
Abstract
This text is written for a course on instrumental methods of quantitative analysis. It summarizes the basic concepts of modern voltammetric techniques of analysis. The guiding concept of this text is to demonstrate how the ratio of faradaic to capacitive currents decides about the sensitivity of the techniques, and how this ratio can be increased by electronics, electrode construction and by chemical means. Finally, the advantages and disadvantages of voltammetric techniques of analysis are briefly described.
Similar content being viewed by others


Introduction
In industry and research, voltammetric techniques (VTs) of analysis have certainly less importance than chromatographic, electrophoretic and spectroscopic techniques. However, for specific analytical tasks, they offer superior solutions, and hence it is important that students become acquainted with their essential features and the fundamentals they are based on. It is not easy to give an overview on voltammetric techniques of analysis, because this topic is very diverse and the theory behind goes deep into thermodynamics and kinetics of electrochemistry, a branch of physical chemistry which also does not find sufficient attention in many curricula. This text is written to give a wide overview on voltammetric techniques so that students get an orientation in what cases they can and should be used with benefit. The text is written for a general course in instrumental analysis. For more detailed information the reader is referred to monographs [1, 2], textbooks on electrochemistry and electroanalysis [3–5], and to the “Electrochemical Dictionary” [6]. There are also electroanalytical techniques which are of immense value in studies of the electrochemical properties of compounds and systems, but of little importance for quantitative analysis, such as cyclic voltammetry, chronocoulometry and impedance spectroscopy. These techniques will not be discussed in this text. Equally, we will not discuss electroanalytical techniques using organic solvents.
Electroanalytical techniques based on measuring charge transport properties of ions
If one understands electroanalysis as all applications of electricity for chemical analysis, the position of voltammetric analysis is that of a subdivision: an overview on electroanalysis is given in Fig. 1. When electrodes (electron conductors, like metals, e.g. Hg, Au, Pt) or carbon (graphite, glassy carbon) are introduced in an electrolyte solution (or a solid electrolyte, i.e., ion conductor) it is possible to study (a) the conduction (charge transport) in the electrolyte, and (b) the charge transfer at the electrode/electrolyte interface. Techniques based on the charge transport are mainly (i) conductometry, i.e., measuring the ionic conductivity of solutions, and (ii) electrophoresis [7, 8], where the variations in charge transport are used to separate ions. Electrophoresis is usually treated in the framework of separation techniques, although it makes use of migration of ions in an electric field, and thus could be taken as an electrochemical technique. Conductometry is well known as a method to indicate the equivalence point of titrations, and also as the basic measuring principle of detectors in ion chromatography.

A possible subdivision of electroanalysis. “Dynamic techniques” are those in which the electrochemical equilibrium is shifted with the help of an excitation (potential changes or current changes), whereas in the “static techniques” the electrochemical equilibrium at the electrode is not affected. Sometimes, dynamic techniques are described as active, and the static techniques as passive techniques
Although electroanalytical techniques based on measuring the transport properties of ions are frequently used, the term electroanalysis is often identified with voltammetric techniques of analysis, simply because voltammetric techniques outnumber all other electroanalytical techniques. Of course, potentiometric techniques, especially, pH measurements with glass electrodes, and also other potentiometric measurements with ion-selective (or ion-sensitive) electrodes are very frequently performed in laboratories, industry and environmental monitoring.
Electroanalytical techniques based on measuring charge-transfer processes at interfaces
When a current passes through an electrochemical cell, the following processes proceed: (a) the electrons have to be transported within the metal wires connecting a current (or voltage) source with the electrodes. This is guaranteed by the free electrons in the metals (metal conduction). (b) when the electrons reach the electrode/electrolyte interface, a process of charge transfer has to guarantee that charge can pass through the interface. In some cases, the electron can be transferred to a species in solution, e.g. an iron(III) ion:
In other cases, a cation may join the metal electrode (e.g. a piece of lead, or a mercury droplet) and uptake electrons to be reduced:
This reaction is equivalent to the passing of electrons through the interface, because the electrons are “neutralized” by the cation which joins the electrode.
The currents flowing in these two reactions are called faradaic currents because they follow Faradays law:
“The number of moles of a substance, m, produced or consumed during an electrode process, is proportional to the electric charge passed through the electrode, q. Assuming that there are no parallel processes, \(q = nFm\), where n and F are the number of electrons appearing in the electrode reaction equation, and the Faraday constant, respectively” [9].
The Faraday constant is the charge of one mol electrons: 9.64853383(83) × 104 C mol−1. The history of the discovery of this law is nicely described in [10].
There is also another pathway of current through an electrochemical cell: The two sides of the electrode/electrolyte interfaces can be charged and discharged like the two plates of a plate condenser. Such currents are called capacitive currents or charging currents. They will be explained in detail further down.
Electroanalytical techniques based on charge-transfer processes at electrode/electrolyte interfaces can be subdivided in static and dynamic techniques. In the cases of static techniques, the currents are ideally zero and no net reaction occurs. In the dynamic techniques, measurable currents flow and the system is dynamic in the sense that a net reaction proceeds.
The most important static technique is potentiometry, where the potential of an electrode is measured relative to the potential of a reference electrode. This technique is also called an equilibrium technique, or I = 0 technique. In the case of pH glass electrodes, the potential drops at the inner and outer glass membrane solution interfaces are measured with two reference electrodes. Membrane electrodes are the most important type of ion-sensitive electrodes.
Dynamic techniques (also called transient techniques, because they record time-dependent phenomena) to measure processes at electrode/electrolyte interfaces can be subdivided in controlled potential and controlled current techniques (see the next paragraph). In controlled potential techniques, devices are used (so-called potentiostats) which allow controlling the potential in a desired way, i.e., keeping it constant at a certain value, or shifting it in a programmed way. The current is then measured as the answer of the system to the potential. This kind of techniques is called voltammetric techniques.
Alternatively, one may use devices that control the current (so-called galvanostats) and measure the potential of the working electrode as the answer to the current.
Controlled potential and controlled current techniques can in principle give the same information; however, with the development of potentiostats, controlled potential techniques have won the race.
What are voltammetric techniques?
The term voltammetry is derived from voltamperometry, and it expresses that the current is measured as a function of voltage, i.e., electrode potential. Since any electrochemical cell needs two electrodes, it would be impossible to extract unambiguous analytical information, if both electrodes would determine the magnitude of the flowing current. Therefore, one electrode is made much smaller than the other, so that the flowing current is limited by this electrode only. This electrode is called the working electrode, and the other (larger) electrode is called the auxiliary electrode.
Although an electrochemical cell needs only two electrodes to operate, it is very beneficial to introduce a third electrode: this third electrode is a reference electrode, i.e., an electrode which has a known and fixed electrode potential. No current should ever pass this electrode, as a current would change its potential, and possibly damage this electrode. The reference electrode is used to control the potential of the working electrode by measuring the voltage between these two electrodes. A cell with a working electrode (WE), an auxiliary electrode (AE) and a reference electrode (RE) is called a three-electrode cell (Fig. 2). The potential difference at the electrolyte/electrode interface of the working electrode is controlled with the help of a potentiostat. Most potentiostats use a feedback circuit that applies such voltage between the auxiliary and the working electrodes, that the potential of the working electrode exactly matches the value it should have. Usually the working electrode is grounded, so that in fact the potential of the solution is controlled.
Voltammetry is now the general term for all techniques in which the current is measured as a function of electrode potential. At the beginning of its development was Jaroslav Heyrovský, the inventor of a voltammetric technique which he named POLAROGRAPHY. (The term polarography was meant to indicate that the polarization of an electrode was graphically recorded as current–potential curves: current-flow means depolarization, and no-current-flow means polarization, meaning that the potential increases without or only with a minor current response). Heyrovský used a dropping mercury electrode (see further down) as working electrode. For some time, the terms voltammetry and polarography have been used as synonyms; however, now polarography is used exclusively for “voltammetry with a dropping mercury electrode”.

Scheme of a three-electrode electrochemical measurement
Faradaic versus capacitive currents
Faradaic currents
The best way to determine the concentration of compounds is to measure faradaic currents which they generate when they are oxidized or reduced at electrodes. The generation of faradaic currents needs the molecules or ions to reach the electrode surface. This can be accomplished only by mass transport toward the electrode surface. There are only three possible mechanisms which can transport them: (a) diffusion in a concentration gradient, (b) migration of ions in a potential gradient, and (c) convection. A concentration gradient will always build up at the electrode/electrolyte interface, when the compound is oxidized or reduced at the electrode surface. Figure 3 visualizes this.

Diffusion layer which forms when the compound A is oxidized to form B on an electrode surface. The (Nernst) diffusion layer thickness d Nernst is the distance from the interface to the intercept of the two (almost) linear parts of the concentration profile
The diffusion profile depends on the diffusion coefficients of diffusing ions (or molecules), and it is a function of time. The diffusion layer thickness grows with time, and the concentration gradients (i.e., the steepness of concentration versus distance function) will decrease with time. This is shown for the compound A only in Fig. 4.

Increase of diffusion layer thickness with time of experiment
The diffusion profile also depends on the electrode geometry and size. When the electrode dimension goes down to the µm range, edge effects become important and the diffusion layer may assume a spherical shape when the electrode is a disc. When the solution is stirred (or the electrode is moved, e.g. rotated, when it is a disc or wire), the diffusion layer shrinks because the solution is mixed.
Since current is the first derivative of charge over time, i.e., \(I = \frac{{{\text{d}}q}}{{{\text{d}}t}}\), the current at a planar electrode (much above µm dimension) decays with time, because of Fick’s law:
J A is the flux of species A in mol cm2 s−1, D A is the diffusion coefficient of A in cm2 s−1, and c A is the concentration of A in mol cm−3, as a function of distance x from electrode surface and time t. Clearly, because the concentration gradient \(\frac{{\partial c_{\text{A}} \left( {x,t} \right)}}{\partial x}\) decreases with time, the current also drops with time. If diffusion is the only process limiting the current, and if the electrode surface is planar and the solution unstirred (and electrode not moving), the current decay is described by the Cottrell equation:
According to this equation, the current decreases with t −1/2. The Cottrell equation can be derived from the following linear differential equation:\(\frac{{\partial c_{\text{A}} (x,t)}}{\partial t} = D_{\text{A}} \frac{{\partial^{2} c_{\text{A}} (x,t)}}{{\partial x^{2} }}\) with appropriate boundary conditions (see, e.g. [11]).
Capacitive currents
The interface of an electron conductor and a solution forms a capacitor because charge can be accumulated on both sides. The side of the electron conductor can be charged negatively by accumulating electrons and positively by an electron deficit. The solution side can be charged by an excess of either anions or cations. In addition to the ions, dipole molecules of the solvent can be oriented on the interface and give an additional contribution to the accumulated charge. Because of the capacitive properties, the interface is also called an electrochemical double layer (EDL). Figure 5 shows a simplified scheme for the case of a metal electrode in contact with an electrolyte solution. The following features are important: (a) at the very interface, there is a layer of adsorbed and oriented dipole solvent molecules. (b) Solvated cations can approach the interface only down to a minimum fixed distance, the so-called outer Helmholtz plane. (c) Some anions can specifically adsorb at the interface (the dark green ball in Fig. 5). The plane passing through the centres of the specifically adsorbed anions is called the inner Helmholtz plane. (d) In the diffuse layer, the excess charge compensating the charge on the metal side is decaying toward the solution bulk, because the numbers of cations and anions are changing in such way that their charges compensate. (In case of unit charges, this happens for equal numbers of cations and anions). In reality, the structure of the EDL is more complicated, on both sides; however, for understanding electroanalytical methods, this is of minor importance. The potential varies in the EDL linearly within the compact layer and up to the outer Helmholtz plane. It then decays exponentially in the diffuse layer. This is important, because outside the EDL, there is no electric field resulting from the charged electrode, i.e., outside the EDL ions are not affected by the electrode potential, and so no migration can happen. The diffuse layer results from the thermal energy of the system, which prevents the ions to be arranged in a fixed distance from the metal, but randomizes their position to some extent. The electrical field acts to fix their position, but the thermal movement counteracts this fixation. The thickness of the EDL depends primarily on the number of ions per volume of solution (i.e., their concentration), the charge of ions, and on the charge density on the metal side, i.e., on the electrode potential. The dielectric constant of the solution is a third factor. The dielectric constant varies in the EDL, due to the non-random orientation of the dipoles. At a certain potential, there is no excess charge on either side of the EDL. This potential is called the “potential of zero charge” (pzc). It depends on the metal, the solvent, and the nature and concentration of the electrolyte.

Scheme of an electrochemical double layer. IHP inner Helmholtz plane, OHP outer Helmholtz plane. ϕ metal and ϕ solution are the inner potentials in the metal and solution, respectively. On the abscissa, the distance r from the interface is plotted and on the ordinate the potential
Capacitive currents flow at electrodes because charge can be accumulated on the two sides of the EDL. To understand the origin of capacitive currents, it is helpful to remember the physics of a capacitor: current is the first derivative of charge over time:
The accumulated charge is the product of surface area A and charge density (charge per unit of surface area) q:
This gives according to the rules of mathematics:
Now we have to introduce a specific parameter of the EDL, the so-called “differential double layer capacity” C dl:
The differential double layer capacity takes into account that the charge density is also a function of electrode potential E. Multiplication of Eq. 8 with the term \(\frac{{{\text{d}}t}}{{{\text{d}}t}}\) leads to:
We can rearrange Eq. 9 as follows:
and insert this in Eq. 7:
Equation 11 is of fundamental importance: It tells us that a capacitive current will always flow when (i) the surface area changes with time (\(\frac{{{\text{d}}A}}{{{\text{d}}t}} \ne 0\)), or (ii) the potential changes with time (\(\frac{{{\text{d}}E}}{{{\text{d}}t}} \ne 0\)), or (iii) both, the surface area and the potential change with time. Since voltammetry means to measure currents as a function of potential, it is clear that \(\frac{{{\text{d}}E}}{{{\text{d}}t}}\) can never be kept at zero, at least not for the entire measurement. We shall later see how one can keep it at least at zero for certain short periods of time. The surface area of an electrode can much easier be kept constant (\(\frac{{{\text{d}}A}}{{{\text{d}}t}} =0\)), as solid metal electrodes of course possess a constant area. However, when a dropping mercury electrode is used (see further down), it needs a valve to stop the outflow of mercury periodically, to keep the surface area constant during certain periods of time.
Why do the capacitive currents limit the sensitivity of voltammetric measurements?
When capacitive and faradaic currents flow at the same time, one can always only measure their sum. Often the analytical signal is a peak-shaped faradaic current, whereas the capacitive current is a sloping line. Further, all measurements involve noise, i.e., stochastic fluctuations which origin in electronics, the electrode, the cell etc. When the concentration of the analyte decreases, also the faradaic currents decrease; however, the capacitive currents are unaffected by the concentration of the analyte. Hence, the ratio \(\frac{{I_{\text{f}} }}{{I_{\text{c}} }}\) decreases with decreasing analyte concentration. The measurement of I f will thus become less and less precise, as the overall current is set by the capacitive behaviour of the EDL. This is easily understandable when one imagines the following situation: the mass of a captain should be measured, and somebody has the idea to put in one measurement the ship with the captain, and in another measurement the ship without the captain on a balance. Clearly, the difference should give the captain’s mass. However, to measure the mass of a ship, a very large balance is needed, which will have a scale in the thousand tons range, but the measuring uncertainty will be so much larger than the captain’s mass, that this experiment must fail. The same is also true for a chemical laboratory: it will be impossible to measure in the milligram range, when the pot for weighing the milligram-sample has the mass of a kilogram (or this would need an extremely sophisticated balance, as it is not available in chemical laboratories). From analytical point of view, current measurements are not different: it will become impossible to distinguish extremely small faradaic currents from large capacitive currents. Here it needs to be emphasized that it is not the absolute smallness of currents which limits the quantitative determination of analytes, but it is their smallness in relation to the capacitive current! The problem is getting even tougher when the base line current is noisy. Noise means the stochastic fluctuation of current which is caused by stochastic perturbations at the electrode, the cell, the electronics, etc. So-called white noise contains all frequencies and one may improve the signal-to-noise ratio by accumulation of voltammograms (like in spectroscopy), but this is another topic.
As we have seen here, it is necessary to improve the ratio of faradaic to capacitive currents \(\frac{{I_{\text{f}} }}{{I_{\text{c}} }}\) if very low analyte concentrations have to be determined. We shall now see that all developments of voltammetric analysis techniques can be seen as a long march to improve the ratio \(\frac{{I_{\text{f}} }}{{I_{\text{c}} }}\).
Electrochemical reversibility and irreversibility
In the following parts, we will need to refer to the reversibility or irreversibility of electrochemical reactions. Therefore, some basic information needs to be given, which relates to the kinetics of electrode reactions. (A more detailed discussion is given in [12] ).
For an electrode reaction to proceed, the educt has to be transported to the interface, and the product has to be transported away from the interface (cf. Figure 6). At the interface, the charge transfer occurs with the rate v ct. The overall rate of the electrode reaction can be limited (i) by the mass transport of educt to the electrode, (ii) by the mass transport of product to the bulk of solution, or (iii) by the rate of charge transfer. We shall discuss here only case (i) and (iii), as case (ii) is less frequently found. When the equilibrium between the oxidized and reduced forms of the electroactive compound is very quickly established at the interface, i.e., when v ct is faster than v mt, the ratio \(\left( {\frac{{a_{\text{ox}} }}{{a_{\text{red}} }}} \right)_{\text{interface}}\) at the interface will assume exactly the value which follows from the Nernst equation (\(E = E_{\text{A/B}}^{\,\bigcirc\!\!\!\!\!\!\!{\rm{-\!\!-\!-}}} + \frac{RT}{nF}\ln \left( {\frac{{a_{\text{ox}} }}{{a_{\text{red}} }}} \right)_{\text{interface}}\)) for this potential. The Nernst equation describes an equilibrium situation, and hence a system where the ratio \(\left( {\frac{{a_{\text{ox}} }}{{a_{\text{red}} }}} \right)_{\text{interface}}\) corresponds to the potential given by the Nernst equation, is called an electrochemically reversible one. However, when the charge-transfer rate v ct is smaller than the mass transport rate v mt, the equilibrium cannot be established with a sufficient rate and the ratio of \(\frac{{a_{\text{ox}} }}{{a_{\text{red}} }}\) at the interface will deviate from the ratio given by the Nernst equation for this potential. Then the electrochemical system has to be regarded as more or less irreversible. One can phrase this also as follows: the current is the observable which tells us that an electrochemical reaction proceeds. If that current is smaller than expected, the reaction is obviously not proceeding at equilibrium and thus not reversible. Figure 7 illustrates this using a rate scale.

The rate of charge transfer v ct, and the rate of mass transport v mt of an electrode reaction

The relation of the rate of charge transfer v ct to the rate of mass transport v mt decides about the reversibility of an electrochemical system. When the charge transfer (v ct) is faster than the transport (v mt), the ratio of surface activities \(\left( {\frac{{a_{\text{ox}} }}{{a_{\text{red}} }}} \right)_{\text{interface}}\) corresponds to the electrode potential E given by the Nernst equation and the system is electrochemically reversible. When the transport is faster than the charge transfer, the Nernst equation is not obeyed and the system is electrochemically irreversible
For voltammetric analysis, it is important to note that the rate of charge transfer v ct, and the rate of mass transport v mt can be, in certain limits, changed by purpose. The transport rate can be changed by the hydrodynamic conditions (rate of stirring the solution, rate of rotating the electrode, drop time of the DME, etc.). The rate of charge transport can be only changed by changing the chemistry (electrocatalysis, see later). From this follows that the characterization of an electrochemical system as reversible or irreversible, is nothing absolute, but it depends on the experimental conditions. We will come back to this at a later stage.
The early times of direct current polarography with the dropping mercury electrode
As already mentioned, the first voltammetric technique for analytical application was polarography, invented by Jaroslav Heyrovský (1890–1967), who has received for this the Nobel Prize for Chemistry in 1959. Heyrovský used a dropping mercury electrode (DME) as working electrode. The DME consists of a glass capillary (about 10 cm length, 0.5 cm outer diameter, 20–50 µm inner diameter) which is fitted to a tube connecting it to a mercury reservoir which was placed about 50 cm above the level of the glass capillary orifice, where the mercury constantly drops out (Fig. 8). Depending on the inner diameter of the glass capillary and the height of the mercury reservoir (i.e., the height of the mercury column), the lifetime of the mercury drops (the time they hang at the orifice) may vary between some fractions of a second up to minutes, but normally 1–5 s. The mercury droplet is connected to the electrochemical circuit by a platinum wire dipping in the mercury reservoir. This kind of free dropping mercury electrodes is now obsolete and was substituted by a new generation of mercury electrodes, which allow controlling the outflow of mercury from the capillary [see further down the paragraph “The Multi-Mode electrode (static mercury drop electrode)”].

The orifice of the glass capillary with a mercury drop
Heyrovský used a two-electrode cell with the DME as working electrode and a mercury pool on the bottom of the cell as auxiliary electrode. The latter has the advantage that it has a rather constant potential when the electrolyte solutions contain halide ions, like chloride. The measuring equipment was assembled from a lead–acid battery (as voltage source), a resistor wire, and a mirror galvanometer for current measurements. The resistor wire was coiled around an insulating cylinder that rotated driven by a clock-work, so that a metal contact moved along the resistor wire as to vary the applied voltage. Coupled with the cylinder holding the resistor was a cylinder holding photographic paper (containing light-sensitive silver halides), so that the light beam of the galvanometer could “write” the measuring curve by passing through a long slit of the housing. The entire instrument, called polarograph, was the first self-recording analytical instrument. Figure 9 shows a schematic drawing of the instrument. The linear function of electrode potential versus time used in this kind of polarography (voltammetry) is shown in Fig. 10. This technique was called direct current polarography (DCP) [or now, more generally, linear scan (or sweep) voltammetry (LSV)].

Scheme of a Heyrovský polarograph: AB is the resistor wire coiled around a cylinder. C is the sliding contact, F is the housing of the cylinder with the photographic paper, G is the mirror of the galvanometer, L is the light source, N is the nitrogen inlet for deaerating the solution (because oxygen must be removed as it is reducible and would produce undesirable currents), K the glass capillary, R is the circuit to connect the galvanometer, S is the slit through which the light beam of the galvanometer passes to the photographic paper. As illustrated, the glass capillary was connected via a tube with the mercury reservoir. Reproduced with permission from [13]

Potential versus time dependence for direct current polarography (voltammetry)
The curves recorded with such polarograph showed characteristic current oscillations due to the dropping out of the mercury: during each drop life the current grows, and abruptly decreases when the drop falls down, because the next drop at the orifice is substantially smaller. This happens both with the capacitive and the faradaic currents. When ions or molecules are present in the solution, which can be reduced or oxidized, wave-like current signals are recorded. Figure 11 shows a historical curve with the waves of several metal ions. For a better understanding of polarographic curves, we shall use the modern notation, i.e., drawing the potential axis from negative potentials on the left side to positive potentials of the right side, and giving reduction currents with a negative sign, and oxidation currents with a positive sign.

A polarographic curve recorded with a Heyrovský polarograph. On top, the voltage between the two electrodes is given. The element symbols stand for the ions giving the respective waves. The potential of the DME is increasingly negative going from left to right. There is no current scale given; the reduction currents are increasing from bottom to top. Reproduced from [14]. (Copied by permission from the “Electrochemistry Encyclopedia” (http://knowledge.electrochem.org/ed/dict.htm) on “March/30/2015.” The original material is subject to periodical changes and updates.)
At a DME, the surface area of the mercury drop grows proportional to t 2/3 (t is time). This follows because the outflow of mercury is constant and the drop has the shape of an almost perfect sphere. The faradaic current at a growing drop is proportional to t 1/6, when it is limited by diffusion only (cf. Fig. 12). This proportionality was derived by Dionýz Ilkovič, a coworker of Heyrovský. The capacitive current at a growing mercury drop is proportional to t −1/3, i.e., the capacitive current drops during the life of a drop, whereas the faradaic current grows (see Fig. 12). When the current is continuously measured during the entire life time of the drop, it is obvious that the sum of both currents has a large capacitive contribution in the early period of drop life, and a relative small capacitive contribution at the end of the life time. This sparked the idea to measure the current only during a relative short time period at the end of the drop life (Fig. 13): the result is that the measured current will then have a more favourable ratio of \(\frac{{I_{\text{f}} }}{{I_{\text{c}} }}\). This was the first instrumental approach to discriminate the capacitive current in a voltammetric measurement. The technique is called “current sampled polarography”. Normally, the average current measured during those time intervals at the end of each drop will produce one data point, so that the entire polarogram consists of a series of data points.

Dependence of the faradaic and capacitive currents on time during the life time of a growing mercury drop

Current sampling at a growing mercury drop for discriminating the capacitive current
Figure 14a shows schematically the polarogram of an acidic solution when using a DME (the oscillation due to the dropping is neglected). At positive potentials, the mercury (electrode) is oxidized and causes an oxidation current. At rather negative potentials the protons of the solution are reduced to hydrogen. These two currents limit the range in which other signals can be measured. Fortunately, the proton reduction (hydrogen evolution) is strongly retarded on mercury, so that the potential window for measurements is rather large (depending on the electrolyte and current range, it can be up to 2.5 V, more realistic is a range of 1 to 1.5 V). Figure 14b shows the polarogram when a substance A is present in the solution, and A can be reduced to B. The wave shape of the reduction signal of A can be understood as follows: when the electrode potential is not sufficiently negative to drive the faradaic reaction, no current can flow. When the potentials reach values at which, according to the Nernst equation

a Schematic drawing of a polarogram showing the base currents in a acidic solution using a DME. b Polarogram of the same solution, when a reducible compound A is present in the solution
A needs to be converted to B, and a reduction current flows. The magnitude of this current increases with decreasing electrode potential because the potential accelerates the reduction. The potential acts like an increasing activation energy. However, the current grows only up to a limiting value (I lim) because each mercury drop is exposed to fresh solution with the bulk concentration of A, and the diffusion layer (see Fig. 4) grows during each drop life to the same thickness. The two effects (i) increasing of current magnitude with decreasing potential, and (ii) diffusion limitation result in a current wave. This is typical for electrodes with a continuous mass transport towards the electrode surface, and the resulting voltammograms are called steady-state voltammograms (polarograms). At half-height of the wave, there is a turning point that is called the half-wave potential. This is the qualitative parameter depending on what compound is reduced (or oxidized). In case of electrochemical reversible systems, it is directly related to the standard potential (respectively, formal potential [15, 16] ) of the compound. In rare cases, it can be almost equal to the standard potential.
The long march to improve the ratio of faradaic to capacitive currents
The introduction of the current sampling technique to polarography with the DME allowed to decrease the detection limit from about 10−5 mol L−1 to around 5 × 10−6 mol L−1. The next important steps in decreasing the detection limits have been achieved (i) thanks to instrumental developments of the measuring techniques, (ii) thanks to electrode developments, and (iii) thanks to ingenious chemical ideas. Without caring for the historical pathways, we will briefly discuss the various approaches here. Figure 15 shows a classification of the different approaches.

Classification of methods to affect the ratio of faradaic to capacitive currents. One arrow up (↑) means that this current is increased; two arrows up (↑↑) means strong increase. Arrows down (↓ and ↓↓) mean in the same way a decrease. A horizontal line ( ) indicates that this current is not (or not much) affected
) indicates that this current is not (or not much) affected
The Multi-Mode electrode (static mercury drop electrode)
An important idea was to exchange the continuously dropping mercury electrode (DME), where the surface area continuously changes (\(\frac{{{\text{d}}A}}{{{\text{d}}t}} \ne 0\)) and prompts rather large capacitive currents, by an electrode with an intermitting dropping. This electrode operates with a valve that controls the outflow of mercury in such way that a drop is quickly formed and then hangs for a fixed time, until it is dislodged and a new drop is formed. Figure 17 shows the dependence of surface area on time for a train of drops. Such electrodes are sold under the name Multi-Mode Electrode (Metrohm) (see Fig. 16), Static Mercury Drop Electrode (Princeton Applied Res.), Controlled Growth Mercury Electrode (BASi).

The Multi-Mode electrode of the Metrohm company. a photo, b cut. This figure has been made using figures provided by Deutsche Metrohm GmbH & Co. KG
The basic idea of this electrode has been published by Gokhshtein [17], a Russian scientist, already in 1961, but it needed some decades and electronic progress to make a commercial system. As Fig. 17 shows, each drop is hanging for a certain period of time having a constant surface area. If a measurement is made during that time span, it has no contribution of \(\frac{{{\text{d}}A}}{{{\text{d}}t}}\) to the capacitive current, as this differential is zero. However, if a linear function of electrode potential versus time (Fig. 10) is still used, there is also still the capacitive current caused by \(\frac{{{\text{d}}E}}{{{\text{d}}t}}\), as this is not zero. If one likes to reduce that capacitive current, a staircase potential ramp (Fig. 18) has to be used. This kind of voltammetry (polarography) is called staircase voltammetry (polarography). When potential steps where \(\frac{{{\text{d}}E}}{{{\text{d}}t}} = 0\) are synchronized with the periods of drop life where \(\frac{{{\text{d}}A}}{{{\text{d}}t}} = 0\), in principle both sources of capacitive currents are suppressed. This is only true to some (although large) extent, because the stepwise growth of potential also means that \(\frac{{{\text{d}}E}}{{{\text{d}}t}}\) is very large from one step to the other (the potential rise happens with a rate around 1 MV s−1). With such combination of a Multi-Mode Electrode with Staircase Voltammetry, very low detection limits can be achieved (around 10−7 mol L−1); however, this depends on a very proper functioning of the electrode and measuring instrument. It should be remarked here, that with the development of digital instrumentation, all linear potential sweeps are realized by staircase functions.

The surface area as function of time for a Multi-Mode electrode (static mercury drop electrode)

Staircase potential ramp for staircase voltammetry
We have now to understand the dependence of the faradaic and capacitive currents following a potential step when the electrode surface area is kept constant. This is essential for all “potential step techniques”, i.e., Staircase Voltammetry and the pulse techniques discussed further down. Figure 19 depicts these dependencies. The t −1/2 dependence (see Eq. 4) is only observed for planar diffusion (planar electrode with dimensions much above the µm range) and for an electrochemical reversible system (see further down).

Dependence of faradaic and capacitive current at an electrode with constant surface area following a potential step. On the right side, the time t m is indicated during which the faradaic current is measured with minimal contribution of capacitive current
On the right side of Fig. 19 is shown, how a proper choice for the period of current measurement minimizes the capacitive current. This current sampling at the end of the step is essential to benefit from the different current transients. When staircase voltammetry (Fig. 18) is performed with a Multi-Mode Electrode (see above) the diminishing of capacitive currents is due to the behaviour shown in Fig. 19. However, one can still improve the discrimination of capacitive currents, especially, by applying pulse techniques:
Pulse techniques
Many different pulse techniques have been developed. Among them differential pulse voltammetry (DPV) is the most important and the most widely applied [18, 19]. The main idea realized in DPV is to subtract from the current measured at the end of a potential step (cf. Figure 20) the current measured before the step. For this, potential pulses are superimposed to a staircase ramp (Fig. 20)

Left side: superposition of a staircase ramp with potential pulses. Right side: the potential–time function resulting from this superposition. Typical parameters are: pulse duration t pulse = 200 ms, pulse height: E pulse = 20 mV, staircase ramp: t step = 500 ms, E step = 5 mV
The potential–time function shown in Fig. 20 (right side) offers the possibility to subtract the current shortly before the pulses from the current sampled at the end of the pulse. This is depicted in Fig. 21. Each pulse will produce one measuring point according to the equation
with n being the number of the individual pulse. The differential pulse voltammogram consists of discrete ΔI n,dp data plotted as a function of potential. This curve resembles the first derivative of the direct current voltammogram (polarogram), but it should be emphasized that it is NOT simply the mathematical first derivative. Figure 22 shows a schematic differential pulse voltammogram. The peak potential E peak of the signal is almost equal to the half-wave potential E 1/2 in DCP (deviates only due to the pulse amplitude E pulse).

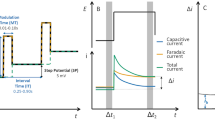
Potential–time function in differential pulse voltammetry. On the right side, the time periods Δt 1 and Δt 2 for current measurements are shown and also the faradaic and capacitive currents before the pulse (dashed lines) and at the end of the pulse (full lines). Note that they have opposite signs because the potential foregoing potential steps go in opposite directions

Differential pulse polarogram (full line) in comparison with the direct current polarogram shown in Fig. 14. The current scale giving ΔI n,dp relates only to the differential pulse voltammogram
Differential pulse voltammetry is so popular because it is possible to determine electroactive compounds down to 10−7–10−8 mol L−1. Here it is appropriate to explain that in voltammetric methods of analysis, detection limits are usually given in mol L−1 because the currents depend on molar concentrations due to diffusion. Therefore, the nature of the electroactive compounds is less important (the different numbers n of electrons involved in the electrode reaction have only a marginal effect on detection limits).
Despite differential pulse voltammetry, so-called normal pulse voltammetry and reverse-pulse voltammetry are other pulse techniques [18, 19]. The latter techniques are very suitable for studies of electrode mechanisms, but less for analytical purposes.
Alternating current voltammetry (ACV)
Alternating current techniques are attractive because one can measure selectively only the capacitive or only the faradaic currents. When an alternating voltage, e.g. a sinusoidal voltage, is superimposed to a linear voltage ramp (or a staircase ramp), the voltage oscillations give rise to an alternating current component. This alternating current can be separated from the direct current, which results from the linear voltage ramp. Measuring the complete alternating current (after rectification) is of no benefit for the detection of faradaic signals, because the ratio \(\frac{{I_{\text{f}} }}{{I_{\text{c}} }}\) is even worse (smaller) than in direct current measurements. However, there are two possibilities to increase \(\frac{{I_{\text{f, ac}} }}{{I_{\text{c, ac}} }}\), i.e. (i) phase-sensitive measurements, and (ii) measurements of higher harmonics.
Phase-sensitive measurements
Figure 23 illustrates the phase relationship between the applied alternating potential and the faradaic and capacitive currents. The faradaic current has a phase lead of 45° with respect to the potential, and the capacitive current has a phase lead of 90°, i.e., between the faradaic and capacitive currents is a phase shift of 45°. The phase shift between capacitive current and potential is well known from the physics of a plate capacitor, where the capacitive current is proportional to the first derivative of potential over time. The phase shift of faradaic current is the result of diffusion (if no reaction rates are involved). Figure 23 shows that a current measurement at 0° is theoretically free of capacitive current; however, the faradaic current is reduced by a factor of \(\frac{\sqrt 2 }{2}\). This reduction of the absolute value of faradaic current is no problem, since the ratio \(\frac{{I_{\text{f}} }}{{I_{\text{c}} }}\) is strongly increased. Figure 24 shows the ac-voltammograms of the reduction of an experimental example (Cd2+ ions) measured at different phase shifts with respect to the applied alternating voltage. At 0°, the faradaic peak is superimposed to an almost zero current background level, which shows that the capacitive current is obviously very small. At 90°, the faradaic peak is superimposed to an appreciable background current, which is clearly the contribution from the capacitive current. Figure 24 also illustrates that real systems do not behave as ideally, as predicted by theory: at 135° and 315°, there should not appear faradaic peaks, as theoretically I f should be zero, but the figure shows small peaks.

Phase shift between faradaic and capacitive currents

Phase-selective measurement of the reduction signal of Cd2+ ions in 5 mol L−1 hydrochloric acid solution. \(c_{{{\text{Cd}}^{2 + } }} = 10^{ - 3} {\text{ mol L}}^{ - 1}\), alternating potential frequency: 100 Hz, alternating potential amplitude: 10 mV. Adopted from [20]
Higher harmonics
Figure 24 shows that the faradaic ac signals have the form of symmetric bell-shaped peaks, i.e., the relation between alternating current and potential is highly non-linear. In cases of non-linear current–potential relationships, there are always current components also at higher frequencies: when the alternating potential has the frequency f (so-called fundamental frequency), the basic ac response is at the same frequency, but there are also currents at 2f, 3f, … etc. The 2f response is called the second harmonic, the 3f response the third harmonic, etc. The shape of the current signals of the \(\left( {n + 1} \right)f\) response ((n + 1)th harmonic) resembles the first derivative of the nf response (nth harmonic). Higher harmonics are well known from acoustics. Many music instruments generate higher harmonics, which are important for the produced sound impression.
Since the first harmonic (f) has the form of a one peak, the second harmonic has two peaks, a maximum and a minimum. The third harmonic has two maxima and one minimum. The higher the harmonic, the smaller are the currents. However, the ratio \(\frac{{I_{\text{f}} }}{{I_{\text{c}} }}\) is more favourable (larger) the higher the harmonics, because the capacitive current produces only very small higher harmonics: The capacitive current is a much more linear function of potential (an almost linear sloping background current) and thus its higher harmonics are exceptional small. It is also possible to perform phase-selective recording of higher harmonics, so that the capacitive currents is reduced by both approaches. Figure 25 depicts the first to third harmonics of phase-sensitive ac-voltammograms of a solution containing cadmium ions: the peak currents decrease with increasing order of harmonics, and the background current reduces tremendously.

Phase-sensitive first to third harmonics of the reduction signal of Cd2+ ions in 1 mol L−1 hydrochloric acid solution. \(c_{{{\text{Cd}}^{2 + } }} = 10^{ - 4} {\text{ mol L}}^{ - 1}\), alternating potential frequency: 156 Hz, alternating potential amplitude: 10 mV. Adopted from [21]
Tensammetry
The relationships depicted in Fig. 23 can also be used to selectively measure capacitive currents, which is very useful in case of compounds which are strongly adsorbing on the electrode surface but not undergoing faradaic transformations (oxidations or reductions). If the compound is hydrophobic (or amphiphilic) it is adsorbed strongly at potentials around the potential of zero charge (pzc), as there the coulombic interaction between the electrode surface and the solution (dipoles and ions) is the weakest. Figure 26 shows schematically the dependence of surface concentration Γ ads of an adsorbed compound on electrode potential. There are two potential ranges (grey underlay in Fig. 26), where the change in surface concentration as function of potential, i.e., \(\frac{{{\text{d}}\varGamma_{\text{ads}} }}{{{\text{d}}E}}\), has maxima. In this ranges, the double layer capacity of the double layer changes dramatically, and this prompts capacitive currents in the form of peaks (Fig. 26, bottom). These peaks can be used to quantify compounds, which adsorb on electrodes. The most suitable electrode for this is a dropping mercury electrode, because (i) mercury has a highly hydrophobic surface at which hydrophobic compounds strongly adsorb, and because (ii) the electrode surface is constantly renewing. The ac voltammetric (polarographic) technique aimed at measuring such adsorption–desorption peaks is called tensammetry, because surface-active compounds (tensides) can be quantified. Although this technique is suitable for some special applications, it suffers from the following drawbacks: First, the peak currents are not a linear function of solution concentration, and, second, when several different adsorbing compounds are present in the solution, there are strong interferences because adsorption is always a competition for a limited number of adsorption sites. Thus, adsorption strength and concentration will affect the results.

Top: dependence of surface concentration Γ ads of an adsorbed compound on electrode potential. Bottom: capacitive currents resulting from adsorption–desorption at potentials where \(\frac{{{\text{d}}\varGamma_{\text{ads}} }}{{{\text{d}}E}}\) has maxima
Square-wave voltammetry (SqWV)
When instead of a sinusoidal alternating potential, a square-wave alternating potential is superimposed to a linear, or better to a staircase ramp, the resulting voltammetry is called square-wave voltammetry [22]. The potential–time function is a train of identical pulses, and by sampling the currents within short periods of all pulses, and subtraction of the currents of the negative pulse from the following positive pulse (or vice versa), again an increase of the \(\frac{{I_{\text{f}} }}{{I_{\text{c}} }}\) ratio results. Square-wave voltammetry is very popular, especially also in stripping voltammetry (next paragraph). Like the ac-voltammetry with sinusoidal alternating potential, it suffers from the fact that irreversible systems give low currents, especially at higher frequencies.
Stripping techniques
Anodic stripping voltammetry (ASV)
The simplest way to increase the ratio \(\frac{{I_{\text{f}} }}{{I_{\text{c}} }}\) is an enrichment (preconcentration) of a compound on the electrode surface. Let us assume copper ions (Cu2+) are present in a solution at a concentration of 10−9 mol L−1. Not any of the previously discussed techniques is able to record a distinct signal of reduction of the copper ions, because that faradaic current is completely obscured by the capacitive current (and noise and other faradaic currents of impurities). The obvious solution of the problem is to electrolyze the solution for some minutes with the working electrode keeping at a potential where the Cu2+ ions are reduced to copper which deposits at the electrode. If the mass transport towards the electrode is kept during reduction at a high rate, e.g. by stirring the solution (i.e., by keeping the concentration gradient steep, as shown in the paragraph “Faradaic currents”), it is possible to accumulate a considerable amount of copper on the electrode. If this copper is then anodically oxidized during a rather fast potential scan, the current (\({d_{\text{q}} }\ /{d_{\text{t}} }\)) is much larger than the non-detectable reduction current. When metals are deposited by reduction and the metal deposit is anodically dissolved, the technique is called anodic stripping voltammetry. Figure 27 shows the potential–time function and the current versus potential curve for the anodic stripping voltammetry of copper ions. In this figure, a simple linear scan is depicted. Due to the enrichment of the oxidizable compound on the electrode, the faradaic current is much higher than in a measurement where the diffusion of the dissolved ions fuels a reduction current. By this simple approach, it is already possible to decrease the detection limit to 10−8 and 10−9 mol L−1. Even lower (10−10 and 10−11 mol L−1) can be reached when the dissolution reaction is recorded with differential pulse voltammetry.

Scheme of the potential–time function and the current versus potential curve for the anodic stripping voltammetry of copper ions
Anodic stripping voltammetry allows easy determination of metals which form amalgams with mercury. In these cases, a stationary mercury drop electrode can be used, or, even with higher sensitivity, so-called mercury film electrodes: when mercury(II) ions are added in rather low concentration to the analyte solution, and when carbon (especially, glassy carbon) is used as working electrode, a “film” of micrometer size mercury droplets is deposited, and simultaneously the analyte metals are deposited on the surface of the mercury droplets, where they usually dissolve and diffuse into the droplets interior. Because these mercury droplets are so small, the metals have short diffusion lengths when oxidized. This enhances the faradaic currents and leads to very narrow peaks of large currents. Anodic stripping voltammetry can be applied especially for the determination of elements, which dissolve in mercury, like Cu, Bi, Pb, Zn, Cd, Sb. However, in recent times, a lot of methods have been developed which use bismuth and carbon electrodes instead of mercury. When metals are deposited in solid form on electrodes, the dissolution signals are often more complicate (multi peaks) because of the different activities of different crystallographic surface planes. However, in recent years, it has been shown that bismuth film electrodes (on glassy carbon) are an attractive alternative to mercury electrodes [23].
Cathodic stripping voltammetry (CSV)
Cathodic stripping voltammetry is a technique where a material deposited on the electrode during the preconcentration step is dissolved by a cathodic, i.e., reductive, process. Ions like chloride, bromide, iodide can be preconcentrated in the form of sparingly soluble salts, like Hg2Cl2, Hg2Br2, Hg2I2, when the potential of the mercury electrode is kept sufficiently positive to oxidize mercury to Hg2 2+ ions, which precipitate these salts:
(Of course, both reactions are equilibria, but the single arrow indicates the dominating direction of the reactions during preconcentration.)
After the preconcentration period, the cathodic reduction of these salts is recorded by scanning the electrode potential from the potential for preconcentration to potentials at which the mercury salt is reduced to mercury and the chloride ions are released to the solution
The sensitivity of these methods depends on the solubility of the deposited salts [24]. The smaller their solubility, the lower are the concentrations which can be detected. This limitation is rather severe, so that only a few cathodic stripping methods are more widely applied: the determination of selenium is a good example. When a solution contains SeO3 2− ions, one has to add Cu2+ ions and can preconcentrate selenium as Cu2Se (copper(I) selenide) because at an appropriate potential the following reactions proceed:
The resulting ions Se2− and Cu+ form the very sparingly soluble salt Cu2Se:
During the reductive scan, a voltammogram is recorded which shows a reduction peak corresponding to the reaction:
i.e., solid Cu2Se is reduced to solid copper metal and the selenide ions are released to the solution.
Adsorptive stripping voltammetry (AdsSV)
The fact that hydrophobic (and amphiphilic) compounds adsorb on electrodes, can be also used for preconcentration. Metal complexes with organic ligands, and also a number of organic compounds can be preconcentrated this way. Usually the adsorbed compounds do not undergo an oxidation or reduction reaction during the preconcentration. Following the preconcentration period, a voltammetric curve is recorded. This has to scan the potentials into positive potentials, when the adsorbed compound can be oxidized, and to negative potentials, when the compounds can be reduced.
As an example, the adsorptive stripping voltammetric determination of Co2+ and Ni2+ ions is given. Every chemistry student learns that dimethylglyoxime (dmg) is a good chelating agent for these ions. The nickel complex is very sparingly soluble in water and used for the gravimetric determination of nickel. At very low concentration, the cobalt and nickel complexes are soluble and, due to the hydrophobicity of the ligand dmg, strongly adsorb on a mercury electrode. The metal(II) ions form a complex in solution according to the reaction:
At the mercury surface, an adsorption equilibrium is established, and adsorption is used to preconcentrate the complex at the surface:
The adsorbed complex is reduced during the voltammetric measurement:
(DHAB = 2, 3-bis(hydroxylamino)butane), In this example [25], both the metal ions and the ligands are reduced. The detection limit is in the nmol L−1 range. It can be decreased by two orders of magnitude by a combination with catalytic currents (see further down).
Another example for the adsorptive stripping voltammetry is the determination of chromium using diethylenetriaminepentaacetic (DTPA) acid as chelating agent (detection limit around 20 nmol L−1 [26]. This method has been later modified, the sensitivity enhanced, and even used for chromium speciation with respect to Cr(III) and Cr(VI) [27].
Although adsorptive stripping methods often allow to achieve extremely low detection limits, they are also prone to severe interferences by other compounds which also adsorb on the electrode.
Cathodic stripping voltammetry following an adsorptive preconcentration
In this technique, metal complexes are preconcentrated on an electrode surface by adsorption, and the metal ions of the adsorbed complexes are then reduced. The latter process may be recorded with differential pulse voltammetry for an additional improvement of the current response (i.e., discrimination of capacitive currents). A good example for this strategy is the determination of copper, lead, and cadmium ions using 8-hydroxyquinoline (oxine) as complexing agent. This allowed to achieve detection limits of 1.2 × 10−10 mol L−1 Cd2+, 3.0 × 10−10 mol L−1 Pb2+ and 2.4 × 10−10 mol L−1 Cu2+ [28].
Adsorptive stripping voltammetry can also be used to determine elements like aluminium(III)—which cannot be reduced in aqueous solutions—when it is bond in a complex by redox active ligands.
Catalytic currents
Catalysis is a very important issue in electrochemistry. As mentioned in the paragraph “Electrochemical reversibility and irreversibility”, there are many electrochemical systems where the rate of electron transfer v ct is smaller than the rate of mass transfer \(v_{\text{mt}}\). When the rate of electron transfer is increased by means of a catalyst, this is called electrocatalysis. There are applications of such kind of electrocatalysis for improving the performance of voltammetric methods of analysis. A nice example is the determination of platinum traces: a platinum complex is adsorbed on a mercury electrode, the platinum ions are reduced to platinum metal, which is a highly active catalyst for the reduction of protons to hydrogen. The resulting current of hydrogen formation is a measure of platinum ions in the bulk solution and allows to analyse Pt concentrations down to 2.5 × 10−13 mol L−1 [29]! This is a result of preconcentration and catalysis.
Catalytic currents can also be generated by homogeneous chemical reactions of the product of electrode reaction so that the educt is recycled. A good example is a solution containing Mo(VI) [30]. Molybdenum(VI) is reduced in acidic solutions first to Mo(V) and at more negative potentials to Mo(III). When the reduction is performed in a solution containing a high concentration of chlorate ions (around 0.1 mol L−1), the following reactions proceed, when the potential range is limited to values of Mo(V) formation:
(For simplicity, we have written here Mo6+ and Mo5+, although ions with such high charges cannot exist as aqua complexes, but only in complexes with oxide ions, hydroxide ions or other ligands.). Equation 24 shows that chlorate is able to oxidize Mo(V) to Mo(VI): from thermodynamic point of view, this means that chlorate must be even easier reducible than Mo(VI), i.e., at even less negative potentials! However, this does not happen for kinetic reasons. The reduction of chlorate is so irreversible, i.e., v ct so slow that it is absolutely not reduced at these potentials. However, in a homogeneous chemical reaction proceeding in the diffusion layer before the electrode, reaction 24 proceeds without any kinetic hindrance. Figure 28 schematically visualizes the situation in the absence and presence of chlorate ions. In the absence of chlorate, Mo5+ diffuses away from the electrode surface, the diffusion layer expands, and the concentration gradient of Mo6+ is not very steep. However, in the presence of chlorate, the Mo5+ is reoxidized to Mo6+ while diffusing away. The result is a very thin diffusion layer, which is called in this case “diffusion and reaction layer” because both processes determine its thickness. The very steep concentration profile of Mo6+ leads to a high current. Although this current originates from the reduction of Mo6+ at the surface, it is in fact fuelled by the high concentration of chlorate ions, which are chemically reduced by Mo5+. One can formulate this as a catalytic cycle, where Mo6+/Mo5+ is the catalyst and chlorate the substrate as shown in Fig. 29. The catalytic current depends on the rate of the reaction given by Eq. 24. When chlorate is at very large excess with respect to the molybdenum concentration, the currents are directly proportional to the molybdenum concentration. This way it is possible to reach detection limits down to 10−10 mol L−1 (and lower). Such catalytic currents are observed especially with transition metal ions, which easily undergo redox reactions, e.g. Mo, W. Ti, Fe, Ni, Pt, etc.

Concentration profile of Mo6+ and Mo5+ at an electrode in the absence of chlorate ions (top graph), and in its presence (bottom graph)

Catalytic cycle showing the regeneration of Mo6+ by oxidation of Mo5+ with chlorate ions
The combination of cathodic stripping voltammetry with catalytic current is also very beneficial in some cases: so it was possible to determine iron down to 8·10−11 mol L−1 in sea water [31]. Similarly, the combination of adsorptive stripping voltammetry and catalytic currents can also be used to decrease the limit of detection: thus, in the presence of the oxidant nitrite (NO2 −), the adsorptive stripping determination of Co2+ (see the paragraph “Adsorptive stripping voltammetry”) has a detection limit in the 10−12 mol L−1 range [32].
Conclusions: the advantages and disadvantages of voltammetric techniques of analysis
This lecture text aimed at demonstrating that voltammetric techniques (VTs) of analysis have one central challenge, i.e., the discrimination of capacitive currents to achieve most favourable ratios of the faradaic to capacitive currents. Only rarely, capacitive currents are used for analytical determinations, in which cases the faradaic currents have to be discriminated.
Combinations of techniques, each discriminating capacitive currents, are especially effective to achieve low detection limits. Examples of such combinations are:
-
(a)
Preconcentration techniques (electrochemical or adsorptive) + differential pulse (or square-wave, or phase-sensitive ac techniques) + static mercury electrode (or mercury film electrode).
-
(b)
Preconcentration techniques (electrochemical or adsorptive) + catalytic currents + differential pulse (or square-wave, or phase-sensitive ac techniques) + static mercury electrode (or mercury film electrode) [33].
Table 2 shows examples of applications of VTs for the determination of elements and compounds.
Speaking about the advantages and disadvantages of VTs, one has to compare them with spectroscopic, chromatographic, electrophoretic, and radiochemical methods, to name just the most important. Then, the following features of VTs should be noted:
-
(i)
VTs can be applied for the quantitative determination of inorganic and organic species (ions and molecules). They are best suited for species which can be either oxidized or reduced on electrodes.
-
(ii)
There are also possibilities (e.g. tensammetry) to determine species, which—under the experimental conditions—do not undergo electron transfer reactions.
-
(iii)
VTs are much better suited to determine inorganic species than organic compounds, because in the analysis of organic compounds only very rarely a single compound is present, and in case of mixtures, the interferences usually circumvent the use of VTs (or need combinations with separation techniques).
-
(iv)
For the determination of inorganic species in aqueous solutions, organic compounds present in the sample have to be completely destroyed (e.g. digested by hydrogen peroxide and UV irradiation). This is especially important in trace analysis using stripping techniques.
-
(v)
It is a great advantage of VTs that they probe the species really existing in solutions. (This is not the case for atomic spectroscopy, where the species present in solution are transferred to a plasma where they are present as atoms.) VTs are therefore suitable for speciation analysis. Speciation analysis determines not only concentration, but also qualitatively identifies different species (different complexes, or different oxidation species); VTs can do that at trace concentrations, e.g. well below 10−6 mol L−1. This is something which only some spectroscopic techniques can achieve in rare cases.
-
(vi)
VTs are primarily used to analyse solutions. Applications to solid samples are possible, but this affords much more sophisticated strategies, and then it is only applicable to main components [34].
-
(vii)
The instrumentation for VTs is rather inexpensive (compared with atomic spectroscopy techniques).
-
(viii)
Instrumentation for VTs can be made so small and lightweight that it can be used in field analysis (portable instruments with batteries)
Abbreviations
- AC:
-
Alternating current
- ACP:
-
Alternating current polarography
- AE:
-
Auxiliary electrode
- ACV:
-
Alternating current voltammetry
- AdsSV:
-
Adsorptive stripping voltammetry
- ASV:
-
Anodic stripping voltammetry
- CSV:
-
Cathodic stripping voltammetry
- DC:
-
Direct current
- DCP:
-
Direct current polarography
- DCV:
-
Direct current voltammetry
- DME:
-
Dropping mercury electrode
- DPP:
-
Differential pulse polarography
- DPV:
-
Differential pulse voltammetry
- EDL:
-
Electrochemical double layer
- IHP:
-
Inner Helmholtz plane
- OHP:
-
Outer Helmholtz plane
- pzc:
-
Potential of zero charge
- RE:
-
Reference electrode
- SHE:
-
Standard hydrogen electrode
- SMDE:
-
Static mercury electrode
- VT:
-
Voltammetric technique
- WE:
-
Working electrode
References
Bond AM (1980) Modern polarographic methods in analytical chemistry. Marcel Dekker, New York
Wang J (2006) Analytical electrochemistry. 3rd edn. WILEY-VCH, Hoboken
Bockris JO’M, Reddy AKN (1970) Modern Electrochemistry. 2nd edn. vol 1, 2a and 2b (with Gamboa-Aldeco M). Plenum Press, New York
Scholz F (ed) (2010) Electroanalytical methods. Guide to experiments and applications, Springer, Berlin
Bard AJ, Faulkner LR (2001) Electrochemical Methods. Fundamentals and applications, 2nd edn. Wiley, New York
Bard AJ, Inzelt G, Scholz F (eds) (2012) Electrochemical dictionary, 2nd edn. Springer, Berlin
Westermann R (2001) Electrophoresis in practice, 3rd edn. WILEY-VCH, Weinheim
Görg A (2012) Elektrophoretische Verfahren, In: Lottspeich F, Engels JW (eds) Bioanalytik. Springer-Spektrum, Berlin, pp 169
Pajkossy T (2012) Faraday’s law. In: Bard AJ, Inzelt G, Scholz F (eds) Electrochemical dictionary, 2nd edn. Springer, Berlin, p 363
Stock JT (1991) Bull Hist Chem 11:86–92
Bard AJ, Faulkner LR (2001) Electrochemical Methods. Fundamentals and applications. 2nd edn. Wiley, New York, p 162
Inzelt G (2010) Kinetics of electrochemical reactions. In: Scholz F (ed) Electroanalytical methods. Guide to experiments and applications. Springer, Berlin, p 33
Heyrovský J (1960) Polarographisches Praktikum, 2nd edn. Springer-Verlag, Berlin, p 33
Heyrovský M. Jaroslav Heyrovský and polarography. In: Electrochemistry encyclopedia. http://knowledge.electrochem.org/ed/dict.htm
Inzelt G (2015) ChemTexts 1:2
Scholz F (2010) Thermodynamics of electrochemical reactions. In: Scholz F (ed) Electroanalytical methods. Guide to experiments and applications. Springer, Berlin, p 11
Scholz F (2012) Gokhshtein Yankel Peysakhovich. In: Bard AJ, Inzelt G, Scholz F (eds) Electrochemical dictionary, 2nd edn. Springer, Berlin, p 425
Stojek Z (2010) Pulse Voltammetry. In: Scholz F (ed) Electroanalytical methods. Guide to experiments and applications. Springer, Berlin, p 107
Molina A, Gonzalez J (2015) Pulse voltammetry in physical electrochemistry and electroanalysis. In: Scholz F (ed) Monographs in electrochemistry. Springer, Berlin
Bond AM (1980) Modern polarographic methods in analytical chemistry. Marcel Dekker, New York, p 327
Bond AM (1980) Modern polarographic methods in analytical chemistry. Marcel Dekker, New York, p 297
Mirčeski V, Komorsky-Lovrić Š, Lovrić M (2007) Square-wave voltammetry. In: Scholz F (ed) Monographs in electrochemistry. Springer, Berlin
Wang J (2005) Electroanalysis 17:1341–1346
Scholz F, Kahlert H (2015) ChemTexts 1:7
Baxter LAM, Bobrowski A, Bond AM, Heath GA, Paul RL, Mrzljak R, Zarebski J (1998) Anal Chem 70:1312–1323
Golimowski J, Valenta P, Nürnberg HW (1985) Fresenius Z anal Chem 322:315–322
Scholz F, Lange B, Draheim M, Pelzer J (1990) Fresenius J Anal Chem 338:627–629
van den Berg CMG (1986) 215:111–121
Wang J, Czae M-Z, Lu J, Vuki M (1999) 62:121–127
Pelzer J, Scholz F, Henrion G, Heininger P (1989) Fresenius Z Anal Chem 334:331–334
van den Berg CMG, Aldrich AP (1998) Electroanalysis 369–373
Vega M, van den Berg CMG (1997) Anal Chem 69:874–881
Czae M-Z, Wang J (1999) Talanta 921–928
Scholz F, Schröder U, Gulaboski R, Doménech-Carbó A (2015) Electrochemistry of immobilized particles and droplets, 2nd edn. Springer, Berlin
Author information
Authors and Affiliations
Corresponding author
Glossary
- Amperometry
-
Measurement of current (usually at a constant electrode potential)
- Analyte
-
Compound to be determined in an analysis
- Auxiliary electrode
-
In an electrochemical cell with a working electrode and an auxiliary electrode (and possibly a reference electrode), the latter only serves to close the electric circuit. The auxiliary electrode does not limit the overall current
- Capacitive current
-
Current due to the charging (or discharging) of the electrochemical double layer
- Charge transfer
-
The crossing of an interface by charge (electrons or ions)
- Charge transport
-
The transport of charges (electrons or ions) inside a phase
- Charging current
-
=capacitive current
- Chronoamperometry
-
Measuring the current at an electrode as function of time (usually at a constant potential)
- Chronocoulometry
-
Measuring the charge flowing through an electrode as function of time (usually at a constant potential)
- Chronopotentiometry
-
Measuring the potential of an electrode as function of time (either at constant current or zero current)
- Conductometry
-
Measurement of the conductivity of a phase
- Coulometry
-
Measurement of the conductivity of a phase
- Cyclic voltammetry
-
Measurement of current through an electrode when the electrode potential is periodically cycled between two potentials (usually with constant potential scan rate)
- Electrode
-
The term “electrode” has different meaning;
(a)The electronic conductor at which electrochemical reactions take place (e.g. a platinum wire)
(b)The half-cell consisting of an electron and an ion conductor having a common interphase (e.g. a silver wire and its adjacent electrolyte solution containing silver ions)
- Electrochemical cell
-
An electrochemical cells consist of at least two electron conductors (usually metals) in contact with ionic conductors
- Electrochemical double layer (EDL)
-
Capacitor formed at the interphase of two phases, esp. of that formed at the interface of an electron conductor and ion conductor (electrode)
- Electrode of second kind
-
Metal electrode (\( {\text{Me}}^{n + } /{\text{Me}} \)) where the activity of \( {\text{Me}}^{n + } \) is fixed by the solubility of a sparingly soluble salt of \( {\text{Me}}^{n + } \)
- Faradaic current
-
Current due to the oxidation or reduction of a compound on an electrode; generally following the Faraday law
- Galvanic cell (galvanic element)
-
An electrochemical cell in which reactions occur spontaneously at the electrodes when they are connected externally by a conductor
- Galvanostat
-
A galvanostat is a constant current device, an amplifier which sends a constant current through an electrochemical cell via a counter electrode and working electrode
- Impedance
-
Complex ratio of the voltage to the current in an alternating current (ac) circuit
- Impedance spectrometry
-
Measurement of (under equilibrium or steady-state conditions) of the complex impedance Z of the electrochemical system as a function of the frequency, f, of an imposed sinusoidal perturbation of small amplitude
- Inner potential
-
\( \phi_{\alpha } \) within the phase α is related to the electric field strength E in the interior of the phase by \( - \Delta \phi = E \)
- Polarography
-
Voltammetry with a dropping mercury electrode
- Potential of zero charge (pzc)
-
The potential corresponding to zero electrode charge
- Potentiometry
-
Measurement of the potential of an electrode against the potential of a reference electrode
- Potentiostat
-
A potentiostat is an electronic amplifier which controls the potential drop between an electrode (the working electrode) and the electrolyte
- Reference electrode
-
Electrode with a very constant and stable potential. Frequently “electrodes of the second kind”
- Potentiometry
-
Measurement of the potential of an electrode against the potential of a reference electrode
- Voltammetry
-
Technique in which the current at an electrode is measured as function of electrode potential
- Working electrode
-
The electrode at which the current is measured
Rights and permissions
About this article

Cite this article
Scholz, F. Voltammetric techniques of analysis: the essentials. ChemTexts 1, 17 (2015). https://doi.org/10.1007/s40828-015-0016-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40828-015-0016-y