
Abstract
Apatite, Ca5(PO4)3F, concentrate from LKAB in Kiruna, Sweden, has been characterized and pyrometallurgically treated using (i) silicon metal and fluxes to extract phosphorus and transition metals at 1600 °C (Part I publication), and (ii) carbon to extract phosphorus without fluxing at temperatures exceeding 1800 °C (Part II publication), with the ultimate objective to recover rare earth elements from the resulting slag/residue phases. A variety of methods (SEM/EDS, EPMA, DTA-TGA, Sessile drop, ICP-MS/OES, and XRD) have been used for the characterization and analysis of both the concentrate and reaction products. Elemental deportment, except for the rare earth metals, is broadly in line with the equilibrium thermodynamic predictions performed using HSC Chemistry®. Phase diagrams were predicted using FactSage® and a custom phosphate database to explain the observed melting/fluxing behaviors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rare earth elements (REEs) are considered by the European Union (EU) to be of strategic significance to the European economy. They are also deemed to possess both important economic value, due to their many applications, and to have the most significant supply risk of all classes of materials evaluated by the “Working Group on Defining Critical Raw Materials” [1]. Areas of importance comprising REEs are 38% metallurgical (magnets 20%, batteries 8%, other metallurgy 10%), 20% catalysts (fluid catalytic cracking 13%, auto-catalysts 6%, other catalysts 1%), and 42% other (polishing 15%, glass 7%, phosphors 7%, ceramics 5%, and not defined 8%) [2]. A detailed breakdown of the REE consumption by metal and application can be found in documents prepared by the U.S. Geological Survey (USGS), such as by Goonan 2011 [3]. The relative abundances of the different REEs within the earth’s crust and the mineralogy of the rare earth minerals have been discussed in detail by Castor and Hedrick [4].
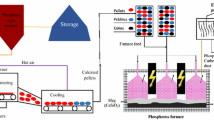
The supply risk for Europe associated with REEs became readily apparent when China in 2010 changed its export quotas, which in turn resulted in a clear spike in market prices as indicated in Fig. 1 [5]. This was not only due to the nonnative REE source in Europe, but also a result of the extremely low recycling rates, which in 2011 was reported to be <1% from end-of-life products [3]. In order to reduce the chances of a similar supply risk reappearing in Europe in the future, as well as to increase the competence in REE metallurgy, the European Commission (EC) financed a number of projects/initiatives focusing on recovery of REEs from waste material, i.e., ERECON [6], Hydro-WEEE [7], EURARE [8], RECLAIM [9], REEcover [10], etc. The REEcover project addresses REE recycling from end-of-life WEEE, as well as increased supply by studying REE recovery from an industrially produced apatite concentrate. The flowchart of the whole process targeted in the project is presented in Fig. 2. The overall objective of the work is to develop a pyrometallurgical process that extracts and purifies low REEs grade raw material into a material stream with high REE contents and purity for further recovery of REEs.

Historic rare earth oxide production and prices (in constant 1998 dollars) [5] (Color figure online)

Flowchart for the whole process targeted in the present study. The red parts are included in the present paper (Part I), while the blue part is included in Part II by Kennedy et al. [12] (Color figure online)
The work described in the present paper is based on an industrially produced apatite concentrate from Luossavaara-Kiirunavaara AB (LKAB) mine tailings originating from the magnetite ore production in Kiruna, Sweden. The mine tailings usually contain 4–8% apatite tailings, which in turn, contain 1200–1300 ppm REEs [11]. With a stockpile of 40 M tonnes of tailings, and an annual production of an additional 2 M tonnes, the site is a significant ‘primary’ REE resource in Europe, with a possible annual production of ~ 4000 tonnes REE/year.
In order to evaluate the viability of extraction of REEs from the apatite concentrate, the following high-temperature analyses and pyrometallurgical experiments, as reported in two interconnected papers (Parts I and II; to be communicated), have been performed, see Fig. 2, i.e.:
-
(1)
Initial characterization of the apatite concentrate to determine its composition and mineralogy before and after heat treatment (Part I).
-
(2)
DTA-TGA analysis of the behavior of the apatite concentrate during heating (Part I).
-
(3)
Metal-extraction experiments in order to concentrate REEs for downstream hydrometallurgical treatment:
-
(a)
Using silicon to remove phosphorus and reducible metal species with and without fluxing (Part I).
-
(b)
Using carbon to remove predominantly phosphorus, and to produce a rare earth oxide (REO) that could potentially be further processed to recover REE (Part II by Kennedy et al. [12]).
-
(a)
Apatite Concentrate Characterization and Behavior During Heating
Chemical and Mineralogical Characterization
The apatite concentrate used in the present study, is an upgraded form of the original apatite tailings, consisting of a mixture of mainly fluorapatite (Ca5(PO4)3F) with carbonates (Calcite/Dolomite), and residual iron containing minerals. In Table 1, the chemical analysis of the apatite concentrate has been expressed as simple oxides, to which loss on ignition (LOI), F− and total REO have been added. The major mineralogical species reflected by the assays in Table 1 are approximately: 77.4% Fluorapatite (Ca5(PO4)3F), 9.1% Calcite (CaCO3), 5.9% Dolomite (CaMg(CO3)2), and 1.4% Quartz (SiO2). To achieve a unique solution, all magnesium was assumed as dolomite, calcite was estimated from LOI minus the dolomite contribution, fluorapatite was calculated from total calcium minus the dolomite and calcite contributions, and all silica was assumed to be quartz. Calculated results are in good agreement with the reported mineralogy by the LKAB lab in Kiruna, Sweden: 83.1% Apatite, 10.1% Calcite, 4.6% Dolomite, 1.6% Clinochlore ((Mg,Fe,Al)6[(Al,Si)4O10](OH)8), and 0.6% Quartz [13].
The rare earth containing species have been found to be monazite, allanite, and apatite, as indicated in Figs. 3 and 4 [11, 17]. More recently, titanite and several other minor species containing REE have also been identified [18]. A number of independent laboratories have analyzed the apatite using five different analytic methodologies in roundrobin testing. It has proven remarkably difficult to analyze these mineral samples containing both silicates and low concentrations of REOs [19]. Alkaline fusion in lithium metaborate, followed by dissolution in dilute nitric acid, was particularly successful when combined with either ICP—OES or MS. A summary of the available lab results for apatite indicating the average, number of samples analyzed, and coefficient of variance of the results, is indicated in Table 2.

EPMA results indicating mainly the presence of Ca5(PO4)3F (fluorapatite), CaCO3 (calcite), and (Ce,Nd)Al2(SiO4)(Si2O7)O(OH) (allanite) [20] (Color figure online)

EPMA results indicating the presence of the (Y,La,Ce,Nd)PO4 monazite phase (large bright particle in red circle and nearby small white dots) [20] (Color figure online)
An SEM image of the concentrate is shown in Fig. 5, and the mineralogical descriptions of the different points are described in Table 3 based on EDS microprobe analyses.

The total percentages of La, Ce, Nd, and Y are much higher (of the order of tens of percent) in the monazite and allanite phases than those in the general apatite phase, raising hopes that it may be possible to melt, cool, precipitate, and grow large separable phosphate- or silicate-based crystals containing high concentrations of REEs for subsequent physical separation and extractive metallurgy. The possibility to grow such crystals was the subject of the slag saturation and slow cooling experiments. The initial results of these have been reported elsewhere [20].
The phases initially present in the apatite concentrate have been verified by XRD as shown in Fig. 6, and found to include in quantities significant enough to be observed: apatite, calcite, dolomite, talc, allanite, and monazite.

XRD results from using a powder diffractometer (Bruker D8 Advance A25) operating in Bragg Bretano mode, and utilizing Cu K-alpha radiation. A 2θ range of 10°–75° using a voltage of 30 kV. Collected data were analyzed using the software DIFFRAC.EVA V3.0 from Bruker with the database, PDF-4+2013 RDB (Color figure online)
High-Temperature Behavior of Apatite Concentrate Under Argon
The concentrate being predominantly apatite was expected to have a melting point close to that reported for synthetic apatites, i.e., of the order of 1608–1622 °C [21]. Initially, phase diagram information for the ‘phosphate’ containing system was lacking, and screening experiments were therefore conducted at the Norwegian University of Science and Technology (NTNU) in Trondheim, Norway, in an electrically heated sessile drop apparatus using a number of substrates in order to observe the melting, as well as the interactions with the substrates. Optical observation of apatite concentrate on various substrates (graphite, molybdenum, and tantalum) in an electrically heated wetting furnace indicated a liquidus point of ~1598±5 °C [20].
DTA-TGA tests were then conducted in a flowing argon stream (12 NL/h) using a molybdenum crucible, which the previous wetting experiments had shown to be a compatible material (graphite, while suitable, reacts to produce P2 gas, and tantalum was more expensive and less readily available than molybdenum). The DTA-TGA results showed that there was a mass loss of ~6.8% in flowing argon in the range of 650–910 °C, as indicated on Fig. 7. The loss of CO2 was verified by mass spectrometer readings, as shown on Fig. 8. During three separate calcining experiments run with 50–100 g, a mass loss of 6.62 wt%, with a standard deviation of 0.027%, was observed in air at 1000 °C. Similarly, a mass loss of 6.8% was found for 20 g calcined at 1450 °C. Thermodynamic analysis using HSC Chemistry® version 7.1 [22] reveals that the dolomite should decompose into the double oxide at 606 °C, and that calcite should decompose at 887 °C. Apparent values will vary due to the dilution from argon, the diffusion time out of the sample crucible, and the plug flow in the flowing argon stream within the DTA-TGA apparatus.

DTA-TGA results of the apatite concentrate using a molybdenum crucible under argon atmosphere. The temperature curve (red dotted line), the thermal gravimetric curve (green dot-dash line), and the differential thermal analysis curve versus time (solid blue line) are presented (Color figure online)

DTA-TGA and mass spectrometer results of the apatite concentrate using a molybdenum crucible under argon atmosphere. The differential temperature curve (blue line), the weight-loss curve (tan line) and the relative CO2 concentration (gray line) versus temperature are presented (cooling period is not shown) (Color figure online)
Using the DTA-TGA, an endothermic reaction (melting) was observed in the temperature range of 1112-1613 °C, as shown in Figs. 7 and 8. For comparison, the published melting point for the most-prevalent apatite in the concentrate (fluorapatite) is 1616 °C [21].
Phosphorus Extraction Experiments Using Silicon
Background for Extraction Experiments Using Silicon
The experimental work was challenging due to the limited information initially available on the behavior and properties of this high phosphorus-containing apatite concentrate. Finding compatible materials and the lack of fundamental thermodynamic information, e.g., phase diagrams and REE behavior in related slag systems, further complicated the work.
Data on rare earth oxides stability were, however, readily available as shown in Fig. 9. The data in Fig. 9 show that all of the REOs are more stable than silica, and that silicon lies below phosphorus and all of the transition elements, as indicated by the most stable transition metal oxides Cr2O3 and MnO. Silicon metal is therefore (in theory) an ideal metal to extract phosphorus and different transition metals (as well as alkali metals) without the removal of any rare earth metals. From the relative positions shown in Fig. 11, one would expect that the REEs would distribute nearly quantitatively to the slag phase. The use of silicon as a reductant adds silica to any resulting slag, and the complete reduction of phosphorus will predominantly leave a calcium silicate slag.

Ellingham diagram indicating the REOs, the phosphorus, the most difficult to reduce transition elements and the possible metallothermic reductants. The diagram is based on the software by Howard [23] (Color figure online)

CaO-SiO2 phase diagram using mole fractions [24] (Color figure online)

CaO-Al2O3-SiO2 phase diagram indicating 3 possible low melting targets for silicothermic reduction of apatite concentrate achievable with the originally planned additions of (1) silica flux (~1465 °C), (2) clay flux (~1324 °C), and (3) alumina flux (~1489 °C), with phase diagram and liquidus values estimated using FactSage®. The targets do not account for the amount of SiO2 produced during the reduction reactions (Color figure online)
Examination of Table 1 shows that complete reduction of silicon metal reducible species including phosphorus, would yield a ‘binary’ slag containing about 43 mol% SiO2 having a liquidus of ~1464 °C (composition calculated by excluding all Ca present as CaF2), as indicted in Fig. 10. A liquidus of ~1379 °C was found for the multicomponent slag using FactSage® calculations to account for the impact of CaF2 and other minor slag components. This would in theory yield a technically feasible process at temperatures easily achievable in the lab; however, the indicated values are only applicable after full removal of phosphorus.
Fluxing to reduce the product slag liquidus is one option to achieve a rapid and efficient phosphorus removal process. Obvious candidates include SiO2 and Al2O3. The CaO-Al2O3-SiO2 phase diagram, calculated using FactSage®, is shown in Fig. 11. The diagram represents the endpoint of the system after complete phosphorus removal, as well as ignoring the small contribution of fluoride present. Three possible low melting point targets are indicated on Fig. 11, which can be achieved using: (i) silica flux, (ii) clay flux (composition showed in Table 4), and (iii) alumina flux. These three approaches were each considered in the extraction experiments conducted by TECNALIA in San Sebastián, Spain (one of the REEcover consortium partners), as indicated on Fig. 11.
As previously mentioned, the phase diagram in Fig. 11 is only applicable after the phosphorus has been removed, and it does not provide information regarding possible difficulties that might be encountered during the process of phosphorus removal from a CaO-P2O5-SiO2-Al2O3 system. The required P2O5 containing phase diagrams were not available at the time of the experimental work; however, the required phosphorus slag database was developed by the EU sponsored project RecoPhos [25]. The necessary phase diagrams have now been generated by InsPyro (a REcoPhos consortium partner), on behalf of the REEcover project, and the implications of these diagrams will be addressed in the present work.
Some of the applicable thermodynamics for the silicothermic reduction of fluorapatite concentrate are explored in Table 5 and Fig. 12 using calculations performed with HSC Chemistry® version 7.1 [22] and assuming ideal solutions. From the reactions listed in Table 5 and the phosphorus distribution shown on Fig. 12, it was expected that substantial quantities of P, K and Na would be distributed to the gas phase in equilibrium with pure silicon at 1600 °C (see reactions (3), (13) and (14)). This is in good agreement with the experiences obtained during industrial pyrometallurgical phosphorus production.

Expected distribution of phosphorus to gas and metal versus temperature at equilibrium, and distribution of phosphorus by species at 1600 °C (estimated using HSC Chemistry® version 7.1 [22] assuming ideal solutions and using the 1.8:1 clay:apatite concentrate fluxing ratio) (Color figure online)

Furnace with the alumina crucibles used for the phosphorus extraction using silicon metal, as well as a closeup (Color figure online)
Apparatus and Procedures
A resistance heated ‘muffle’ furnace rated up to 1700 °C was used for the silicon extraction experiments. This furnace had dimensions of 10″ × 10″ × 10″ (L × W × H) and used eight MoSi2 heating elements. To control the operating temperature, a type-S thermocouple was inserted through the end wall of the furnace. Argon was injected, through a ceramic tube introduced through a hole made at the bottom of the furnace, in order to create a low oxygen atmosphere. Another ceramic probe was installed through the furnace roof so that some of the atmosphere could be extracted and analyzed/logged for oxygen concentration. A high flowrate of argon, 60 NL/h, was required to keep the muffle furnace at <2.5% O2, and under these conditions, the furnace could only achieve a stable operating temperature of 1600 °C. The temperature of the furnace was ramped according to the program plotted in Fig. 14, which also indicates the typical oxygen versus time profile. A photo of the furnace and a close-up of one crucible containing the blended charge are shown in Fig. 13.

Furnace temperature and oxygen profile versus treatment time for the silicothermic extraction experiments (Color figure online)
Alumina crucibles of 99.96% purity were used during these experiments. Previous work at NTNU [20] and TECNALIA had shown that high-purity alumina was the most suitable crucible material for potentially aggressive and unusual slags, as expected from these apatite experiments.
Experimental procedures were developed progressively through a total of 33 experiments. Six experiments were conducted using the final procedure: 10 g of apatite, 18 g of clay flux and 15 g of silicon, i.e., weight ratios of 1:1.8:1.5 at 1600 °C, and using an argon atmosphere.
Results and Discussion
In order for silicon to react to extract phosphorus from the apatite at a reasonable rate, both the silicon and apatite should be present in the liquid state in order to maximize the interfacial areas. As the extraction reaction produces additional silica, it would be advantageous for the added silica to further depress the melting point, or at least not increase it. At the same time, a low viscosity would also be advantageous in order to promote rapid diffusion. As a result, the use of a very low basicity slags (e.g., <0.5) were considered to be counterproductive.
As seen in Fig. 11, it was initially expected that silica addition alone would be sufficient to produce a reasonably fluid, low-viscosity and low-melting slag with the apatite; however, the addition of 1.8 silica to 1.0 weight of apatite proved insufficient to result in a fully liquid slag at temperatures of up to 1550 °C, see Fig. 15a through c.

CaO-P2O5-SiO2 phase diagrams showing a liquidus and two-phase slag surface; blue-dot represents the initial apatite concentrate particle composition (excluding Ca present as CaF2), and the dotted-red line represents the reduction path using Si as a reductant in the absence of flux; b phase fields and tie-lines at 1600 °C; and c phase fields and tie-lines at 1800 °C (Color figure online)
The blue dot seen in Fig. 15a represents the initial concentration of the apatite concentrate. The dotted red line indicates the reduction path using silicon metal without fluxing, which follows a line of approximately constant CaO (56 wt%). The liquidus quickly increases beyond 2000 °C with increasing degree of reduction, indicating that the silicothermic reduction would likely be very slow even at 1600 °C, with the apatite/silica product being solid in the absence of fluxing.
From Fig. 15a, it is also clear that the initial addition of SiO2 flux to the apatite concentrate will result in a dramatic increase in liquidus requiring temperatures >2000 °C to ensure rapid melting. Once sufficient silica is added to the initial apatite concentrate, the total slag will enter a multiphase region as indicated in Fig. 15a through c. Figure 15b shows that at 1600 °C (the maximum possible temperature under argon for the current experimental apparatus), the slag would quickly saturate with solid SiO2. Even at 1800 °C the slag would enter a two-phase slag region, as shown in Fig. 15c), where one of the slags produced would be nearly pure SiO2 and therefore of extremely high viscosity. It is therefore concluded that silica fluxing alone is not practically feasible with high phosphorus-containing apatite concentrates.
As previously mentioned, an alternative approach to fluxing with silica would be to use alumina flux. If the chemical reaction at the surface of a fluorapatite particle is considered, molten silicon metal will react to form P2 gas and a P-reduced calcium silicate (also containing small amounts of CaF2). As shown in Fig. 16, the blue point corresponds to a surface ‘phosphate’ content of 25% P2O5, representing a P removal of ~38%.

CaO-Al2O3-SiO2 phase diagram with constant 25 wt% P2O5. The blue dot indicates the composition at the surface of an apatite particle, which has been reacted with Si by 38% to achieve a local P concentration equivalent to 25 wt% P2O5. The Ca present as CaF2 is excluded (Color figure online)
The location of point on Fig. 16 indicates that the initial fluxing with alumina would require more than 2000 °C to achieve complete melting. Experiments held at 1450 °C for 60 min, using an apatite concentrate to alumina ratio of 1:0.4, sintered. In other experiments, complete melting of the apatite was not achieved even with a final holding time of 140 min at 1550 °C. It was subsequently decided to use clay flux as the main fluxing agent. Clay flux has the advantage that it is effectively ‘self-fluxed’ with a liquidus of ~1782 °C relative to pure Al2O3 (m.p. 2054 °C) making the melting/dissolution process easier and faster to achieve.
Using clay, it was quickly found that the optimal results (complete melting of the charge with an intact crucible until cooling) could be achieved using an apatite concentrate to clay ratio of 1:1.8, which was double the initially assumed target (the results for this final ratio are the focus of the present study). The final composition of the slag, expected to be produced at 1:1.8:1.5 apatite concentrate:clay:silicon ratio, was established to consist of 58.8% SiO2, 18.9% CaO and 22.3% Al2O3. This would be achieved by fully removing the phosphorus, sodium, potassium as well as all transition metals from the slag, and excluding the calcium present as CaF2. This composition has been indicated on the phase diagram presented in Fig. 11. The initial (assuming full charge melting with no P-removal) and final slag composition, accounting for the maximum possible increase of SiO2 due to the reduction reactions, are shown in Table 6. Results are compared to the actual concentration after the experiments. The results from 3 experiments have been averaged.
The slag assay comparison in Table 6 indicates that the assumption of close to complete reduction seems reasonable. Initial and final masses for the 6 runs executed at 1600 °C are listed in Table 7, and representative images are shown in Fig. 17. Note that runs 1 and 2 were held at full temperature for 60 min, while the remainder of the runs were held for 90 min. The average mass loss for the 60 min runs was 6.7%, while the average mass loss for the 90 min runs was 7.4%, with an overall average of 7.2%. Average metal and slag assays, percent mass distributions to metal and to slag, and mass distribution coefficients to slag, i.e., LS/M = (wt% in slag)/[wt% in metal] are given in Table 8.

Representative images of the experimental crucibles after use showing a a slag (i) with a silicon-based metal phase (ii), and a coating of white powder (>96% SiO2) (iii); b detail of a section showing the slag (i) and the metal phase (ii); c a small amount of condensate on the crucible lid after use (iv), and no damage to the crucible bottom which cracked first after cooling (v) (Color figure online)
Elemental accountability has been estimated using the initial masses and assays, accounting for silicothermic reduction of species above silicon in Fig. 9, as well as the loss to the gas phase of both LOI (crystal water from clay and CO2 from apatite) and a fraction of the Na, K and P due to their volatility. A CO2 mass loss due to carbonates of 6.8% was assumed. Allowance was also made for a clay mass loss of 9.2% (equal to the L.O.I. measured at 1000 °C). After 3 iterations using the “Heat and Material Balance” module of HSC Chemistry® 7.1, the masses of the slag (58.1% of total input), the metal (33.1%), the gas (7.4%) and the salt (1.4%), as well as the relative mass distributions to each phase converged to the values shown in Fig. 18. The values also corresponds to the experimental results in Table 8. The estimated mass loss to gas was 7.4% using this method, versus the average measured mass loss of 7.2%. The iterative procedure was required to estimate the mass deportment, as it was not possible to separately measure the masses of slag, metal, and salt at the completion of the experiments. This was mainly due to a partially dispersed metal phase, as well as the slag adherence to the crucible. Based on thermodynamic equilibrium calculations, the salt phase was assumed to be simply CaF2, as this should be the predominant fluoride at 1600 °C.

Calculated average distribution of elements by phase for silicon-extraction experiments at 1600 °C (Color figure online)
As shown in Table 8, the distributions between the slag phase and the metal phase of typical slag species like K, Na, Al, Mg, and Ca highly favored the slag phase, as indicated by the mass distribution coefficients (values vary from 127 for Al to 793 for Ca). As shown in Fig. 9, one would expect that the distribution coefficients for the REEs would favor the slag to even a greater extent; however, this is not the case. The distribution coefficients for REEs were surprisingly low varying from only 2.2 for Tb to 30 for Y (the least and the most stable REEs depicted on Fig. 11). Evaluation of the apparent ‘equilibrium’ constant for Nd reduction by silicon, i.e., Nd2O3 + 1.5 Si = 2 Nd + 1.5 SiO2, would indicate a 5–6 order of magnitude difference between theoretical (2 × 10−12 at 1600 °C and assuming ideal solutions) and observed value (8 × 10−7 and assuming SiO2 and Si activities estimated using FactSage 7.0®). This suggests that REEs are highly nonideal in either metal, slag or both, which is not surprising considering the ppm levels of the species involved. In other words, the assumption of ideal behavior was invalid.
As shown in Fig. 18, the distribution of species to the ‘balance’ represents the experimental and/or analytic error, as well as the losses to the gas phase. It is, however, impossible to separate the different parts of the ‘balance’ when using the applied calculation methodology. As expected, the phosphorus shows a high ‘balance,’ i.e., distribution to gas. The distribution of phosphorus obtained from experiments matched well with the calculated results: for example, 46.7% obtained vs. 55.5% predicted and 51.3% obtained versus 44.5% predicted for gas and metal, respectively, as shown in Fig. 12. Less than 2% of the phosphorus remained in the slag phase. Also showing high distributions to the gas phase were K (30.4% vs. 98% expected), Mn (21.6% vs. 1.4% expected) and Mg (13.7% vs. 0.25% expected); however less Na than expected evaporated (9.5% vs. 95% expected). Al is more than 100% accounted for, due to gain from the alumina crucible. The ‘balances’ for the REEs vary both positively and negatively, which most likely are due to random variation in the analytic results for these low concentration species. Given the simplifying assumption of ideal solutions, substantial variation between expected and actual distributions are not surprising, particularly for elements with low initial concentration.
It should be noted that the analytic results of the slag and metal used to generate Fig. 18 were only analyzed at a single laboratory, so the sample/sampling variance is not known in comparison with the initial apatite analytic results obtained via roundrobin testing, see Table 2.
SEM/EDS and XRD were performed on the metal and slag phases. Phosphorus was only detected by SEM/EDS in the metal phase and not in the slag phase. Different crystalline phases are formed in the metal, as shown in Fig. 19:

Different phases identified in solidified metal after phosphorus extraction: Spot (1) P 41.4 wt% and Ti 58.6 wt%, Spot (2) Si 98.3 wt % and P 1.7 wt%, and Spot (3) Si 39.1 wt%, Fe 17.6 wt%, and P 43.3 wt% (Color figure online)
-
(a)
The major crystalline phase contains only Si 98.3 wt% and P 1.7wt% (Spot 2).
-
(b)
Other minority phases contain P 41.4 wt% and Ti 58.6 wt% (Spot 1), and
-
(c)
Si 39.1 wt%, Fe 17.6 wt% and P 43.3 wt% (Spot 3).
The metal-phase spectrum measured by XRD suggests Si, FeSi2, and SiP as the main components, but not all the peaks of the spectrum were identified.
Phosphorus was not detected in the slag by SEM/EDS. Using EDS the slag was found to assay: Si 13.9 wt%, Ca 4.5 wt%, Al 7.5 wt% and O 74.1 wt%, and the XRD analysis showed the presence of Anorthite/Dmisteinbergite (CaAl2Si2O8) and Corundum (Al2O3) as the main phases.
Conclusions
Phosphorus extraction is not feasible directly from the apatite concentrate at 1600 °C using only silicon as solvent; the addition of flux is required in order to avoid high melting and high viscosity compositions. A clay flux proved to give the best results.
REEs distribute more than 80% to the slag, while P is nearly completely removed from the slag, being fumed or dissolved into the silicon metal phase. The distribution of REEs were far from the expected equilibrium values, being approximately five orders of magnitude higher in the metal phase than what was expected based on the thermodynamic calculations assuming ‘ideal solution’ thermodynamic calculations (apparent equilibrium constants of 8 × 10−7 versus the expected 2 × 10−12).
The use of flux and Si in the REE extraction process results in a net decrease in the concentration of REEs in the final slag compared to the original apatite concentrate; however, the absence of phosphorus in the obtained slag opens a route for further processing by phase reformation.
Future Work
The results of the present study show that there are numerous gaps in the availability of thermochemical data for REO-containing systems, making it very challenging to propose the “best” process route for REO containing minerals and waste slag. As such, development of key REO-containing phase diagrams is recommended. In terms of apatite concentrate, dephosphorized material will be further pyrometallurgically treated by doping to grow larger, physically separable crystals containing the sought-after REO minerals.
References
E.U. Commission (2014) Report on Critical Raw materials for the EU:1–41
E.U. Commission (2015) A Report by the European Rare Earths Competency Network (ERECON) Strengthening the European Rare Earths Supply-Chain Challenges and policy options:1–104
Goonan TG (2011) Rare earth elements–end use and recyclability: US Department of the Interior. US Geological Survey:1–22
Castor SB, Hedrick JB (2006) Rare earth elements. Industrial minerals volume, 7th edn. Society for Mining, Metallurgy, and Exploration, Littleton, pp 769–792
Rare earths statistics. http://minerals.usgs.gov/minerals/pubs/historical-statistics/ds140-raree.xlsx. Accessed 27 Feb 2016
European Rare Earths Competency Network (ERECON). https://ec.europa.eu/growth/sectors/raw-materials/specific-interest/erecon_en. Accessed 28 Nov 2016
HydroWEEE report summary. http://cordis.europa.eu/result/rcn/45388_en.html. Accessed 28 Nov 2016
EURARE homepage. http://www.eurare.eu/. Accessed 28 Nov 2016
RE-CLAIM Homepage. http://www.re-claim.eu/. Accessed 28 Nov 2016
REEcover homepage. http://www.reecover.eu/. Accessed 28 Nov 2016
Pålsson BI, Fredriksson A (2012) Apatite for extraction. II. Flotation of apatite and rare earth elements from old tailings dumps. In: XXVI International Mineral Processing Congress (IMPC), New Dehli, India, pp 04064–04075
Kennedy MW, Sun T, Yurramendi L, Arnout S, Aune RE and Tranell G (2017) Pyrometallurgical treatment of apatite concentrate with the objective of rare earth element extraction. Part II. J Sustain Metal. https://doi.org/10.1007/s40831-017-0148-y
Sarasola LY (2015) REEcover WP3—Task 3.4-silicon as solvent and separator tests for pyrometallurgical upgrade of apatite tailing. Tecnalia report:1–139
Palsson BI, Martinsson O, Wanhainen C, Fredriksson A (2014) Unlocking rare earth elements from european apatite-iron ores. In: 1st European rare earth resources conference (ERES), pp 211–220
(2016) D6.2 Round Robin test of selected analytical technique and/or material (final update). Tecnalia Report:1–64
Peelman S, Sun ZHI, Sietsma J et al (2016) Hydrometallurgical extraction of rare earth elements from low grade mine tailings. In: Alam S, Kim H, Neelameggham NR, Ouchi T, Oosterhof H (eds) Rare metal technology 2016. TMS, Warrendale
Pålsson BI, Martinsson O, Wanhainen C, Frederiksson A (2014) Unlocking rare earth elements from European apatite-iron ores. In: Proceedings 1st European Rare Earth Resources Conference (ERES2014), pp 211–220
Wanhainen C, Pålsson BI, and Eriksson AE (2016) New REE Mineralogy for LKAB Apatite-Iron Ores. Presented at Mineral Teknik 2016
ICP-MS method—trace element analyses of rocks and minerals by ICP-MS. http://cahnrs.wsu.edu/soe/facilities/geolab/technotes/icp-ms_method/. Accessed 28 Nov 2016
Sun T, Kennedy MW, Tranell G et al (2015) Apatite concentrate, a potential new source of rare earth elements. In: Neelameggham NR, Alam S, Oosterhof H, Jha A, Dreisinger D, Wang S (eds) Rare metal technology 2015. TMS, Warrendale
Pandya JR, Mehta BJ (1969) The melting point of synthetic apatites. Miner Mag 37:527–528
https://www.outotec.com/products/digital-solutions/hsc-chemistry/hsc-sim-process-simulation-module/. Accessed 15 Sept 2016
Howard SM. EllinghamMaker_v12-5.xls. http://showard.sdsmt.edu/MET320/Handouts/EllinghamDiagrams/EllinghamMaker_v12-5.xls. Accessed 24 Mar 2016
FactSage Database Documentation. http://www.crct.polymtl.ca/fact/documentation/. Accessed 28 Nov 2016
Arnout S, Nagels E (2016) Modelling thermal phosphorus recovery from sewage sludge ash. Calphad 55(1):26–31
Acknowledgements
The authors would like to thank their coparticipants in this EU-funded project (Grant FP7-603564—REEcover) for their assistance during the performance of the present work. Concerning apatite concentrate characterization in Chapter II, thanks are offered to: J.R. Tolchard for his help during the XRD analyses; Y. Yu and M.P. Raanes for their advice and work with SEM–EDS and EPMA; T. Anzjøn and S. Bao for their help with the wetting and DTA-TGA-MS analyses; and T. Schanche and D. Leroy for their experimental assistance. The primary author also would like to offer his thanks to the Chinese Scholarship Council (CSC) for funding his Ph.D. research.
Author information
Authors and Affiliations
Corresponding author
Additional information
The contributing editor for this article was Bernd Friedrich.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sun, T., Kennedy, M.W., Yurramendi, L. et al. Pyrometallurgical Treatment of Apatite Concentrate with the Objective of Rare Earth Element Extraction: Part I. J. Sustain. Metall. 3, 829–845 (2017). https://doi.org/10.1007/s40831-017-0140-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40831-017-0140-6