
Abstract
Introduction
Ewing sarcoma family of tumour represents a group of bone and soft-tissue tumours with variable neuroectodermal differentiation. Extraosseous Ewing sarcoma (EES) commonly arises in soft tissues of trunk or extremities with a few reported rare sites include larynx, nasal cavity, neck, lung, retroperitoneum, mediastinum and genital tract. Till now 28 cases of extraosseous Ewing sarcoma of vulva and vagina have been reported in the literature.
Case Report
We report 29th case of Ewing sarcoma vulva in sixth youngest, 16-year-old patient with third largest size tumour. Her all routine blood reports were found to be normal, and MRI showed highly vascular vulval mass with terminal urethral involvement. Tru-cut biopsy revealed uniform sheets of small round cells with geographical necrosis and perivascular clusters of tumour cells. Immunohistochemical staining demonstrated that the cells were focally positive for CK and vimentin and showed membranous positivity for CD99 and nuclear positivity for FLI1, whereas all other IHC markers to rule out differentials of small round cell tumour were negative. Based on HPE and IHC, which is polyimmunophenotypic, has confirmed the diagnosis of an ES/PNET. After ruling out for the metastatic lesions, 14 cycles of chemotherapy with compressed VDC/IE were planned, but after receiving her 1st cycle itself, she expired on 10th day of post-chemotherapy.
Conclusion
EES is a rare fast growing aggressive tumour requiring ancillary techniques for exact diagnosis and multimodality treatment for better survival.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ewing sarcoma family of tumour (ESFT) represents a group of bone and soft-tissue tumours with variable neuroectodermal differentiation where Ewing sarcoma (ES) is typically undifferentiated, and primitive neuroectodermal tumour (PNET) shows a clear evidence of neural differentiation. Though they are aggressive round cell tumour, they are highly chemosensitive. Hence, their identification by pathologist is crucial with the help of immunohistochemistry (IHC) and molecular ancillary techniques.
To our knowledge, very few cases of extraosseous Ewing sarcoma (EES)/PNET have previously been reported in the English literature and they behave not in a different way from skeletal ESFT. We present a case of primary Ewing sarcoma of vulva of an adolescent.
Case Report
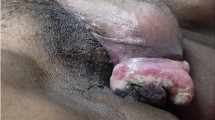
A 16-year-old adolescent girl came to our OPD with a rapidly growing vulval mass for the last 1 month with severe pain. On examination a 15 × 10 cm vulval mass seen on the left side with no ulceration and intact skin. On investigations all her blood reports were found to be normal, and MRI showed highly vascular vulval mass with terminal urethral involvement. FNAC was inconclusive. She was then taken up for tissue biopsy. Histopathological examination showed uniform sheets of small round cells with scanty, indistinct cytoplasmic outline, round nuclei with fine chromatin and inconspicuous nucleoli. Geographical necrosis and perivascular clusters of tumour cells are seen. A diagnosis of round cell tumour of vulva was offered, and IHC was recommended. Immunohistochemical staining demonstrated that the cells were focally positive for CK and vimentin and showed membranous positivity for CD99 and nuclear positivity for FLI1, while negative for terminal deoxyribonucleotidyl transferase (TdT), myogenin, desmin and CD34. This combination of HPE and IHC which is polyimmunophenotypic has confirmed the diagnosis of an ES/PNET (Figs. 1, 2, 3).

Sixteen-year-old girl presented with huge vulval mass

H&E, 40X, sheets of small blue round cell tumour

IHC, 40X, nuclear positivity of FLI1
The patient was referred for treatment to a paediatric oncologist. CT and MRI studies were negative for metastasis. A bone marrow biopsy was performed that was negative for ES. Fourteen cycles of chemotherapy with compressed VDC/IE (vincristine, doxorubicin, cyclophosphamide/ifosfamide and etoposide) were planned, but after receiving her 1st cycle, she expired on 10th day of post-chemotherapy.
Discussion
Ewing sarcoma is the second most common bone and soft-tissue (15%) sarcoma in children. Extraosseous Ewing sarcoma (EES) commonly arises in soft tissues of trunk or extremities, especially chest wall and paravertebral region, but a few reported rare sites include larynx, nasal cavity, neck, lung, retroperitoneum, perineum and mediastinum [1–3].
The first case series of round cell neoplasms mimicing Ewing sarcoma of bone was documented by Tefft et al. (1969) in paravertebral region, but later in 1975 Angervall and Enzinger termed these type of tumour as extraskeletal Ewing sarcoma (EES) [4].
EES/PNET of the female genital tract (FGT) is very unusual, and reported most common sites in FGT were the ovaries [5] followed by the uterine body [6] and less commonly the cervix [7], vagina and vulva [8, 9].
After thorough literature search, we could identify 28 cases of extraosseous Ewing sarcoma of vulva and vagina with molecular diagnosis in seven to eight cases [10].
The median age of the reported cases was 27 year with an age range from 10 to 52 years. The average size of the tumour was 5.8 cm (SD 5.03 cm). Eleven cases presented with metastatic disease and had poor survival.
The present case was supposed to be the third largest in size, in sixth youngest patient who was clinically diagnosed as rhabdomyosarcoma. The swelling was painful and fast growing like most of the reported cases.
The differential diagnosis of these tumours includes other small round blue cell tumours such as rhabdomyosarcoma, neuroblastoma (in children), lymphoma, small cell carcinoma (primary or metastatic), melanoma, cutaneous adnexal tumours and Merkel cell carcinoma.
The exact diagnosis of EES relies on histopathology along with ancillary techniques like immunohistochemistry and cytogenetic analysis, reverse transcriptase polymerase chain reaction and fluorescence in situ hybridisation.
Immunohistochemical demonstration of CD99/MIC 2, which is a cell membrane glycoprotein and intranuclear FLI1, was indicative of ES/PNET.
While negative desmin and MyoD1 ruled out a rhabdomyosarcoma, negative epithelial and neuroendocrine marker rules out possibility of small cell carcinoma and merkel cell carcinoma, and lacking of LCA positivity rules out lymphoma.
Demonstration of t(11;22)(q24;q12) chromosomal translocation (EWS-FLI1 gene rearrangement) is highly specific for ES/PNET since it is seen in more than 90% of the tumours.
It would be worth mentioning about the finding of Xiao et al. [9] that serum CA125 may be an important marker for prognosis and follow-up of PNET of the female genital tract as amongst their 11 cases, the CA125 levels of 8 patients were elevated before treatment and fell markedly to the normal when the disease was controlled although the level does not correlate with the severity of the disease.
Literature suggested that EES had more aggressive behaviour and a worse prognosis than the bony counterpart, with reported five-year survival rate for skeletal Ewing sarcoma around 75% as compared to 38% in the extraskeletal counterpart. But there were available literature reporting relatively favourable outcome for extraosseous ES/PNET arising in cutaneous and other superficial site. Hence, prognosis basically depends on tumour size, superficial location, early detection, metastasis and complete surgical removal of the lesion [1].
Since the present case had very large mass with involvement of urethra, surgery was not possible, and after post-chemotherapy, patient died of tumour lysis syndrome although there was no obvious metastasis at the time of presentation.
Being so aggressive in nature, these tumours would require multimodal treatment strategies. Current treatment modality for PNET includes surgical resection whenever feasible and adjuvant chemotherapy, which was thought to play an important role in the management [9]. Role of radiotherapy is not known for the reported cases at these sites.
Conclusion
EES is a rare fast growing aggressive tumour affecting young females. Exact diagnosis requires ancillary techniques such as IHC and molecular cytogenetics along with routine H&E. Early diagnosis and multimodality treatment are necessary along with follow-up for survival benefit. CA-125 may play a role in prognosis and follow-up, for that more and more studies are required.
References
Fong EY, Terrada DL, Zhai QJ. Primary Ewing sarcoma/peripheral primitive neuroectodermal tumor of the vulva. Hum Pathol. 2008;39:1535–9.
Khoury JD. Ewing sarcoma family of tumors. Adv Anat Pathol. 2005;4:212.
Weiss SW, Goldblum JR. Primitive neuroectodermal tumors and related lesions. In: Weiss SW, Goldblum JR, editors. Enzinger and Weiss’s soft tissue tumors. Chap 32. 4th ed. Mosby; 2001. p. 1289–308.
Vang R, Taubenberger JK, et al. Primary vulvar and vaginal extraosseous Ewing’s sarcoma/peripheral neuroectodermal tumor: diagnostic confirmation with CD99 immunostaining and reverse transcriptase-polymerase chain reaction. Int J Gynecol Pathol. 2000;19:103–9.
Kleinman GM, Young RH, Scully RE. Primary neuroectodermal tumors of the ovary. A report of 25 cases. Am J Surg Pathol. 1993;17:764–78.
Snijders-Keilholz A, Ewing P, Seynaeve C, et al. Primitive neuroectodermal tumor of the cervix uteri: a case report: changing concepts in therapy. Gynecol Oncol. 2005;98:516–9.
Li B, Ouyang L, Han X, et al. Primary primitive neuroectodermal tumor of the cervix. Onco Targets Ther. 2013;6:707–11.
Yang J, Guo Q, Yang Y, et al. Primary vulvar Ewing sarcoma/primitive neuroectodermal tumor: a report of one case and review of the literature. J Pediatr Adolesc Gynecol. 2012;25:e93–7.
Xiao C, Zhao J, Guo P, Wang D, Zhao D, et al. Clinical analysis of primary primitive neuroectodermal tumors in the female genital tract. Int J Gynecol Cancer. 2014;24:3.
Tunitsky-Bitton E, et al. Primary Ewing sarcoma presenting as a vulvar mass in an adolescent: case report and review of literature. J Pediatr Adolesc Gynecol. 2015;2(e179e):e183.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Human and Animal Rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Concent
Informed concent has been obtained.
Conflict of interest
None.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kakoti, L.M., Sharma, J.D., Kataki, A.C. et al. Primary Ewing Sarcoma of Vulva: A Case Report and a Review of Literature. Indian J Gynecol Oncolog 15, 15 (2017). https://doi.org/10.1007/s40944-017-0103-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40944-017-0103-7