
Abstract
The recycling of lead acid batteries (LABs) comprises relevant concerns on the suitable methodologies to recover lead. In this investigation, two electrorefining processes, by using acidic and alkaline electrolytes, have been compared to determine the most significant results of both methodologies. Acidic electrolytes used 200 g/L HBF4, 1.2 g/L H3PO4, 10 g/L H3BO3, and 100 g/L PbO, while the alkaline ones employed 120 g/L NaOH, 75 g/L PbO, 50–92 g/L glycerol, and 2 g/L gelatin. All the solutions were studied by varying temperature and current density (CD) to determine remarkable changes on current efficiency (CE), cell voltage (CV), specific energy consumption (SEC), and on the deposit quality. The results highlighted that by using the acidic electrolyte containing dextrin it is possible to obtain a compact deposit even using high CDs. Acidic solution without dextrin addition allows to obtain good quality deposits by working at 40 °C and 100 A/m2 CD. After testing the behavior of different alkaline electrolytes, it is possible to observe that the solution containing the highest glycerol concentration allows to obtain, for intermediate valued of CD, CE higher than 97% with a SEC of about 0.37 kWh/kg. By comparing the results it is evident that the acidic electrolyte is the one that allows to reach higher productivity with lower SEC. In the selected conditions, lead deposits appear pure and compact.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
One of the oldest types of rechargeable batteries are lead acid batteries (LABs). Ever since its invention in 1859, LABs production increased because of their applications in automotive, chemical, energy storing, transportation, and telecommunication sectors [1]. LABs present a configuration constituted by sealed cases containing the acidic electrolyte and the lead electrodes. Each electrode is electrically distanced by separators in order to avoid short circuits. Thus, the current supply is finally achieved through two metallic terminals that connects the inner electrode system to the external terminal arrangement. From the recycling point of view, lead can be recovered from poles, grids, and lead paste that contains on average about 22.6% PbSO4, 16.5% PbO·PbSO4, and 10.1% PbO2.
With the increasing demand and utilization, the quantity of spent LABs is also increasing rapidly [1]. Currently, about 85% of the total LABs are recycled to recover lead. Despite the broad availability of mineral ores, secondary lead production has indicated a sharp increase since the second half of the twentieth century. This growth is basically due to the expanding car market in the emerging countries. Based on this accelerated demand, studying suitable methodologies for lead recycling is becoming important for facing economic and environmental issues.
Most of the 85% of the total recycled LAB is currently processed by pyrometallurgical processes, which are certainly well-developed processes, but have at least three main drawbacks: the treatment of liquid waste from batteries, which produces a considerable quantity of sludges that cannot be discharged into common landfills, due to the presence of heavy metals; the carbonation of the sulfates that, if done before the furnace, improves the subsequent operations, but produces sodium sulfate which is difficult to sell and difficult to landfill due to the presence of heavy metals; the treatment of gaseous effluents that can represent a significant fraction of the plant cost. Currently, there are two prevailing techniques for the recovery and recycling of lead from LABs: pyrometallurgy and hydrometallurgy. Over the last decades, the most used processes were the pyrometallurgical ones. Their advantages include simple operation processes, fast reaction rates, and ability to run large-scale production. However, they have some drawbacks such as atmospheric pollution due to SO2 and lead particles emissions and high operative costs [2, 3]. On the other hand, hydrometallurgical processes offer more flexibility, lower operational costs, and milder process conditions.
Lead sulfate, which accounts for about 65% of exhausted lead paste, is very stable. For this reason, desulfurization that transforms lead sulfate into other lead compounds is a common pretreatment of pyrometallurgical recycling process. Na2CO3, NaHCO3, K2CO3, (NH4)2CO3, and NaOH are used for the PbSO4 conversion. The desulfurization efficiency reaches values varying over the range 92.4–99.7% [4,5,6,7,8].
The proposed hydrometallurgical processes can be divided in several subcategories. One of the novel methods is the “paste to paste” process in which an organic leaching is followed by a calcination stage that allows to obtain lead oxide used as active materials for battery production [9,10,11]. Another proposed process is the one involving alkaline leaching followed by crystallization [12]. In this process, the spent lead paste is dissolved in NaOH solution. After filtering the insoluble impurities, the solution that contains Na2SO4 and NaHPbO2 is cooled and PbO product is obtained by crystallization. Most of the research is focused on processes including leaching followed by electrowinning. There are many acidic leaching–electrowinning processes [13,14,15,16,17]. In the PLACID process, for example, lead chloride is obtained by the reaction of spent lead paste with hydrochloric acid and sodium chloride. After solid–liquid separation, the PbCl2 solution is purified and pure lead is obtained by electrowinning. Although this process allows to obtain lead with a purity of 99.995%, the specific energy consumption (SEC) can reach 1.3 kWh/kg Pb [13]. Another commonly used leaching solution is the one containing fluoroboric or fluorosilicic acid [14, 15]. A leaching–electrodeposition process using HClO4 has been proposed by some authors [16]. In this process, the desulfurized spent lead paste and lead grids are dissolved in the HClO4 solution. Metallic lead is recovered by this solution by means of electrowinning. The obtained SEC is 500 kWh/t Pb, much lower than other acid leaching–electrowinning processes. Despite this process allows to recover more than 98.5% of lead, environmental concerns and corrosion issues are drawbacks difficult to overcome.
Alkaline leaching–electrowinning processes have been also proposed for recovering pure lead [4, 18]. Lead paste is first desulfurized by NaOH to produce PbO. Then the obtained lead compounds are leached by using a NaOH–KNaC4H4O6 solution and the obtained lead may have a purity of 99.99%. Other processes have been studied to recover lead and lead compounds from spent lead batteries following different routes [11, 12, 19,20,21,22].
In this paper, a novel process has been proposed with the aim of recovering pure lead with an environmentally friendly hydrometallurgical process. This process includes the production of anodes from lead and lead paste, simply by melting them at temperatures for which SO2 does not form, followed by direct electrorefining [23]. In fact, the most relevant innovation is the fact that we perform a “direct recovery” of lead from an anode produced directly by melting components of spent batteries. In order to obtain good results, selecting the electrolyte composition and the most suitable process parameters is of paramount importance. This is a novel concept in the recycling of LABs. In this work, electrorefining tests with acidic and alkaline solutions have been carried out and the results, in terms of current efficiency (CE), SEC, and deposit quality have been discussed.
Materials and Methods
The lead electrorefining tests have been carried out in a Perspex cell having cathodic and anodic compartments separated by a polypropylene membrane. This membrane avoids that solid particles, which may be generated by the anode crumbling during electrorefining, could be mechanically embedded in the lead deposit. The anolyte after purification was continuously fed to the cathodic compartment by using a peristaltic pump, as it can be seen in Fig. 1. In this investigation, two different electrolytes were used: an acidic one containing HBF4, H3PO4, H3BO3, and PbO, and an alkaline one containing NaOH, PbO, glycerol, and gelatin. Each solution was prepared by using Carlo Erba high-purity reagent. The pH adjustment was realized by using H2SO4 or NaOH solutions. A 316L stainless steel cathode and an anode prepared by melting the exhausted LABs were spaced 30 mm apart. The anode preparation was made by melting at 967 °C lead-bearing components of the battery and casting the liquid metal into a titanium support. The crystallographic phases were identified by X-ray diffraction (XRD) spectrometer equipped with a CuKα source.

Electrochemical cell design used to carry out lead recovery
In the voltammetry tests, a Pt electrode was used as the counter electrode, the lead compound electrode as working electrode, and the saturated calomel as reference electrode. The test was performed at 20 mV/s scanning rate in the 1.00 and − 1.20 V range for three cycles.
Electrorefining processes were carried out using AMEL galvanostat (Amel Instruments Galvanostat/potentiostat 2053) to supply current, while bath temperature and stirring were controlled by FALC F70 hot plate magnetic stirrer. The deposits were analyzed by SEM–EDS (Hitachi S-2500) to identify the surface morphology and the lead deposit purity.
Results and Discussion
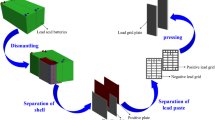
Figure 2 shows the whole process for obtaining pure lead from exhausted batteries. In this paper, we focused on electrorefining operation. Lead compounds have been used as anode in an electrowinning process to obtain pure lead, as shown in Fig. 2. In the flowsheet proposed in this investigation, the exhausted LABs initially undergo a separation stage to separate lead components (metal, grids, and paste) from other materials (plastics and other metals).

Flowsheet of the lead recovery from lead exhausted batteries by using electrorefining
Connectors, polypropylene pieces, and acidic electrolyte were separated and disposed. As it is well known, the electrorefining process is key to carry out the deposition of pure lead from lead bullion taking advantage of low temperature and the absence of toxic vapors and particle emission. Although we named the electrolytic stage electrorefining, the anode is not constituted by impure lead, but by lead oxidized species.
Considerations on Process Reactions
The used anode in the electrorefining process was prepared, as reported in a previous work [23], by using exhausted lead batteries. The XRD pattern shown in Fig. 3 highlights the presence of four oxidized lead compounds.

X-ray diffraction pattern of the used anode
Therefore, at the anode a mixed mechanism should be considered:
a) with acidic electrolyte, chemical dissolution of the bivalent species occurs at the anode [24]:
To verify if there could also be an anodic dissolution reaction, a voltammetry was carried out using a fluoroboric acid solution without dissolved Pb as electrolyte and using the cast lead compound anode [23]. In Fig. 4, the cathodic part of the voltammetry for the first and the third cycle is reported. From Fig. 4 it is evident the lead reduction peak is in the third cycle, while it is not present in the first cycle. It is therefore evident that in the first cycle there is no lead reduction peak, because the electrolyte does not contain any lead, while in the third cycle the lead reduction peak is present and this shows that there has been an anodic dissolution of lead.

Cyclic voltammetry performed at 20 mV/s on lead compound working electrode by using the acidic solution without lead (Sln A) at 25 °C
The anodic reaction is
and the cathodic reaction is
The global reaction is Reaction (7),
Reaction (7), considering the Reaction (1) can be written as
(b) with alkaline electrolyte, dissolution/complexation of the lead bivalent species occurs:
PbO obtained with the Reactions (9), (10), and (11) could then be complexed similarly to the solid PbO in the anode [25]
where OH− comes from the NaOH dissociation.
The anodic reaction is
The cathodic reaction, as highlighted in literature [25], is the following one:
The global reaction is
That considering the Reaction (12) can be written as
Lead Electrorefining Using Acidic Electrolyte With and Without Dextrin
The acidic electrolyte (200 g/L HBF4, 1.2 g/L H3PO4, 10 g/L H3BO3, and 100 g/L PbO) was used to carry out the lead electrorefining process at 100 A/m2. These operative conditions were initially considered because of the interesting results in terms of CE and SEC obtained from preliminary studies performed on exhausted LABs recycled by combining pyrometallurgical anode preparation and electrorefining. Figure 5 shows the temperature effect on cell voltage (CV), CE, and SEC. By increasing the temperature, CV shows a slight drop from 0.33 V at 25 °C to 0.23 V at 40 °C, then it raises progressively until reaching 0.43 V at 60 °C. When electrolytic bath is heated at 40 °C, the CV decreases as it is expected since temperature affects the electrolyte ohmic drop. However, at higher temperatures, a progressive decrease of CV is not observed, instead an increasing trend in CV occurs. A reasonable explanation could be found in the contemporary decrease of CE in the same temperature range. This decrease in CE indicates an increasing rate of the parasitic hydrogen evolution reaction. Thus, increasing the temperature, the hydrogen diffusion rate toward the cathode is higher than that of lead. The partial insulation of the cathodic surface due to hydrogen evolution and the lower lead concentration close to the electrode surface increase the cathodic overpotential and thus the CV. As far as the CE is concerned, a progressive drop from about 98 to 96% is observed as the temperature increases. This trend suggests that room temperature is the most suitable thermal condition, where lead can be effectively reduced minimizing parasitic reactions. Particularly, a different behavior is noticed for the SEC during the whole temperature range. Differently from CV and CE, SEC remains almost constant showing a value of about 0.06 kWh/kg with a slight variation at 60 °C where it reaches around 0.13 kWh/kg.

Effect of temperature on CE, CV, and SEC using the acidic electrolyte, 100 A/m2 and without the dextrin addition
Figure 6a, b shows a comparative analysis of the lead electrorefining process varying the CD between 100 and 500 A/m2 at room temperature by using the same electrolyte without (a) and with dextrin (b). The electrolyte without dextrin has been already considered at 100 A/m2 and the results suggested that room temperature is the best temperature that can be used at that CD (Fig. 6). The CD variation between 100 and 500 A/m2 (Fig. 6a, b) shows that CE remains above 98%. The maximum CE values, about 100%, are reached by working at 500 A/m2. The CV increases, as expected, by increasing CD, while SEC shows small variations with an almost constant trend.

Effect of current density on CE, CV, and SEC in the lead electrorefining using the acid electrolyte a without and b with dextrin at room temperature
It is interesting to note that the use of dextrin, which should have its greatest effect on the deposit quality, also shows a moderate influence on the CV showing a decrease of about 200 mV, at least for the lowest and the highest current densities (CDs) used in this work. As a consequence of this, SEC also shows slight lower values. As known from literature dextrin acts not only as electroplating brightener and surfactant, but also as carriers of ions: this reduces the concentration overpotential and then depolarizes the cathodic reaction [26].
Dextrin has been added to the acidic solution because this additive allows to increase smoothness and compactness of the deposited metal. Figure 7 shows that, without dextrin addition, the lead deposit is very porous and it can embed electrolytic solution that decreases the process efficiency during remelting. By increasing the test temperature, the deposit morphology changes and at 50 °C it is characterized by a coarse microstructure with visible cavities.

SEM micrographs showing the morphology of lead deposits obtained at 100 A/m2 without dextrin addition at a 25, b 40, and c 50 °C
By observing the lead deposits obtained at room temperature with dextrin addition (Fig. 8), it is possible to see that, at the three considered CDs, the deposits appear compact. This highlights that adding dextrin is essential to increase both the remelting and thus the global process efficiency. At 300 A/m2 it is possible to observe a platelet-like growth with evident edges. At 250 A/m2 edges become blunt and grains appear bigger. At 100 A/m2 the microstructure appears finer with dendrites having rounded arm tips.

SEM micrographs showing the morphology of lead deposits obtained at room temperature and with dextrin addition at a 300, b 250, and c 100 A/m2
In order to highlight the effect of temperature, the deposits obtained at room temperature and at 50 °C have been compared. Figure 9 shows that, by increasing the temperature, the deposit is more porous and that, without dextrin in the electrolyte, it shows many germination nuclei on the dendrite arms. Lead obtained at 50 °C grows by forming dendrites, cuboids, and thin foils, while at room temperature it appears very compact. It is evident from Fig. 9 that dextrin allows to obtain more compact deposits at each tested temperature.

SEM micrographs showing the morphology of lead deposits obtained at 250 A/m2, with (lower) and without (upper) dextrin addition at 50 °C (a–c), and at room temperature (b–d)
Lead Electrorefining Using Alkaline Electrolyte
The lead electrorefining test results, coming from tests performed by using alkaline electrolytes, can be seen in Fig. 10. On the left side of Fig. 10, a sharp increase of the CE can be noticed when the used CD is 70 A/m2. From the room temperature, the CE grows and it reaches around 99% at 70 and 150 A/m2 when temperature is fixed at 40 and 30 °C, respectively. The CE at 100 A/m2 indicates a slight change over the tested temperature range.

Effect of temperature on CE and SEC of the lead electrorefining using 120 g/L NaOH, 75 g/L PbO, and 92 g/L GL as the alkaline electrolyte
On the other hand, SEC values remain almost constant over the considered temperature range with some small variations when higher CD values are used. Independently from the selected temperature, SEC values are about 0.250 kWh/kg at 70 A/m2. A similar behavior was highlighted by using 150 A/m2 CD with a mean SEC value of 0.400 kWh/kg and a slight SEC drop at 30 °C. By using 100 A/m2, SEC changes from 0.400 kWh/kg at room temperature to 0.325 kWh/kg at 50 °C (right side of Fig. 10).
By considering different electrolyte compositions, Fig. 11 (left side) shows that a significant change of CE can be achieved by adding 2 g/L of gelatin (electrolyte C). Although the CE value reaches around 99% at 100 A/m2, a fall for higher CD values is also observed. In the case of the other two electrolytes (A and B), where only the concentration of glycerol (GL) is changed, CE has a similar trend for every CD value. High GL concentrations show CE higher than 97% for 100 and 150 A/m2. Electrolyte B shows slightly lower CE values.

Effect of CD on CE and SEC of the lead electrorefining using as the alkaline electrolyte at 25 °C: (A) 120 g/L NaOH, 75 g/L PbO, and 92 g/L GL; (B) 120 g/L NaOH, 75 g/L PbO, and 50 g/L GL; and (C) 120 g/L NaOH, 75 g/L PbO, 50 g/L GL, and 2 g/L gelatin
On the right side of Fig. 11, the SEC for three composition seems to show a similar trend. For CD higher than 150 A/m2, SEC for all the three electrolytes shows a sharp increase, although it is more evident when electrolyte A and B are used. A comparison among the results obtained with the three electrolyte indicates that the presence of gelatin affects both the CE and SEC even at 250 A/m2, where high CE and low SEC at high CD indicate a notable improvement in productivity. As highlighted by other authors [27] gelatin, which is usually added to improve the deposit quality, is chemisorbed on the cathodic surface and increases limiting CD of metal ion and CE.
Alkaline electrolytes are interesting from the point of view of environmental protection and for their low corrosiveness compared to acidic electrolytes, but from the process point of view, it is important to evaluate the deposit morphology. First the effect of process temperature has been evaluated. As it can be observed in Fig. 12, at 250 A/m2, by increasing the temperature the structure becomes more heterogeneous with the presence of fine crystallites with a cuboidal shape. Among the other process parameters also the CD affects not only the CE, but also the deposit growth. In Fig. 13 it is possible to observe that the deposit obtained at 50 A/m2 is characterized by grains that are bigger than the ones obtained at 250 A/m2, and on their surface it is possible to observe the presence of very small germination nuclei.

SEM micrographs showing the morphology of lead deposits obtained from an alkaline solution at 250 A/m2 at room temperature (a) and at 50 °C (b)

SEM micrographs showing the morphology of lead deposits obtained from an alkaline solution containing 120 g/L NaOH–75 g/L PbO–92 g/L Glycerol at room temperature and at 50 A/m2 (a) and at 250 A/m2 (b)
When lead is deposited from alkaline solutions different additives can be added to the electrolyte either to decrease the SEC or to improve the deposit quality. Figure 14 shows the deposit morphology as a result of a process carried out at 100 A/m2 and at room temperature with three different solutions. From the figure it is apparent that, by adding glycerol, which is usually added to the solution with the aim of increasing the metal smoothness, the deposit compactness is good. By decreasing the glycerol concentration from 92 to 50 g/L it is possible to see that the lead deposit is characterized by formation of small 2D lead dendrites on the surface. The addition of 2 g/L of animal gelatin (Fig. 14c) avoids dendrite formation and promotes the growth of quite regular grains, although the lead deposit is not extremely compact.

SEM micrographs showing the morphology of lead deposits obtained at 100 A/m2 and at room temperature by using the basic solution containing 92 g/L of glycerol (a), 50 g/L of glycerol (b), and 50 g/L of glycerol + 2 g/L of animal gelatin (c)
Conclusions
An anode constituted by bivalent compounds of lead has been subjected to electrorefining to recover metallic lead by using acidic and alkaline electrolytes. The compound constituting the anode were PbO, PbSO4, PbOPbSO4, and PbSO4(PbO)2.
By using the acidic electrolyte, the anodic dissolution takes place both chemically, due to the reaction between fluoroboric acid and anodic compounds, and by electrochemical dissolution, as shown by the voltammetry. The cathodic reaction is the divalent lead reduction.
The test results highlighted that by using the acidic electrolyte containing dextrin it is possible to obtain a compact deposit even using high CDs. When dextrin is not added to the solution it is possible to obtain good quality deposits by working at 40 °C and 100 A/m2 CD. As far as the alkaline electrolytes is concerned, it is possible to observe that the best results in terms of CE and SEC are obtained by using the solution containing the highest glycerol concentration, an intermediate value of CD and room temperature. By comparing the results obtained by using the two electrolytes it is evident that the acidic electrolyte is the one that allows to reach higher productivity with lower SEC. In fact it is possible to recover lead with CEs close to 100% by using 500 A/m2 CD and 0.2 kWh/kg. In the selected conditions lead deposits appear pure and compact.
References
Zhang W, Yang J, Wu X et al (2016) A critical review on secondary lead recycling technology and its prospect. Renew Sustain Energy Rev 61:108–122. https://doi.org/10.1016/j.rser.2016.03.046
Li M, Liu J, Han W (2016) Recycling and management of waste lead-acid batteries: a mini-review. Waste Manag Res 34:298–306. https://doi.org/10.1177/0734242X16633773
Ramachandran P, Nandakumar V, Venkateswaran KV (1998) Recovery of lead from battery sludge by leaching and electrowinning. Trans Indian Inst Met 51:389–391
Li M, Yang J, Liang S et al (2019) Review on clean recovery of discarded/spent lead-acid battery and trends of recycled products. J Power Sources 436:226853. https://doi.org/10.1016/j.jpowsour.2019.226853
Li L, Zhu X, Yang D et al (2012) Preparation and characterization of nano-structured lead oxide from spent lead acid battery paste. J Hazard Mater 203–204:274–282. https://doi.org/10.1016/j.jhazmat.2011.12.021
Yanakieva VP, Al Haralampiev G, Lyakov NK (2000) Desulphurization of the damped lead battery paste with potassium carbonate. J Power Sources 85:178–180. https://doi.org/10.1016/S0378-7753(99)00398-5
Yu W, Zhang P, Yang J et al (2019) A low-emission strategy to recover lead compound products directly from spent lead-acid battery paste: key issue of impurities removal. J Clean Prod 210:1534–1544. https://doi.org/10.1016/j.jclepro.2018.11.133
Zhang W, Yang J, Hu Y et al (2016) Effect of pH on desulphurization of spent lead paste via hydrometallurgical process. Hydrometallurgy 164:83–89. https://doi.org/10.1016/j.hydromet.2016.05.012
Zhu X, Li L, Sun X et al (2012) Preparation of basic lead oxide from spent lead acid battery paste via chemical conversion. Hydrometallurgy 117–118:24–31. https://doi.org/10.1016/j.hydromet.2012.01.006
Zhu X, He X, Yang J et al (2013) Leaching of spent lead acid battery paste components by sodium citrate and acetic acid. J Hazard Mater 250–251:387–396. https://doi.org/10.1016/j.jhazmat.2013.02.018
Sonmez MS, Kumar RV (2009) Leaching of waste battery paste components. Part 1: lead citrate synthesis from PbO and PbO2. Hydrometallurgy 95:53–60. https://doi.org/10.1016/j.hydromet.2008.04.012
Pan J, Zhang X, Sun Y et al (2016) Preparation of high purity lead oxide from spent lead acid batteries via desulfurization and recrystallization in sodium hydroxide. Ind Eng Chem Res 55:2059–2068. https://doi.org/10.1021/acs.iecr.5b04221
Andrews D, Raychaudhuri A, Frias C (2000) Environmentally sound technologies for recycling secondary lead. J Power Sources 88:124–129. https://doi.org/10.1016/S0378-7753(99)00520-0
Ferracin LC, Chácon-Sanhueza AE, Davoglio RA et al (2002) Lead recovery from a typical Brazilian sludge of exhausted lead-acid batteries using an electrohydrometallurgical process. Hydrometallurgy 65:137–144. https://doi.org/10.1016/S0304-386X(02)00087-7
David Prengaman R (1995) Recovering lead from batteries. JOM 47:31–33. https://doi.org/10.1007/BF03221127
Zhang X, Sun Y, Pan J (2017) A clean and highly efficient leaching-electrodeposition lead recovery route in HClO4 solution. Int J Electrochem Sci 12:6966–6979. https://doi.org/10.20964/2017.08.26
Lupi C, Pescetelli A (2008) Treatment of some liquid waste associated with lead battery recycling. In: Proceedings of the 2008 global symposium on recycling, waste treatment and clean technology, REWAS 2008, Cancun, Mexico, pp 807–813
Chen CS, Shih YJ, Huang YH (2016) Recovery of lead from smelting fly ash of waste lead-acid battery by leaching and electrowinning. Waste Manag 52:212–220. https://doi.org/10.1016/j.wasman.2016.03.056
Brandon NP, Pilone D, Kelsall GH, Yin Q (2003) Simultaneous recovery of Pb and PbO2 from battery plant effluents. Part II. J Appl Electrochem 33:853–862. https://doi.org/10.1023/A:1025824130320
Volpe M, Oliveri D, Ferrara G et al (2009) Metallic lead recovery from lead-acid battery paste by urea acetate dissolution and cementation on iron. Hydrometallurgy 96:123–131. https://doi.org/10.1016/j.hydromet.2008.09.001
Ma Y, Qiu K (2015) Recovery of lead from lead paste in spent lead acid battery by hydrometallurgical desulfurization and vacuum thermal reduction. Waste Manag 40:151–156. https://doi.org/10.1016/j.wasman.2015.03.010
Liu WF, Deng XB, Zhang DC et al (2018) A clean process of lead recovery from spent lead paste based on hydrothermal reduction. Trans Nonferr Met Soc China (Engl Ed) 28:2360–2367. https://doi.org/10.1016/S1003-6326(18)64881-2
Ciro E, Lupi C, Mondal A, Pilone D (2021) Novel lead battery recycling process combining pyrometallurgical anode preparation and electrorefining. J Sustain Metall 7:1727–1735. https://doi.org/10.1007/s40831-021-00447-y
Ping Z, Liu X, Tao Q et al (2019) Mechanism of dissolving tin solders from waste printed circuit board assemblies by cyclic fluoboric acid composite system. Environ Eng Sci 36:903–911. https://doi.org/10.1089/ees.2018.0308
Tan S, Payne DJ, Hallett JP, Kelsall GH (2019) Developments in electrochemical processes for recycling lead-acid batteries. Curr Opin Electrochem 16:83–89. https://doi.org/10.1016/j.coelec.2019.04.023
Tipson RS, Horton D (1989) Advances in carbohydrate chemistry and biochemistry. Academic, New York
Afifi SE, Elsayed AA, Elsherief AE (1987) Additive behavior in copper electrorefining. JOM 39:38–41. https://doi.org/10.1007/BF03259469
Funding
Open access funding provided by Università degli Studi di Roma La Sapienza within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
The contributing editor for this article was Hongmin Zhu.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mondal, A., Ciro, E., Lupi, C. et al. Lead Electrorefining Process from Exhausted Lead Acid Batteries by Using Acidic and Alkaline Electrolytes. J. Sustain. Metall. 8, 1744–1755 (2022). https://doi.org/10.1007/s40831-022-00598-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40831-022-00598-6