J Cell Biol 187:135–148
Piperno G, LeDizet M, Chang XJ (1987) Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J Cell Biol 104:289–302
Portran D, Schaedel L, Xu Z, Théry M, Nachury MV (2017) Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat Cell Biol 19:391–398
Praetorius HA, Spring KR (2001) Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol 184:71–79
Praetorius HA, Spring KR (2003a) Removal of the MDCK cell primary cilium abolishes flow sensing. J Membr Biol 191:69–76
Praetorius HA, Spring KR (2003b) The renal cell primary cilium functions as a flow sensor. Curr Opin Nephrol Hypertens 12:517–520
Prota AE, Magiera MM, Kuijpers M, Bargsten K, Frey D, Wieser M, Jaussi R, Hoogenraad CC, Kammerer RA, Janke C, Steinmetz MO (2013) Structural basis of tubulin tyrosination by tubulin tyrosine ligase. J Cell Biol 200:259–270
Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA (2007) HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 129:1351–1363
Ran J, Yang Y, Li D, Liu M, Zhou J (2015) Deacetylation of alpha-tubulin and cortactin is required for HDAC6 to trigger ciliary disassembly. Sci Rep 5:12917
Rao KN, Anand M, Khanna H (2016) The carboxyl terminal mutational hotspot of the ciliary disease protein RPGRORF15 (retinitis pigmentosa GTPase regulator) is glutamylated in vivo. Biol Open 5:424–428
Redeker V, Levilliers N, Schmitter J, Le Caer J, Rossier J, Adoutte A, Bre M (1994) Polyglycylation of tubulin: a posttranslational modification in axonemal microtubules. Science 266:1688–1691
Redeker V, Levilliers N, Vinolo E, Rossier J, Jaillard D, Burnette D, Gaertig J, Bre MH (2005) Mutations of tubulin glycylation sites reveal cross-talk between the C termini of alpha- and beta-tubulin and affect the ciliary matrix in Tetrahymena. J Biol Chem 280:596–606
Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig J, Verhey KJ (2006) Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol 16:2166–2172
Regnard C, Audebert S, Desbruyères É, Denoulet P, Eddé B (1998) Tubulin polyglutamylase: partial purification and enzymatic properties. Biochemistry-US 37:8395–8404
Regnard C, Desbruyeres E, Denoulet P, Edde B (1999) Tubulin polyglutamylase: isozymic variants and regulation during the cell cycle in HeLa cells. J Cell Sci 112(Pt 23):4281–4289
Regnard C, Desbruyeres E, Huet JC, Beauvallet C, Pernollet JC, Edde B (2000) Polyglutamylation of nucleosome assembly proteins. J Biol Chem 275:15969–15976
Regnard C, Fesquet D, Janke C, Boucher D, Desbruyeres E, Koulakoff A, Insina C, Travo P, Edde B (2003) Characterisation of PGs1, a subunit of a protein complex co-purifying with tubulin polyglutamylase. J Cell Sci 116:4181–4190
Robison P, Caporizzo MA, Ahmadzadeh H, Bogush AI, Chen CY, Margulies KB, Shenoy VB, Prosser BL (2016) Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science 352:0659
Rocha C, Papon L, Cacheux W, Marques Sousa P, Lascano V, Tort O, Giordano T, Vacher S, Lemmers B, Mariani P, Meseure D, Medema JP, Bièche I, Hahne M, Janke C (2014) Tubulin glycylases are required for primary cilia, control of cell proliferation and tumor development in colon. EMBO J 33:2247–2260
Rogowski K, Juge F, van Dijk J, Wloga D, Strub JM, Levilliers N, Thomas D, Bre MH, Van Dorsselaer A, Gaertig J, Janke C (2009) Evolutionary divergence of enzymatic mechanisms for posttranslational polyglycylation. Cell 137:1076–1087
Rogowski K, van Dijk J, Magiera MM, Bosc C, Deloulme JC, Bosson A, Peris L, Gold ND, Lacroix B, Grau MB, Bec N, Larroque C, Desagher S, Holzer M, Andrieux A, Moutin M-J, Janke C (2010) A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell 143:564–578
Rosenbaum JL, Witman GB (2002) Intraflagellar transport. Nat Rev Mol Cell Biol 3:813–825
Rüdiger M, Plessman U, Klöppel K-D, Wehland J, Weber K (1992) Class II tubulin, the major brain β tubulin isotype is polyglutamylated on glutamic acid residue 435. FEBS Lett 308:101–105
Ru M, Plessmann U, Ru AH, Weber K (1995) β tubulin of bull sperm is polyglycylated. FEBS Lett 364:147–151
Sadoul K, Wang J, Diagouraga B, Vitte AL, Buchou T, Rossini T, Polack B, Xi X, Matthias P, Khochbin S (2012) HDAC6 controls the kinetics of platelet activation. Blood 120:4215–4218
Schaedel L, John K, Gaillard J, Nachury MV, Blanchoin L, Thery M (2015) Microtubules self-repair in response to mechanical stress. Nat Mater 14:1156–1163
Scholey JM (2008) Intraflagellar transport motors in cilia: moving along the cell's antenna. J Cell Biol 180:23–29
Scholey JM, Anderson KV (2006) Intraflagellar transport and cilium-based signaling. Cell 125:439–442
Schroder HC (1985) Purification of brain tubulin-tyrosine ligase by biochemical and immunological methods. J Cell Biol 100:276–281
Sergouniotis PI, Chakarova C, Murphy C, Becker M, Lenassi E, Arno G, Lek M, MacArthur DG, Bhattacharya SS, Moore AT, Holder GE, Robson AG, Wolfrum U, Webster AR, Plagnol V (2014) Biallelic variants in TTLL5, encoding a tubulin glutamylase, cause retinal dystrophy. Am J Hum Genet 94:760–769
Sharma N, Bryant J, Wloga D, Donaldson R, Davis RC, Jerka-Dziadosz M, Gaertig J (2007) Katanin regulates dynamics of microtubules and biogenesis of motile cilia. J Cell Biol 178:1065–1079
Shashi V, Magiera MM, Klein D, Zaki M, Schoch K, Rudnik-Schöneborn S, Norman A, Lopes Abath Neto O, Dusl M, Yuan X, Bartesaghi L, De Marco P, Alfares AA, Marom R, Arold ST, Guzmán-Vega FJ, Pena LD, Smith EC, Steinlin M, Babiker MO, Mohassel P, Foley AR, Donkervoort S, Kaur R, Ghosh PS, Stanley V, Musaev D, Nava C, Mignot C, Keren B, Scala M, Tassano E, Picco P, Doneda P, Fiorillo C, Issa MY, Alassiri A, Alahmad A, Gerard A, Liu P, Yang Y, Ertl-Wagner B, Kranz PG, Wentzensen IM, Stucka R, Stong N, Allen AS, Goldstein DB, Schoser B, Rösler KM, Alfadhel M, Capra V, Chrast R, Strom TM, Kamsteeg E-J, Bönnemann CG, Gleeson JG, Martini R, Janke C, Senderek J (2018) Loss of tubulin deglutamylase CCP1 causes infantile-onset neurodegeneration. EMBO J 37(23):e100540. https://doi.org/10.15252/embj.2018100540
Shida T, Cueva JG, Xu Z, Goodman MB, Nachury MV (2010) The major alpha-tubulin K40 acetyltransferase alphaTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc Natl Acad Sci USA 107:21517–21522
Shin SC, Im SK, Jang EH, Jin KS, Hur EM, Kim EE (2019) Structural and molecular basis for katanin-mediated severing of glutamylated microtubules. Cell Rep 26(1357–1367):e1355
Sirajuddin M, Rice LM, Vale RD (2014) Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat Cell Biol 16:335–344
Song Y, Brady ST (2015) Post-translational modifications of tubulin: pathways to functional diversity of microtubules. Trends Cell Biol 25:125–136
Soppina V, Herbstman JF, Skiniotis G, Verhey KJ (2012) Luminal localization of alpha-tubulin K40 acetylation by cryo-EM analysis of fab-labeled microtubules. PLoS ONE 7:e48204
Stepanek L, Pigino G (2016) Microtubule doublets are double-track railways for intraflagellar transport trains. Science 352:721–724
Sun X, Park JH, Gumerson J, Wu Z, Swaroop A, Qian H, Roll-Mecak A, Li T (2016) Loss of RPGR glutamylation underlies the pathogenic mechanism of retinal dystrophy caused by TTLL5 mutations. Proc Natl Acad Sci USA 113:E2925–E2934
Suryavanshi S, Edde B, Fox LA, Guerrero S, Hard R, Hennessey T, Kabi A, Malison D, Pennock D, Sale WS, Wloga D, Gaertig J (2010) Tubulin glutamylation regulates ciliary motility by altering inner dynein arm activity. Curr Biol 20:435–440
Szyk A, Deaconescu AM, Piszczek G, Roll-Mecak A (2011) Tubulin tyrosine ligase structure reveals adaptation of an ancient fold to bind and modify tubulin. Nat Struct Mol Biol 18:1250–1258
Szyk A, Deaconescu AM, Spector J, Goodman B, Valenstein ML, Ziolkowska NE, Kormendi V, Grigorieff N, Roll-Mecak A (2014) Molecular basis for age-dependent microtubule acetylation by tubulin acetyltransferase. Cell 157:1405–1415
Thazhath R, Jerka-Dziadosz M, Duan J, Wloga D, Gorovsky MA, Frankel J, Gaertig J (2004) Cell context-specific effects of the beta-tubulin glycylation domain on assembly and size of microtubular organelles. Mol Biol Cell 15:4136–4147
Torres VE, Harris PC (2006) Mechanisms of disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol 2:40–55
Tort O, Tanco S, Rocha C, Bieche I, Seixas C, Bosc C, Andrieux A, Moutin MJ, Aviles FX, Lorenzo J, Janke C (2014) The cytosolic carboxypeptidases CCP2 and CCP3 catalyze posttranslational removal of acidic amino acids. Mol Biol Cell 25:3017–3027
Valenstein ML, Roll-Mecak A (2016) Graded control of microtubule severing by tubulin glutamylation. Cell 164:911–921
van Dijk J, Miro J, Strub JM, Lacroix B, van Dorsselaer A, Edde B, Janke C (2008) Polyglutamylation is a post-translational modification with a broad range of substrates. J Biol Chem 283:3915–3922
van Dijk J, Rogowski K, Miro J, Lacroix B, Edde B, Janke C (2007) A targeted multienzyme mechanism for selective microtubule polyglutamylation. Mol Cell 26:437–448
Vogel P, Hansen G, Fontenot G, Read R (2010) Tubulin tyrosine ligase-like 1 deficiency results in chronic rhinosinusitis and abnormal development of spermatid flagella in mice. Vet Pathol 47:703–712
Walter WJ, Beranek V, Fischermeier E, Diez S (2012) Tubulin acetylation alone does not affect kinesin-1 velocity and run length in vitro. PLoS ONE 7:e42218
Weber K, Schneider A, Muller N, Plessmann U (1996) Polyglycylation of tubulin in the diplomonad Giardia lamblia, one of the oldest eukaryotes. FEBS Lett 393:27–30
White JG, Rao GH (1998) Microtubule coils versus the surface membrane cytoskeleton in maintenance and restoration of platelet discoid shape. Am J Pathol 152:597–609
Wloga D, Dave D, Meagley J, Rogowski K, Jerka-Dziadosz M, Gaertig J (2010) Hyperglutamylation of tubulin can either stabilize or destabilize microtubules in the same cell. Eukaryot Cell 9:184–193
Wloga D, Joachimiak E, Louka P, Gaertig J (2017) Posttranslational modifications of tubulin and cilia. Cold Spring Harb Perspect Biol 9:a028159
Wloga D, Rogowski K, Sharma N, Van Dijk J, Janke C, Edde B, Bre MH, Levilliers N, Redeker V, Duan J, Gorovsky MA, Jerka-Dziadosz M, Gaertig J (2008) Glutamylation on alpha-tubulin is not essential but affects the assembly and functions of a subset of microtubules in Tetrahymena thermophila. Eukaryot Cell 7:1362–1372
Wloga D, Webster DM, Rogowski K, Bre MH, Levilliers N, Jerka-Dziadosz M, Janke C, Dougan ST, Gaertig J (2009) TTLL3 is a tubulin glycine ligase that regulates the assembly of cilia. Dev Cell 16:867–876
Wolff A, Denechaud B, Chillet D, Mazarguil H, Desbruyeres E, Audebert S, Edde B, Gros F, Denoulet P (1992) Distribution of glutamylated alpha-tubulin and beta-tubulin in mouse-tissues using a specific monoclonal-antibody, Gt335. Eur J Cell Biol 59:425–432
Wu HY, Wei P, Morgan JI (2017) Role of cytosolic carboxypeptidase 5 in neuronal survival and spermatogenesis. Sci Rep 7:41428
Xia L, Hai B, Gao Y, Burnette D, Thazhath R, Duan J, Bré M-H, Levilliers N, Gorovsky MA, Gaertig J (2000) Polyglycylation of tubulin is essential and affects cell motility and division in tetrahymena thermophila. J Cell Biol 149:1097–1106
Xia P, Ye B, Wang S, Zhu X, Du Y, Xiong Z, Tian Y, Fan Z (2016) Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat Immunol 17:369–378
Xu Z, Schaedel L, Portran D, Aguilar A, Gaillard J, Marinkovich MP, Thery M, Nachury MV (2017) Microtubules acquire resistance from mechanical breakage through intralumenal acetylation. Science 356:328–332
Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M, Cao C, Li N, Cheng HL, Chua K, Lombard D, Mizeracki A, Matthias G, Alt FW, Khochbin S, Matthias P (2008) Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol 28:1688–1701
Zhao Z, Xu H, Gong W (2010) Histone deacetylase 6 (HDAC6) is an independent deacetylase for alpha-tubulin. Protein Peptide Lett 17:555–558
Zhou X, Fan LX, Li K, Ramchandran R, Calvet JP, Li X (2014) SIRT2 regulates ciliogenesis and contributes to abnormal centrosome amplification caused by loss of polycystin-1. Hum Mol Genet 23:1644–1655
Zilberman Y, Ballestrem C, Carramusa L, Mazitschek R, Khochbin S, Bershadsky A (2009) Regulation of microtubule dynamics by inhibition of the tubulin deacetylase HDAC6. J Cell Sci 122:3531–3541
Acknowledgements
We thank our many colleagues who have contributed to the work described herein, and apologize to those whose work we could not cite due to the space issue. We are grateful for funding from the National Institutes of Health (R01DK090038, 1R01DK099160), and P30 center grant (P30DK90728).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Kai He, Kun Ling, and Jinghua Hu declare that they have no conflict of interest.
Human and animal rights and informed consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, K., Ling, K. & Hu, J. The emerging role of tubulin posttranslational modifications in cilia and ciliopathies. Biophys Rep 6, 89–104 (2020). https://doi.org/10.1007/s41048-020-00111-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41048-020-00111-0