
Highlights
-
The latest advancements in Cu-based catalysts for photocatalytic and electrocatalytic CO2 reduction into C2+ products are reported.
-
The relationship between the Cu surfaces and their efficiency in photocatalytic and electrocatalytic CO2 reduction is emphasized.
-
The opportunities and challenges associated with Cu-based materials in the CO2 catalytic reduction applications are presented.
Abstract
Carbon dioxide conversion into valuable products using photocatalysis and electrocatalysis is an effective approach to mitigate global environmental issues and the energy shortages. Among the materials utilized for catalytic reduction of CO2, Cu-based materials are highly advantageous owing to their widespread availability, cost-effectiveness, and environmental sustainability. Furthermore, Cu-based materials demonstrate interesting abilities in the adsorption and activation of carbon dioxide, allowing the formation of C2+ compounds through C–C coupling process. Herein, the basic principles of photocatalytic CO2 reduction reactions (PCO2RR) and electrocatalytic CO2 reduction reaction (ECO2RR) and the pathways for the generation C2+ products are introduced. This review categorizes Cu-based materials into different groups including Cu metal, Cu oxides, Cu alloys, and Cu SACs, Cu heterojunctions based on their catalytic applications. The relationship between the Cu surfaces and their efficiency in both PCO2RR and ECO2RR is emphasized. Through a review of recent studies on PCO2RR and ECO2RR using Cu-based catalysts, the focus is on understanding the underlying reasons for the enhanced selectivity toward C2+ products. Finally, the opportunities and challenges associated with Cu-based materials in the CO2 catalytic reduction applications are presented, along with research directions that can guide for the design of highly active and selective Cu-based materials for CO2 reduction processes in the future.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Over the past few years, there has been a significant increase in the global energy consumption rate, resulting in massive use of fossil fuels such as natural gas, oil, and coal, which presently account for approximately 85% of primary energy supply [1,2,3]. Since fossil fuels, a non-renewable resource, are finite, their overconsumption has caused energy shortage issues. Moreover, the burning of fossil fuels is connected with excessive emissions of hazardous gases, including NOx, SO2, and CO2. CO2 is one of the primary gases that contribute to greenhouse effect, which results in rising global temperatures, ocean acidification, climatic variation, and so on [4,5,6]. Therefore, the mitigation of CO2 emissions is critical in tackling the issues of global warming and energy depletion. In this context, numerous research studies have focused on developing effective ways for carrying out the artificial conversion of CO2 in order to address these issues. It is intended to utilize CO2 as a carbon feedstock in order to produce more value and useful hydrocarbon products through catalytic reactions. These products may then be used in a variety of industrial processes. These initiatives are crucial for promoting a sustainable approach toward energy production while also lowering the harmful impacts of carbon emissions on the environment.
As a chemical feedstock, CO2 has attracted the attention of researchers across several fields, including thermocatalytic, electrocatalytic, and photocatalytic CO2 conversion. However, the high cost of thermochemical carbon dioxide conversion limits its practical use. As a consequence, researchers are investigating alternative approaches for catalytically converting CO2 into valuable products while reducing the cost of CO2 conversion, such as wind and solar energy. Sustainable CO2 conversion methods that use renewable energy sources at room temperature, such as photocatalytic CO2 reduction reactions (PCO2RR) and electrocatalytic CO2 reduction reactions (ECO2RR), have the potential to be more practical and cost-effective alternatives for CO2 conversion [7, 8]. These methods differ in the source of electrons involved in the catalysis process, and the mechanisms by which this conversion is achieved are distinct: Photons are the primary source of electrons in photocatalysis, while an external electric field drives electrons in electrocatalysis.
Although the methodologies and underlying principles of PCO2RR and ECO2RR processes are different, their nature is essentially identical. These approaches employ catalysts to transform CO2 into high value-added products; nevertheless, significant challenges remain in terms of CO2 conversion efficiency and the selectivity of resulted products due to the inert nature of CO2 molecules and the intricate nature of the process. This outcome arises from the thermodynamic stability and chemical inertness of CO2, which is a linear molecule with completely oxidized carbon and an average C=O double bond energy of up to 804.4 kJ mol−1 (at 298 K) that requires substantial energy to break its carbon–oxygen (C–O) bonds [9, 10]. The thermodynamically stable nature of CO2 makes it challenging to catalyze the CO2 conversion process, and the inertness of its molecular structure limits the number of catalytic sites and affects the reaction selectivity, leading to low yields of value-added products. Additionally, the complexity of the CO2 conversion process makes it difficult to optimize conditions for high activity and product selectivity, which limits the overall CO2 conversion efficiency. These challenges underline the need for improvements in CO2 conversion technologies, particularly in respect to developing efficient catalysts that enhance CO2 adsorption and activation to promote higher activity and selectivity of the process. Meanwhile, finding the right catalyst for each method requires a deep understanding of the underlying mechanism of each approach.
In general, the CO2 reduction reaction (CO2RR) is a complex chemical process that consists of a series of electron transfer steps, hydrogenation, C–C bond coupling, and intermediate compounds. At present, products obtained from CO2RR are often classified into C1 and multi-carbon C2+ compounds. The representative C1 compounds such as methane (CH4), carbon monoxide (CO), methanol (CH3OH), formaldehyde (CH2O), and formic acid (HCOOH) have been extensively researched, whereas the formation of C2+ products, including but not limited to ethylene (C2H4), ethane (C2H6), ethanol (C2H5OH), and propanol (C3H7OH), poses a significant challenge because of the complex reaction pathways along with competitive reactions. Table 1 summarizes the possible reactions of CO2RR to C1 and C2+ products and their market prices [11,12,13,14]. Based on the market price point of view, C2+ products are more attractive in comparison with C1 products. Thus, it is effective to produce C2+ through a single process via the CO2RR. Figure 1 displays the annual count of scholarly research articles and review papers published within the timeframe of 2012–2022. The data collected from the Web of Science database indicates an increasing interest among the scholarly community in the topic of photocatalytic and electrocatalytic CO2 conversion into highly valuable C2+ compounds. This highlights the significance of research in this area as it could potentially offer solutions for combating climate change and energy shortage.

The number of published articles between 2012 and 2022 retrieved from the Web of Science database: a photocatalytic CO2 reduction to C2+ products and b electrocatalytic CO2 reduction to C2+ products
Over the recent decades, significant efforts have been made to enhance the selectivity of C2+ products and investigate the various factors that influence their distribution. Copper (Cu) displayed distinctive properties in converting CO2 into C2+ chemicals such as ethylene, ethane, and ethanol, in both photocatalytic and electrocatalytic CO2 reduction [15,16,17,18,19]. Copper is a readily available and cost-effective element that is abundant in the earth’s reserves. It possesses multiple oxidation states, which allow for the formation of various copper-based materials such as cuprous oxide (Cu2O) and copper oxide (CuO). CuO and Cu2O semiconductors possess narrow bandgaps of 1.7 and 2.2 eV, respectively, making them efficient in capturing visible light energy [20, 21]. Additionally, the sufficiently negative conduction band (CB) position and the ability to effectively adsorb CO2 make these materials efficient photoactive catalysts for CO2 reduction [22, 23]. For instance, several structural factors, including surface states, surface defects, particle size, morphology, and crystal facet have been identified to affect the catalytic performance of Cu-based materials in PCO2RR and ECO2RR [24,25,26,27]. However, the synergy between these factors further complicates the understanding control of CO2RR product selectivity. According to a recent report, the valance state of the Cun+ (0 < n < 2) that is present on the surface of catalysts is a crucial factor in governing C–C coupling in PCO2RR and ECO2RR processes. A recent study by Zhao et al. [27] demonstrated that Cu2+ species present on the catalyst’s surface underwent reduction to Cu+ by photoinduced electrons, creating active sites that captured the in situ generated *CO intermediate and thereby facilitating the subsequent C–C coupling reaction. Nevertheless, maintaining the stability of Cu+ species in aqueous solutions remains a challenging problem, as it is important for prolonging the lifetime of *CO intermediate and enhancing the CO2 reduction into C2+ products. Therefore, understanding the links between the selectivity of end products and the valence state/coordination environment of copper species has been a subject of significant interest. To date, numerous electrocatalysts have been explored for the ECO2RR, each yielding specific reduction products. The types and the number of desired products can be adjusted by fine-tuning the binding energy of adsorbed intermediates, including *CO, *COOH, *CHO, and *COH. For example, when the interaction between the electrocatalyst surface and reduction intermediates is relatively weak, the primary products are CO and formate ions (HCOO−). This occurs because, in cases of weak binding, the C–O bond in these intermediates. Conversely, if electrocatalysts strongly bind *CO intermediates, the production of CO and HCOO− is limited. This is because the *CO intermediate remains attached to the catalyst surface for a more extended period, allowing it to undergo further reduction into other products. Among several catalysts investigated, Cu stands out as a unique metal because it can efficiently produce C2+ products including hydrocarbons and alcohols [28, 29]. The basic explanation for its ability to produce C2+ products is that Cu binds *CO neither too weakly nor too strongly [30, 31]. Nevertheless, the selectivity of bare Cu electrodes for particular products is generally poor, leading to the simultaneous formation of a range of reduction products. On a microscopic level, the underlying reason for Cu’s poor selectivity lies in its moderate binding affinity for most reaction intermediates. Cu2O exhibits similar characteristics to metallic copper in terms of its ability to adsorb and activate CO2 that leads to the promotion of C–C bond coupling and the generation of C2+ chemical compounds. During the ECO2RR process, Cu2O shows rapid surface reconstruction which converts a portion of Cu+ to Cu0. Cu0/Cu+ pairs exhibit a synergistic effect that enhances the *CO adsorption on Cu2O surface, improving the selectivity toward C2+ products [32,33,34].
Despite the significant progress made in photocatalytic and electrocatalytic CO2 reduction, the factors that influence the ability of copper containing catalysts to tune the reaction mechanism and the selectivity toward C2+ products are not yet well understood. Herein, we report the latest advancements in Cu-based materials as catalysts for PCO2RR and ECO2RR with the aim of gaining a deeper comprehension of the structure–activity relationships. The schematic illustration of this review is shown in Fig. 2. The review begins with the fundamentals of photocatalytic and electrocatalytic CO2RR. In particular, we summarize the detailed mechanisms and possible reaction pathways for CO2 reduction to C2+ products on various Cu-based materials. We then introduce the application of Cu-based materials in PCO2RR and ECO2RR to C2+ products. To gain an in-depth knowledge of the factors that impact the catalytic performance of Cu, we have classified copper-based materials into various categories including Cu metal, Cu oxides, Cu alloys, and Cu SAs species. Finally, the main challenges to be resolved and the future perspective for Cu-based materials in converting CO2 into C2+ products are envisioned.

Schematic illustration of the review article
2 Fundamental Understanding of the Mechanisms of CO2 Reduction Reaction
2.1 Basic Principles of Photo- and Electro-catalytic CO2 Reduction
2.1.1 Photocatalytic CO2 Reduction Reaction
PCO2RR presents a promising method for directly using solar energy to transform CO2 into valuable products. Photocatalytic reactors are generally configured in two ways: either using particulate semiconductor photocatalysts suspended in a CO2 gas-saturated solution or using uniformly distributed and immobilized photocatalysts mixed with CO2 gas and H2O vapor on a substrate. In principle, PCO2RR process involves three stages: absorption of light (photons), separation and transfer of charges, and subsequent reactions that take place at the interface (Fig. 3). The overall efficiency of the process is determined by multiplying the efficiency of each step. The application of PCO2RR process faces several challenges. In particular, the simultaneous absorption of light across a wide range of solar spectrum and carrying out oxidation–reduction reactions with a single semiconducting material is difficult. Materials with wide bandgaps, such as titanium dioxide (TiO2) and zinc oxide (ZnO), exhibit photoactivity within the ultraviolet (UV) spectrum, while narrow bandgap semiconductors such as Cu2O show activity in the visible NIR range; however, the band potentials of Cu2O semiconductors are not conducive to mediating both reduction and oxidation reactions simultaneously. As a result, single-component systems have lower efficiency for photocatalysis. In order to address this issue, researchers have attempted to develop heterostructures (in forms of nanowires, nanobelts, nanotubes, nanorods, etc.) [35,36,37,38]. In addition, it is crucial to achieve separation and transfer of spatial charges from the catalyst surface to the reactants for CO2 reduction. Nevertheless, photogenerated electrons and hole may recombine rapidly due to the coulombic attraction and the absence of charge trapping states on the surface of the catalyst, which would lower the charge separation efficiency and consequently hinder the reaction process. Therefore, a small proportion of separated charges will move toward the reactive sites located on the catalyst surface and will take part in the redox reaction, which involves the conversion of CO2 and H2O into diverse oxygenated and carbon-containing compounds. The resulting products are ultimately released from the surface of the semiconductor and separated. Apart from these issues, limitations such as insufficient capacity of active sites to adsorb CO2 inhibit charge accumulation and CO2 activation. Moreover, PCO2RR to C2+ products are hindered by the relatively slower rate of electron transfer, along with the sluggish kinetics involved in the formation of carbon–carbon (C–C) bonds, which may lead to the release of *CO from the surface of the catalyst. The release of electrons occurs before *CO can effectively accept the subsequent electrons, which are necessary for its further reduction into C2+ hydrocarbon products. Therefore, the attainment of efficient synthesis of C2+ hydrocarbons in a photocatalytic system remains a significant challenge. It was reported that Cu2O undergoes a preferential reduction to Cu0 during the photocatalytic process, serving as the dominant Cu species involved in the CO2 reduction reaction [39]. Furthermore, due to excellent electrical conductivity of Cu0, it facilitates the accumulation of photogenerated electrons, thereby increasing the electron concentration. Additionally, the higher-energy electrons generated through localized surface plasmon resonance (LSPR) of Cu can activate the typically unreactive and stable chemical bonds present in CO2. Nonetheless, despite the effectiveness of Cu0 as an active site for photocatalytic CO2 reduction and C–C coupling, the recombination of charge carriers on the semiconductor photocatalyst continues to hinder the accumulation of photogenerated electrons on the Cu0 surface. This limitation ultimately leads to relatively low photocatalytic CO2 reduction activity. Up to the present time, addressing this challenge and effectively bridging the gap between the lower efficiency of multielectron transfer and the sluggish kinetics associated with *CO coupling remains a persistent challenge. Researchers continue to explore and develop strategies to improve the effectiveness of charge transfer and the kinetics of C–C bond formation such as metal/non-metal doping, cocatalyst deposition, heterojunction construction, etc.

Photocatalytic CO2 reduction mechanism on a catalyst surface
2.1.2 Electrocatalytic CO2 Reduction Reaction
ECO2RR, which can be powered by renewable energy sources, is an effective way to achieve carbon neutrality. An electrocatalytic CO2 reduction system typically operates in a three-electrode mode. Specifically, the working electrode and reference electrode are situated at the cathode, while the counter electrode is located at the anode. To isolate the half-reactions and enable ion migration, the two electrodes are separated by an ion exchange membrane. On the anode, H2O molecules are oxidized to produce oxygen and protons, with oxygen being collected as a gas product and the protons traverse the membrane to reach the cathode. The CO2 molecules that are present in the electrolyte are transported to the cathode surface through a combination of convection and diffusion. At the cathode, multiple steps of proton and electron transfer convert the CO2 into desired products through reduction potential (Fig. 4). Nevertheless, CO2 dissolved in the electrolytes has high ohmic and mass transfer resistance because of the distance between cathode and anode, while the cathode has limited CO2 diffusion due to its low solubility. An often employed and advantageous electrolyte is an alkaline aqueous solution, primarily because it exhibits a lower overpotential when compared to its neutral counterpart. When various alkaline aqueous solutions were examined, it was noted that higher current densities were achieved with increasing solution concentrations [40]. Electrochemical impedance spectroscopy further revealed a reduction in cell resistance as a result of increased ionic conductivity with rising concentrations. In their study, Sinton et al. [41] observed a notable 240-mV positive shift of the onset potential when they employed a 10 M KOH electrolyte solution instead of a 1 M KOH solution.

Typical electrocatalytic CO2 reduction system
In general, CO2 solubility can be enhanced by employing organic solvents instead of aqueous solutions as electrolytes. Despite CO2 being a nonpolar molecule, it exhibits considerable polarizability and can form hydrogen bonds with compatible donor solvents. The majority of organic electrolytes are polar solvents, which permits electrocatalytic CO2 reduction over a broader potential range. In addition, using an aprotic solvent-like acetonitrile or dimethylformamide could promote the dimerization of *CO–CO, whereas the production of CH4 is favored in a protic solvent. There has also been research exploring the use of ionic liquids, added to either aqueous or organic solvents, which exhibit good thermal stability, high CO2 solubility, and high conductivity; nevertheless, their high cost and the possibility of cathodic corrosion are the challenges that should be considered.
In electrocatalytic CO2 reduction experiments, some researchers have introduced gaseous CO2 to the cathode, ensuring that an ample amount of CO2 is delivered to the catalyst, even when operating at high current densities. Certain research teams have achieved noteworthy enhancements in the current density for CO2 reduction to C2+ products by employing gas diffusion electrodes (GDEs) [42, 43]. Nonetheless, a notable challenge associated with GDE-type electrolyzers is their reliance on aqueous electrolytes for the collection of liquid products. The inclusion of these aqueous electrolytes can result in the dilution of the liquid products, leading to increased expenses in terms of product recovery and separation. To address the challenge posed by GDE-type reactors, several research groups have investigated the electrocatalytic CO2 reduction in a zero-gap type electrolyzer, which eliminates the requirement for an aqueous electrolyte [44, 45]. For example, Lee et al. introduced an effective strategy called catholyte-free electrocatalytic CO2 reduction (CF-CO2R) and designed to circumvent solubility limitations by incorporating an appropriate amount of water vapor along with gaseous CO2 as a cathode reactant [45]. In this CF-CO2R electrolyzer, water vapor serves as a carrier, facilitating the supply of dissolved CO2 to the cathode by creating a CO2-saturated aqueous film on the catalyst surface. This approach offers the advantage of replenishing consumed CO2 in the film directly from the bulk gas stream, thereby enhancing CO2 mass transfer and improving reaction kinetics. However, it is noteworthy that this configuration only permits the detection of gas products. Therefore, it is crucial to carefully choose a suitable cellular arrangement based on the properties of the catalyst and the nature of the desired products.
In general, electrocatalytic systems have a more compact and flexible structure compared to photocatalytic systems. They also have higher catalytic efficiency due to the continuous transfer of electrons to the working electrode driven by an external bias. Nevertheless, the direct redox reaction on the electrode requires a higher overpotential, resulting in relatively high-energy consumption. Additionally, adjusting the strength and form of the external potential allows for easy control of the selectivity of products derived from CO2 reduction. Meanwhile, the ECO2RR process is not as favorable in aqueous electrolytic systems when compared to competing reactions like the hydrogen evolution reaction (HER) [46]. To improve the selectivity toward C2+ compounds, it is necessary to supress the HER pathway and reduce the rates of C1 products. However, similar to the photocatalytic CO2 reduction systems, the electrocatalytic CO2 reduction mechanism is complex, and it results in the production of C1 hydrocarbons, leading to selectivity issues. The diverse final products arising from this complicated pathway make it challenging to obtain the desired product with good selectivity [12, 14, 47].
2.2 The Pathway of CO2 Reduction to C2+ Products over Cu-based Materials
2.2.1 CO2 Adsorption/Activation and Different Models
To optimize the CO2RR process for increased C2+ product formation, it is essential to understand the reaction intermediates and pathways that are involved. As is well known, the CO2RR processes typically involve three main steps. The initial step entails the adsorption/activation of CO2 on the surface of the catalyst, resulting in the production of CO2· ̵ anion radicals (Eq. 1 in Table 1). The second involves a sequence of complex reactions that are dependent on the transfer of e−/H+, leading to the production of precursors such as *CO, *COH, and *CH2, or effecting C–C coupling to produce C2+ chemicals as listed in Table 1 (Eqs. 2–11 in Table 1). Finally, in the third step, the formed products are desorbed from the surface of the catalyst. CO2 is a highly stable molecule with a linear symmetry structure and its direct reduction in an aqueous solution to form CO2· ̵ anion radical through single-electron transfer process with a standard redox potential of − 1.9 V versus NHE is thermodynamically unfavorable. Unlike the activation of CO2 molecule over heterogeneous catalysts through surface atom interactions can effectively minimize the energy required to accept an electron by lowering the lowest unoccupied molecular orbital (LUMO) level of CO2. This can be achieved through the alteration of the molecular properties, including the elongation of C–O bond length and bending of O–C–O angle [48,49,50,51]. Generally, the CO2 adsorption/activation step to form CO2· ̵ anion radical is considered the rate-limiting step for CO2RR process. As of now, several models for CO2 adsorption on catalyst surfaces have been reported using different types of catalysts, including two main theories that focus on the use of either metals or metal oxides as catalysts [52,53,54]. At the molecular level, CO2 activation occurs through a partial transfer of electrons into the LUMO, resulting in the generation of partially charged CO2δ− species [55, 56]. Based on metal catalysts, five configurations for CO2 adsorption and activation were proposed as depicted in Fig. 5a. The structure of CO2δ− species varies according to the adsorption mode, namely, carbon coordination, oxygen coordination, and mixed (carbon/oxygen) coordination. Carbonate-like species ensue from the carbon binding mode wherein the carbon atom serves as electron acceptor for Lewis base centers. Dual bidentate species emerge through oxygen coordination, with two alternative structures. In this model, the oxygen atoms act as electron donor for surface Lewis acid centers owing to the pair electrons. Meanwhile, structures of both species occur in the mixed coordination mode. According to this model, CO2 molecules serve as both an electron donor and acceptor by oxygen and carbon atoms, respectively.

The possible configurations of adsorbed CO2 on: a metal catalysts and b metal oxide catalysts
As for the metal oxide surfaces, CO2 activation can be accomplished by forming coordination bonds with adjacent metal sites, either through the carbon atom or the terminal oxygen atoms of the CO2 molecule, resulting in the formation of monodentate or bidentate carbonate species (Fig. 5b) [48, 57]. The first two configurations show the formation of monodentate species, in which either the oxygen or carbon atom coordinates with a metal atom of the metal oxide by forming M–O or M–C bonds. In the third configuration, the CO2 molecule binds to the metal oxide surface through interactions between both oxygen with metal atom and carbon with oxygen atom of the metal oxide. The fourth configuration depicts a mixed oxygen/carbon coordination, resulting in a bridged carbonate geometry, in which two oxygen atoms bind with two metal atoms, and the carbon atom of CO2 molecule points downward. The last configuration presents a bridging geometry through oxygen coordination, where two oxygen atoms interact with two metal atoms and the carbon atom points upward. These differed binding configurations might result in many intermediates, leading to various reaction pathways. This highlights the importance of understanding the CO2 adsorption/activation steps in CO2RR processes.
The surface structure of metal oxides plays a key role in CO2 adsorption/activation process. Different structures may have different active sites and surface energies that can affect the strength of the interaction between CO2 molecules and metal oxides. As example, the CO2 adsorption on CuO oxide surfaces, namely, (0 1 1), (1 1 1), and (− 1 1 1), was notably strong only on the (0 1 1) surface. Conversely, a weak CO2 adsorption was observed on CuO (1 1 1) and CuO (− 1 1 1) surfaces [58]. As reported in the literature, oxygen vacancies Vo could change the physico-chemical and electronic properties of metal oxides such as TiO2 [59,60,61], CeO2 [62], In2O3 [63], Zn2GeO4 [64], and Cu2O [65, 66] leading to improved surface adsorption and the creation of additional active centers. Zheng and co-workers reported that copper oxide nanodendrites with partially reduced surfaces and abundant Vo (CuOx–Vo) exhibited improved CO2 adsorption and electroreduction abilities when compared to pure Cu and Vo-free CuOx (Fig. 6a and b). The surface Vo has been identified as effective Lewis base sites for the enhancement of CO2 adsorption. Theoretical calculations showed that CuOx–Vo provides strong binding affinities toward *COH and *CO intermediates, while displaying weak affinity toward *CH2, resulting in efficient production of C2H4 with high Faradaic efficiencies reaching 63% (Fig. 6c–e). Moreover, it was demonstrated that the Faradaic efficiency for C2H4 production is greatly influenced by the density of Vo in CuOx [67]. This highlighting the potential of controlling and engineering Vo defects to create more effective catalytic materials for CO2 adsorption/activation and to adjust the selectivity toward desired products in CO2RR. Aside from surface defects, the deposition of basic sites such as alkaline or alkali-earth metals on the catalyst surface can promote the CO2 adsorption owing to the strong interaction with acidic CO2 molecules. Also, increasing the catalyst’s surface area could provide more active sites for CO2 adsorption [68]. Furthermore, surface modification involving the addition of functional groups, such as hydroxyl (OH) or amino (NH2) groups, has been observed to exert significant effects on the interaction between CO2 and the surface, thereby improving its adsorption [69, 70].

a VO-rich CuOx surface via electrochemical control of oxygen vacancies. b Schematic illustration of ECO2RR into C2H4 on Vo-rich CuOx–Vo surface. DFT calculation results with three ECO2RR intermediates (*CO, *COH, and *CH2) of c pure Cu, d Vo-free CuO, and e CuOx–Vo. Reproduced with permission [67]. Copyright 2018, Wiley–VCH
2.2.2 The Pathway of CO2 Reduction to C2+ Products
As illustrated in Table 1, the CO2RR process includes a complex reaction mechanism, which gives rise to the production of multiple products. Consequently, obtaining the desired product becomes challenging due to selectivity concerns. The determination of the favored reaction pathway leading to the wanted product is intricately dependent on the adsorption energy of the intermediates at the active site of the catalyst. As an example, the formation of HCOOH from the key intermediate *OCHO or *COOH depends on the type of adsorption model type resulted from the hydrogenation of the CO2· ̵ anion radical adsorbed onto the catalyst surface [71]. As shown in Fig. 5, *COOH is also a key intermediate in the generation of *CO, which subsequently undergoes desorption to yield the gaseous product CO [72]. The formation of C2+ chemicals requires the exchange of a greater number of electrons in comparison with C1 products, and the subsequent coupling of C–C bonds is deemed to be the step that governs the reaction rate, making this process difficult to occur kinetically. *CO is a key intermediate as it plays a significant role in the C–C coupling reaction, which leads to the generation of C2+ products [7, 73]. It has been reported that *COCHO plays a crucial role as an intermediate in the production of C2+ products, which may be formed through various coupling reactions, such as the coupling of CO and *CHO (*CHO + CO → *COCHO), the coupling of CO and *COH (*COH + CO → *COCOH), or coupling *CO (*COCO + H+ → *COCHO) [73, 74]. Compared to *COCOH, the *COCHO intermediate is more stable as it lacks a double bond to the active site or a free radical on the carbon atom. As illustrated in Fig. 7, the ethylene pathway involves the hydrogenation of *COCHO to *COCHOH, followed by its conversion to *OCH2COH. This ultimately yields both and ethylene (C2H4) and acetic acid (CH3COOH). In the ethanol pathway, the reaction mechanism involves the conversion of *COCHO to glyoxal (C2H2O2) through a one-step hydrogenation process. This glyoxal can be further transformed into either acetaldehyde (CH3CHO) or ethanol (CH3CH2OH) based on the potential applied to the reaction. A lower potential favors the production of acetaldehyde, whereas at higher potential, the reaction yields ethanol [74, 75]. Furthermore, it is possible to form ethylene, ethane, ethanol, and acetaldehyde products through the carbene route, which entails the generation of CO* as a major intermediate compound. The CO intermediate is subsequently fully reduced to “C” and further reduced to form CH2· and CH3· radicals. If the photocatalyst surface can stabilize the CH2· and CH3· radicals, then they are more likely to couple and form C2+ products. However, if the surface cannot stabilize these radicals, then they will desorb as methane.

General pathways for CO2 reduction reactions. Reproduced with permission [22]. Copyright 2023, Wiley–VCH
A reaction pathway involving the coupling of three carbon atoms is required for the production of C3 products. García et al. utilized DFT calculations to conduct an in-depth analysis of the mechanism involved in CO2 conversion to C3 products. According to their study, C3 products are formed by combining C2 and C1 intermediates [76]. These interactions might occur between C1 (CHxO*, CHx*), and C2 intermediates, which may include hydrocarbons (CHyCHz*) or oxygenates (CHyOCHzO*, CHyOCHz*, and CHyCHzO*). However, the current selectivity for C3 products is still low. Thus, further investigations of the reaction pathways are needed design effective catalysts that can promote the formation of C3 products.
While numerous reaction pathways have been confirmed by theoretical and experimental approaches, the actual CO2RR processes are influenced by many factors, and the products selectivity is more complex. In the case of PCO2RR process, the selectivity to C2+ is particularly intricate and largely influenced by the photoinduced e−/h+ density as well as the stability of the generated intermediates. The C1 products are rapidly generated as the intermediate can easily combine with hydrogen atoms, while the opposite charge of intermediates can obstruct the C–C coupling steps. On the other hand, several factors significantly influence the ECO2RR process’s selectivity, including operating conditions, which consist of the electrolyte pH, the type of electrolyte cation/anion, and the applied overpotential [74]. In addition, both systems can be influenced by the catalyst surface properties. These properties, including surface adsorbates, defects, structure, morphology, and facets, can substantially alter the adsorption energies of critical intermediates as well as the kinetic barriers of reactions, resulting in different reaction routes [77]. Despite the fact that PCO2RR and ECO2RR have certain distinctions, their practical implementation is hampered by comparable hurdles and comparable strategies aimed at increasing the efficiency and the C2+ products selectivity as well as the stability may be used in both systems to overcome the challenges.
3 Fine-Tuning Surface Structure of Cu-Based Catalysts for Improving the Activity/Selectivity of CO2RR Toward C2+ Compounds
3.1 Photocatalytic CO2 Reduction Reaction
Despite the potential of PCO2RR as a promising and sustainable approach to synthesize C2+ compounds, the current literature points out a relatively low efficiency and selectivity of this process in C2+ product formation [78, 79]. This is due to limitations on light absorption, rapid charge carriers recombination that results in low electron concentrations on the photocatalyst’s surface, high kinetic barriers, as well as desorption of C1 intermediates. It is well known that Cu-based photocatalysts are effective at multi-electron transfer and can make use of weakly bound d-band electrons. They also have a short bandgap, allowing them produce sufficient charges through the absorption of sunlight within the range of visible light. Moreover, their strong CO2 adsorption and their ability to effectively stabilize the reaction intermediates contribute to their potential for C2+ product generation. Nonetheless, the development of Cu-based photocatalysts that are highly efficient and selective in generating C2+ products remains a significant challenge. Researchers have been exploring different approaches such as crystal phase/morphology optimization, metal doping, defect engineering, heterostructure fabrication, and bimetallic synergies to enhance the activity/selectivity of PCO2RR toward C2+ compounds; nevertheless, there is still debate surrounding the origin of C2+ selectivity enhancement. In this section, we provide basic comprehension and discussions on these strategies to help with further enhancement of the activity/selectivity toward C2+ compounds in PCO2RR. To attain a more comprehensive understanding of the structure–activity–selectivity relationships, we have categorized Cu-based photocatalysts into four groups: Cu oxides/sulfides, Cu alloys, Cu-based single-atom catalysts (Cu SACs), and Cu-based heterojunctions.
3.1.1 Cu Oxides/Sulfides
Copper oxide photocatalysts are p-type semiconductors with narrow bandgap energy and elevated conduction band values enabling them to convert CO2 into various hydrocarbons [80,81,82]. However, the high charge recombination rate and the poor stability of Cu oxides result in a low efficiency of photocatalytic CO2 reduction [83]. The nano-level structural modification has been widely adopted for Cu oxides to overcome these drawbacks. Xue et al. prepared a dendritic 3D porous Cu2O structure via a method involving electrodeposition as well as a subsequent thermal oxidation [84]. The findings indicated that the 3D porous Cu2O exhibited highly effective photocatalytic performance, with a CO yield of 26.8 μmol g−1 h−1, which was 24 times greater than the CO yield obtained from the non-porous Cu2O structure. The 3D porous Cu2O with nano-sized dendrite structure was demonstrated to promote the separation of charge carriers and transport efficiency as well as the overall mass transfer efficiency of CO2 gas, boosting the photoreduction of CO2 and the anti-photocorrosion properties. Importantly, CH4 and C2H4 products were observed for the 3D porous structure, owing to longer retention time of gas adsorption, as well as abundant active sites and high electron transfer rate offered by the porous structure. The conversion pathways of CO2 to CO, CH4, and C2H4 products are depicted in Fig. 8a. The formation of *COOH intermediates occurred initially from CO2 reduction, which further resulted in the generation of *CO intermediates upon dehydration. The *CO intermediates desorb rapidly to generate CO products, while longer *CO adsorption time favors the production of CH4 and C2H4 compounds. Thus, the distribution of the final product depends on the stability of the *CO intermediate. The absorption of *CO intermediates was facilitated by the presence of Cu+ species, which further undergo C–C coupling steps to produce C2H4 product. This work provided insight for studying the morphology control effect for PCO2RR.

a Schematic illustration of CO2 photoreduction mechanism on 3D porous Cu2O. Reproduced with permission [84]. Copyright 2022, Elsevier. The free energy profiles of CO2 reduction to CO* and HCOOH* on b δ-Cu2S, c β-Cu2S and d relative energy diagram of the CHO*–CO* coupling on δ-Cu2S and β-Cu2S. Cu, orange; S, yellow; C, gray; O, red; and H, white. Reproduced with permission [85]. Copyright 2021, The Royal Society of Chemistry
Another strategy for adjusting the C2+ product selectivity of CO2 reduction is crystal phase control. Recently, Wang et al. demonstrated through DFT calculations that the conversion of CO2 into ethanol (C2H5OH) can be efficiently achieved by both 2D β-phase Cu2S bilayer and δ-phase Cu2S monolayers [85]. Figure 8b and c illustrates the free energy profiles of CO2 reduction on δ-Cu2S and β-Cu2S. Two different pathways were examined, namely, CO2–COOH*–CO* and CO2–HCOO*–HCOOH*. Based on the results, the CO* intermediate was more likely to be stabilized on both surfaces. This occurred because the CO2–COOH* route has a lower energy barrier than the HCOO*–HCOOH* pathway. Specifically, the energy barrier for CO2–COOH* pathway was 0.21 eV on δ-Cu2S and 0.08 eV on β-Cu2S, while the energy barrier for HCOO*–HCOOH* pathway was 0.46 eV on δ-Cu2S and 0.43 eV on β-Cu2S. The results of Bader charge analysis show a significant charge transfer between Cu and S atoms on the surface resulting in the generation of Cu+ sites. This led to the adsorption of CO* and the subsequent coupling of CO* and formyl (CHO*) species, as illustrated in Fig. 8d. Notably, the kinetic barriers associated with CO*–CHO* coupling have been determined to be 0.3 eV, confirming the possibility of CO2 to C2H5OH conversion. These findings offer important theoretical insights for future experimental development and synthesis of photocatalysts aiming at producing C2+ products from CO2.
3.1.2 Cu Alloys
Recent studies have reported that Cu alloys have superior photocatalytic properties compared to single Cu catalysts [15, 37, 86,87,88,89]. The use of Cu alloys can improve the CO2 activation and optimize the binding strength of the key intermediates on catalysts surface, which ultimately promotes the efficient formation of C2+ chemicals via C–C coupling. Therefore, by altering the catalyst composition, the reaction mechanism can be fine-tuned and tailored to C2+ production. For instance, Shankar et al. synthesized Pt–Cu alloys supported on TiO2 nanotubes for photocatalytic CO2 reduction. The hydrocarbon production rate was maximized over Cu0.33–Pt0.67/TiO2 photocatalyst, and CH4, C2H4, and C2H6 products were obtained at rates of 2.60, 0.33, and 0.47 mL g−1 h−1, respectively. Whereas, the monometallic Cu/TiO2 or Pt/TiO2 catalysts and the other Cu–Pt compositions resulted in limited C2+ formation, with CH4 being the major reaction product [37]. This is possibly attributed to the ability of Pt to boost the photocatalytic reduction rates and the role of Cu in promoting the C2+ products selectivity owing to the higher reactivity and strong adsorption of CO on copper surface. This finding underscores the importance of carefully controlling the composition of the photocatalyst to tune the selectivity toward C2+ products. In et al. observed a shift in selectivity from CH4 to C2H6 by CO2 photoreduction under artificial sunlight (AM1.5) using bimetallic Cu–Pt alloys deposited reduced blue titania (Cu–Pt/BT) catalysts. The enhanced C2H6 selectivity was attributed to the effective transfer of high density of electrons from BT to Cu nanoparticles through Pt, along with the high concentration of stabilized CH3· intermediates [78]. The formation of C2H6 product occurs through the reaction of two CH3· radicals via a self-reaction process.
Apart from the previously mentioned limitations regarding the production of C2+ compounds during photocatalytic CO2 reduction, the repulsion between reaction intermediates generated during the process also impede C–C coupling reaction required for the generation of C2+ compounds. To minimize the inter-adsorbate repulsive forces, one can establish nearby reaction sites with opposing charges. In this regard, Shankar et al. synthesized large-sized AgCu nanoparticles supported on TiO2 nanotube array (AgCu-TNTA) [15]. The obtained photocatalyst exhibited total rate of hydrocarbon production (CH4 + C2H6) of 23.88 μmol g−1 h−1 with C2H6 selectivity of 60.7%. In comparison, the Ag-TNTA and Cu-TNTA catalysts showed C2H6 selectivity of 15.9% and 10%, respectively, indicating that the presence of both Ag and Cu in the AgCu bimetallic alloy resulted in a synergistic effect, enhancing the production of C2H6 in PCO2RR process. The synergistic effect was related to the multipolar resonances in large plasmonic AgCu nanoparticles, which enabled the creation of active sites with opposite charges, thus reducing the repulsion between reaction intermediates. Additionally, the stabilization of CH3· radicals was observed due to the ability of both Ag and Cu to stabilize radicals through the promotion of C–C coupling on their surfaces. This occurred through charge transfer, where plasmonic electrons were injected into the TNTAs, leaving behind holes that gave the metals a positive charge. This positive charge increased the lifespan of CH3· radicals. Moreover, the researchers noted that a high concentration of hot spots, where the electric field is particularly strong, may increase the polarization of CO2 molecules and promote the production of C2+ compounds.
Numerous studies reported that defect engineering played an important role in governing both the interface electronic structure and active sites of catalysts. This, in turn, can significantly impact the photocatalytic process [90,91,92]. For instance, Yu et al. fabricated a 2D ultra-thin CuGaS2/Ga2S3 (CGS/GS) with S vacancy, which showed unprecedented selectivity toward C2H4 (≈ 93.87%) with a production rate of 335.67 µmol g−1 h−1 [93]. They found that the mechanism for the multi proton–electron pathway of CO2 reduction reaction is altered by the existence of S vacancy. This occurs because S vacancy triggers a highly delocalized electron distribution, leading to a local metallization between Cu and Ga in the vicinity of the S vacancy and resulting in the formation of Cu–Ga metallic bond (Fig. 9a and b). These bimetallic Cu–Ga dual sites could facilitate the C–C coupling and stabilize the formed intermediates, thereby lowering the energy barrier for C2H4 formation. Also, they noted that the photocatalysts’ selectivity is dependent on the Cu oxidation state. As shown in Fig. 9c, the increase in Cu+/Cu2+ ratio resulted in an increase in C2H4 yield and selectivity owing to the improved thermodynamics of *CO dimerization by Cu+ species. However, when the Cu+/Cu2+ ratio exceeded 2, the yield and selectivity of C2H4 decreased significantly, which was attributed to insufficient Cu2+, resulting in a notable reduction in the adsorption capacity of *CO intermediate. In other words, the catalyst surface underwent a charge distribution rearrangement as a result of introducing S vacancies, which dominantly affects the chemical state of Cu ions. These effects are likely to be beneficial for the production of C2+ compounds. The reaction mechanism has been investigated by in situ Fourier transform infrared spectroscopy (FTIR). Figure 9d shows the presence of signals relative to *COOH, *CO, and *OCCHOH intermediates, indicating that *OCCHOH was a key intermediate of coupling of C–C for C2H4 formation. The DFT results reveal that *COOH intermediates were generated from CO2 reduction, which subsequently underwent coupling with H+/e− pairs to yield CO molecules. Eventually, *CO intermediates transform into C2H4 through complex processes involving electron and proton transfer (Fig. 9e–i). In addition, the formation of *CHOHCO by coupling *CO and *CHOH appears to be the most thermodynamically favorable pathway for C–C bonding formation when compared to other pathways (Fig. 9j). This study uncovered a novel approach to optimize the geometric distance of reactive sites through vacancy engineering for increasing the efficiency/selectivity of CO2 photoreduction into C2+ products.

a ELF of CGS/GS (Left) and ultra-thin CGS/GS with S vacancy (Right). b The calculated Bader charge of CGS/GS (Left) and ultra-thin CGS/GS with S vacancy (Right). c The correlation of C1/C2, C2H4, and selectivity with Cu+/Cu2+, d in situ FTIR spectra on ultra-thin CGS/GS. e Free energy of CO2 on different active sites. f Free energy of H2O on different active sites. g Free energy of CO2 photoreduction to CO. h, i Free energy of different intermediates. j Schematic diagram of CO2RR. Reproduced with permission [93]. Copyright 2023, Wiley–VCH
3.1.3 Cu SACs
Single-atom catalysts (SACs) have become a prominent area of research as they allow the utilization of almost all active metal sites [94,95,96,97]. To achieve this, metal particles are reduced in size and aggregation to form individual atoms with low coordination states. This leads to single-atom SAs having exclusive electronic properties that distinguish them from corresponding bulk materials [98, 99]. For PCO2RR, metal SAs doping into substrates has been studied extensively to alter photocatalysts properties. Metal ions can act as hole trappers which, in turn, facilitate water oxidation and produce a significant amount of protons that can form specific C1 intermediates. This can promote the C–C coupling and help in regulating the selectivity toward C2+ compounds by altering pathway the CO2 photoreduction pathway [100,101,102,103,104,105,106,107].
For instance, Cu-doped semiconductors such as TiO2 and g-C3N4 have been employed as catalysts for PCO2RR. Huang et al. synthesized a catalyst composed of Cu–N4 sites supported by phosphorus-modulated g-C3N4, denoted as CuACs/PCN. The CuACs/PCN photocatalyst exhibited high efficiency in producing C2H4, demonstrating a selectivity of 53.2% and a yield rate of 30.51 µmol g−1. Experimental and theoretical investigations revealed that C–C coupling intermediates could be formed on Cu–N4 sites, and the presence of P in the surrounding microenvironment of CuACs/PCN lowered the energy levels of intermediate reactions [100]. It was found that CuACs/PCN exhibited lower energy barrier in almost all of the stepwise reactions, indicating the significant role played by P in enhancing C2H4 product selectivity. This research study emphasizes the importance of fine-tuning the coordination environment and the surrounding microenvironment of Cu SAs for the efficient formation of C2H4. Moreover, it provides a promising strategy that could be utilized to achieve selective C2H4 production in photocatalytic systems. Wang et al. conducted a study where they integrated Cu SAs into a UiO-66-NH2 support for PCO2RR [101]. They found that the Cu SAs and UiO-66-NH2 interact through the –NH2 groups. The resulting catalyst has the ability to transform CO2 into liquid-phase products, including CH3OH at a rate of 5.33 µmol h−1 g−1 and C2H5OH at a rate of 4.22 µmol h−1 g−1. According to DFT calculations, the integration of Cu SAs resulted in a downshift of the HOMO and LUMO energy levels of UiO-66-NH2, resulting in a narrowed bandgap of the catalyst. Moreover, the Fermi level of Cu/UiO-66-NH2 was lower than the LUMO level of UiO-66-NH2, which enhances the electron transfer efficiency. By comparing the partial density of states (PDOS) of UiO-66-NH2 and Cu/UiO-66-NH2, it was found that the introduction of Cu single atoms can decrease the bandgap of UiO-66-NH2 by shifting its CBM to the Fermi level, which explain the C2+ products formation over Cu/UiO-66-NH2.
Recent research suggested that bimetallic SACs exhibit superior photocatalytic performance compared to monometallic SACs for PCO2RR. This is can be attributed to the synergistic effect between the two metals, which promotes the activation of CO2 and stabilizes the reaction intermediates, thereby promoting the C–C coupling process that ultimately leads to the generation of C2+ products [102,103,104]. In this regard, Huang et al. developed a tandem photocatalysis strategy by combining rhenium-(I) bipyridine fac-[ReI(bpy)(CO)3Cl] (Re-bpy) and copper-porphyrinic triazine framework [PTF(Cu)] to create synergistic dual sites [91]. Under visible light irradiation, this approach effectively generated C2H4 at a rate of 73.2 μmol g−1 h−1. However, using only Re-bpy or PTF(Cu) catalysts resulted in generation of CO under similar conditions, and C2H4 could not be obtained. The tandem photocatalytic system allowed for the CO produced at the Re-bpy sites to be adsorbed by neighboring Cu SAs in PTF(Cu), leading to a synergistic C–C coupling process that resulted in C2H4 formation. Liu et al. used MIL-125(Ti) metal organic framework as a precursor and template to create a cake like porous TiO2with doping of Cu and Co. Results showed that after 3 h of simulated sunlight irradiation in water vapor, CO and CH4 were the main products for both pure TiO2 and 1%Cu/TiO2 photocatalysts [103]. The activity of 1%Cu/TiO2 catalyst was observed to be superior to that of pure TiO2, which can be ascribed to the reduction in the bandgap, thereby facilitating the separation of photoinduced charge carriers. Meanwhile, the introduction of trace Co ions through doping resulted in a shift of main products from CO and CH4 to C2H6, with a small amount of C3H8 also detected. The results showed CH3· radical enrichment over Co–Cu/TiO2 in comparison with Cu/TiO2 sample. Thus, doping with Co ions led to a substantial enhancement in the selectivity of C2+ products. It is noteworthy that the Co/TiO2 photocatalyst did not produce any C2H6, indicating that the cooping of Cu and Co in the catalyst was responsible for the formation of C2+ products. Guo et al. conducted a study wherein they synthesized a photocatalyst composed of polymeric carbon nitride anchored with atomically dispersed Cu and In metals, referred to as InCu/PCN. The samples were synthesized by thermal polymerization method starting from mixed “CuCl2 + urea + In-MOF” precursor as illustrated in Fig. 10a [104]. The findings from the SEM and STEM analyses indicated that the InCu/PCN composed of Cu and In atoms that are uniformly dispersed at the atomic level on PCN nanosheets. Additionally, the results revealed the presence of distinctively paired In-Cu configurations (Fig. 10b–d). Interestingly, a remarkable ethanol production rate of 28.5 μmol g−1 h−1 with a high selectivity of 92% was achieved by InCu/PCN photocatalyst, which is 2.4 times greater than that by Cu/PCN (Fig. 10e–g). The theoretical and experimental results indicated that the interaction between In and Cu serves to improve the separation of charges by accelerating the transfer of charges from PCN to the metallic sites. Additionally, the existence of Cu–N–In bonds allowed In to transfer electrons to Cu sites, resulting in a higher electron density at the copper active sites. The interaction between Cu and In atoms also enhanced the adsorption of *CO intermediates and reduced the energy required for C–C coupling. The improved selectivity toward C2+ products in CO2 reduction reaction can be attributed to the synergistic effects of In–Cu dual-metal sites.

a Schematic illustration of the synthetic process of InCu/PCN, b TEM image with SEM image inset of InCu/PCN sample, c, d AC-HAADF-STEM images of InCu/PCN, CO2 photoreduction over InCu/PCN, Cu/PCN, In/PCN, and PCN, e ethanol production rate, f gas products generation rate, and g product selectivity. Reproduced with permission [104]. Copyright 2022, Wiley–VCH
The strong metal–support interaction can enhance the dispersity and stability of SAs, thus leading to better performance of photocatalysis [108,109,110]. With this thought in view, Li et al. have synthesized Cuδ+ sites atomically dispersed on a CeO2–TiO2 support comprising of widely dispersed CeO2 nanoparticles on a porous TiO2 substrate via the pyrolysis of a metal–organic framework (MIL-125-NH2) impregnated with Cu2+ and Ce3+ ions (Fig. 11a). The obtained catalyst displayed increased activity toward CO2 reduction to C2H4 under simulated sunlight with production rate of 4.51 μmol g−1 h−1 and 47.5% selectivity, which are 2.36 and 1.32 times those over the Cuδ+/TiO2 sample (Fig. 11b) [105]. In situ FTIR presented the characteristic spectral peaks of *COOH, *CO, and *COCO groups (Fig. 11c), suggesting that the CO2 reduction pathway for producing C2H4 on Cuδ+/CeO2–TiO2 photocatalyst involved CO2 → *CO2 → *COOH → *CO → *COCO → C2H4 (Fig. 11d). In addition, according to the Gibbs free energy calculations, the whole process of CO2-to-C2H4 conversion on Cuδ+/CeO2–TiO2 catalyst is more thermodynamically favorable than on Cu/TiO2, as shown in Fig. 11e. This is due to the presence of Cu–Ce dual active sites, which enables the efficient generation of key intermediate *CO and support the *CO → *COCO coupling reaction. This demonstrated a synergistic effect between the active sites of Cu and Ce, which optimize the rate-limiting steps and enhance the overall CO2-to-C2H4 conversion. More recently, Wang et al. conducted a study exploring how the valence state and coordination environment of SAs active sites affect the C2+ products selectivity using 2D WO3 catalyst modified by depositing SAs Cu and Pt (CuPt/WO3). The CuPt/WO3 photocatalyst was found to be much more efficient than pristine 2D WO3, Cu/WO3, and Pt/WO3 in producing acetic acid (CH3COOH), with a production rate of 19.41 μmol g−1 h−1 and a selectivity of 88.1%. It has been demonstrated that stabilization of Cu+ species by forming a coordinated complex with Cl in aqueous solution is the key to attain superior efficiency and selectivity toward C2+ products [106]. All the components in CuPt/WO3 photocatalyst work synergistically toward the production of C2+ products. Cu+ species coordinated with Cl enhanced the CO adsorption capacity and increased the lifespan of CO* intermediate, both of which aid in the C–C coupling reaction. On the other side, SAs Pt active sites located in close proximity provided protons for the hydrogenation of CO* intermediate, ultimately leading to the formation of C2+ products.

a Schematic illustration of the steps for Cuδ+/CeO2-TiO2 preparation, b production rates over different photocatalysts, c in situ DRIFTS spectra of Cuδ+/CeO2–TiO2 photocatalyst, d proposed reaction pathway for CO2 photoreduction to C2H4, and e calculated free energy diagrams for CO2 reduction over Cu-TiO2 and Cu–Ce2–TiO2 slabs. Reproduced with permission [105]. Copyright 2022, American Chemical Society
Based on the amount of studies published on photocatalytic CO2 reduction to C2 vs C3 products, it is obvious that direct conversion of CO2 into C3 products is much more difficult [107, 111]. This is due to the fact that the process requires a higher-order reaction pathway involving the creation of multiple C–C bonds. This, in turn, necessitates the integration of two distinct catalytic steps of CO2-to-CO and CO-to-C2+ at different active sites [112,113,114]. Creating these C–C bonds is particularly challenging as it involves energy-intensive endothermic reactions with significant uphill energy barriers, primarily due to the high-energy levels of the critical *C2 and *C3 intermediates. This is mainly due to the lack of efficient catalytic active sites to stabilize these intermediates and reduce energy barriers. To address this concern, Xiong et al. recently developed an efficient photocatalyst for the direct CO2 conversion into C3H8 [107]. The catalyst consisting of Cu SAs implanted on Ti0.91O2 atomically thin single layers (Cu–Ti–VO/Ti0.91O2-SL), which showed superior efficiency toward CO2 photoreduction to C2+ products with high selectivity of 50.2% for C2+ products and 32.4% for C3H8 as shown in Fig. 12a–c. As a comparative experiment, on Cu–O/Ti0.91O2–SL without VO only showed the detection of CO and CH4, while Ti0.91O2–SL showed mainly CO2 reduction to CO product. Notably, the presence of VO resulted in a strong coordination interaction between Cu SAs and neighboring Ti atoms, leading to high electron accumulation at copper sites and electron depletion at Ti sites. Conversely, Cu–O/Ti0.91O2–SL catalyst exhibited isolated single-metal structures with minimal interactions between Cu SAs and Ti atoms. In situ DRIFTS analysis confirmed the generation of the intermediate species (*COOH, *CO, *CHO, and *CHOCO) on Cu–Ti–VO/Ti0.91O2–SL catalyst (Fig. 12d). DFT calculations showed that the pristine Ti0.91O2–SL *CO exhibited a high CO selectivity due to the facile desorption of *CO rather than undergo subsequent hydrogenation or C–C coupling (Fig. 12e). Moreover, C–C coupling on the Cu–O site in the absence of Vos was challenging because of the high uphill energy changes, with hydrogenation of *CO into CH4 being more favorable (Fig. 10f). Figure 10g shows the possible pathway of obtaining C3H8 product Cu–Ti–VO unit. The initial step involved the transformation of absorbed CO2 to *CHO via *COOH and *CO intermediates. Next, *CHO at Cu–Ti–VO unit could react with CO coming from adjacent Ti0.91O2 units, resulting in the formation of *CHOCO intermediates. The subsequent C1–C2 coupling (*CH2OCO + *CO → *CH2OCOCO) was found to be a thermodynamically favorable exothermic process. The DFT results revealed that Cu–Ti–VO units have the ability to stabilize *CHOCO and *CH2OCOCO intermediates, leading to a reduction in their energy levels. This stabilization was probably due to the largely alleviation of electron accumulation and relaxation of the intermolecular and intramolecular electrostatic repulsion within Cu–Ti–VO units. This mechanism can potentially enable both C1–C1 and C1–C2 coupling processes to become favorable exothermic reactions.

a The evolution of photocatalytic production as a function of light irradiation times on Cu–Ti–VO/Ti0.91O2–SL, b production rates, c electron-based selectivity, d in situ DRIFTS spectra of CO2 reduction on Cu-Ti-VO/Ti0.91O2-SL photocatalyst, gibbs free energy diagrams of CO2 reduction on e Ti0.91O2 matrix, f Cu–O site, and g Cu–Ti–VO unit. Reproduced with permission [107]. Copyright 2023, Nature
3.1.4 Cu-Based Heterojunctions
The construction of heterojunctions is regarded as one of the most effective strategies for enhancing the charge separation and transfer processes, consequently improving overall photocatalytic efficiency [115,116,117,118]. Copper-based heterojunctions may involve copper in various forms, such as Cu NPs or CuO, combined with other semiconductor materials, in a way that forms a junction between them. For instance, the introduction of Cu (metal, oxide, and quantum dots) as cocatalyst for heterostructures with various semiconductors (such as g-C3N4, TiO2, ZnV2O4, etc.) was demonstrated to effectively broaden the photoresponse range and effectively improve the PCO2RR activity/selectivity toward C2+ compounds [119,120,121,122,123,124,125,126]. It is worth noting that despite metal copper itself does not have photocatalytic activity, it has high electrical conductivity, which may enhance the effectiveness of other photocatalysts when combined with them. For instance, Zhao et al. successfully constructed a metal–semiconductor (m–s) heterojunction of Cu-dispersive protonated g-C3N4 (PCN) through a thermal reduction process of Cu2O/PCN, where the total conversion rate of CO2 to CH3OH and C2H5OH reached 25.0 μmol g−1 under UV–Vis irradiation for 4 h. This value was 4.18 and 1.84 times higher than those obtained from PCN and Cu2O/PCN, respectively [123]. The Cu/PCN heterojunction demonstrated a selectivity of 51.42% for CH3OH and 46.14% for C2H5OH. These outcomes indicated that the heterojunction effectively facilitates the separation of charge carriers and inhibits their recombination, resulting in high yield of C1 and C2 products. In another study, Zhao et al. reported the preparation of metal–semiconductor Cu/ZnV2O4 heterojunction for the photocatalytic CO2 reduction to CH3OH and C2H5OH [124]. They found that composite Cu0-ZnV2O4 increased the surface area and adjusted the energy band position in a way that matched with the reaction potential toward CH3OH and C2H5OH. The improved photocatalytic activity over Cu/ZnV2O4 was due to the heterojunction interface’s ability to facilitate rapid transmission and hinder the recombination of the photogenerated charges.
In a study conducted by Yu et al., they investigated the impact of depositing CuOx onto BiVO4 for the photocatalytic conversion of CO2 into hydrocarbons [127]. They synthesized monoclinic BiVO4 crystals with a truncated tetragonal bipyramidal shape, allowing for controlled ratios of exposed {010} and {110} facets. Notably, they observed that CuOx NPs were selectively deposited onto the {010} facets of the BiVO4 crystals. Compared to pure BiVO4, the CuOx/BiVO4 catalysts, which maintained a uniform truncated tetragonal bipyramidal morphology, exhibited a higher rate of hydrocarbon fuel formation, including CH4, C2H6, and C3H8. The improved photocatalytic activity was attributed to the enhanced efficiency of charge carrier separation, facilitated by the presence of a Z-scheme junction at the interface between α-CuOx and BiVO4. In another study [128], Z-type Cu2O-modified BiOI microspheres were synthesized through chemical deposition. The incorporation of Cu2O onto the surface of BiOI served to enhance the specific surface area of BiOI, providing more exposed active sites. Additionally, the close interaction between Cu2O and BiOI facilitated the efficient separation and migration of photogenerated carriers, as well as the use of sunlight. Compared to pristine BiOI, the Cu2O/BiOI heterojunction photocatalyst exhibited superior photocatalytic activity. Notably, it resulted in higher yields of CH3OH and C2H5OH CO2 photoreduction, with yields reaching 609.05 and 273.96 μmol gcat−1, respectively.
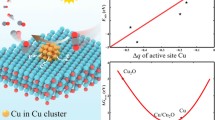
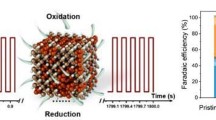
Besides, Zhao et al. reported the construction of hybrid photocatalyst (CuOX@p-ZnO) in which CuOX is evenly distributed throughout polycrystalline ZnO. This photocatalyst demonstrated the ability to reduce CO2 to C2H4, achieving a production rate of 22.3 μmol g−1 h−1 and a selectivity of 32.9% [27]. The combination of X-ray absorption fine structure spectra and in situ FT-IR studies demonstrated that Cu was predominantly present as CuO (Cu2+) in the initial catalyst. Nevertheless, during the photocatalytic process, a distinctive surface layer of Cu+ emerged over the CuO matrix, which served as the active site for capturing in situ generated CO and promoting its transformation into C2H4 through C–C coupling. In situ FT-IR analysis successfully identified *OC–COH intermediate during the PCO2RR, marking the first experimental observation of this intermediate. Additionally, theoretical calculations revealed the significant contribution of Cu+ sites in improving the binding of *CO and enhancing the stabilization of the *OC–COH intermediate (Fig. 13a). This study reveals how the Cu valence state could affect the reaction pathway of CO2 reduction to produce C2+ compounds. The same group recently successfully synthesized a π–π stacking hybrid structure between g-C3N4 and 2D MOF of Cu-CuTCPP [125]. The resulting catalyst was able to convert CO2 to C2H6 and achieving a C2H6 selectivity of 44% (Fig. 13b and c). Interestingly, they identified a light-driving reconstruction of Cu-CuTCPP moiety (CuII2(COO)4 → Cu1+δ2(COO)3) by the photoinduced electrons from excited g-C3N4 as depicted in Fig. 13d. The self-reconstruction mainly improved the stabilization of *CO intermediates as well as the synergistic effect of the dual-Cu site, leading to efficient C–C coupling to produce C2H6.

Copyright 2021, American Chemical Society. b PCO2RR results on g-C3N4, Cu-CuTCPP, Cu-CuTCPP/g-C3N4, Zn-CuTCPP/g-C3N4, and Zn-ZnTCPP/g-C3N4, c photocatalytic CO2 reduction on Cu-CuTCPP/g-C3N4, and d the self-reconstruction of paddle-wheel CuII2(COO)4 during the PCO2RR. Reproduced with permission [125]. Copyright 2022, Elsevier
a Theoretical calculations of the adsorption energy of *CO and first-principles calculations of the C–C coupling process on Cu2O and Cu2O@CuO. Reproduced with permission [27].
As mentioned above, the selectivity of Cu-based photocatalysts depends on the oxidation state of Cu. However, the use of Cu in photocatalysis is not yet fully developed as it exhibits poor stability caused by the variations in its oxidation states by the photoinduced charges. Therefore, it is crucial to ensure the stability of Cu to fully utilize its intrinsic photocatalytic properties. To date, the formation of heterojunctions with other photocatalysts is a widely employed approach to improve the stability of Cu. For example, Liu et al. employed a complexation oxidation approach for the encapsulation of CuO QDs in the pores of metal organic framework of MIL-125(Ti) and further combined it with g-C3N4 to construct a composite photocatalyst, (g-C3N4/CuO@MIL-125(Ti)), as illustrated in Fig. 14a [126]. This encapsulating structure ensured high stability and reusability of the catalyst. Furthermore, the obtained composite facilitated an efficient electron transfer from MIL-125(Ti) and g-C3N4 nanosheets to the CuO QDs, boosting the density of electrons over the QDs (Fig. 14b). Consequently, the initial composition of the product shifted from C1 (CH3OH, CO) to mainly C2+ compounds (CH3CH2OH, CH3CHO) for g-C3N4/CuO@MIL-125(Ti), representing about 77% of the total formed products as shown in Fig. 14c.

a Schematic illustration for the synthesis of g-C3N4/CuO@MIL-125(Ti) composite photocatalyst, b the charge transfer process, and c the proposed reaction pathway over g-C3N4/CuO@MIL- 125(Ti) photocatalyst. Reproduced with permission [126]. Copyright 2020, Elsevier
In summary, several approaches such as optimising crystal phase and morphology, introducing metal doping, engineering defects, fabricating heterostructures, and utilizing bimetallic synergies have been shown to enhance the efficiency and selectivity of Cu-based materials toward the formation of C2+ products in PCO2RR. These approaches impact the reaction routes primarily by stabilizing the crucial C1 or C2 intermediates, decreasing the reaction barrier, offering more active sites, as well as increasing the electron and proton density to facilitate the C–C coupling for the C2+ production. Table 2 lists the photocatalytic systems that have been reported for PCO2RR into C2+ products.
3.2 Electrocatalytic CO2 Reduction Reaction
The selection of the right electrocatalyst is a critical factor that determines the efficacy ECO2RR process. It impacts the reaction kinetics, selectivity of possible products, and the required overpotential. So far, copper-based materials are the most effective electrocatalysts for ECO2RR to C2+ products, but they are still relatively unselective. This is owing to the moderate binding affinity of copper to carbon monoxide which allows the generation of a wide range of products including, but not limited to, methane, methanol, ethylene, ethane, ethanol, and propanol [129,130,131,132,133,134,135,136]. Nevertheless, the origin of the enhanced activity/selectivity of Cu-based materials in ECO2RR toward C2+ chemicals production is not easily identified. To date, many factors have been shown to affect the overall mechanism to C2+ products including the experimental setup conditions (pH, cation/anion of electrolyte, temperature, pressure, and applied overpotential) [137,138,139,140,141,142,143] and the catalyst surface properties (morphology, oxidation states, exposed facets, and defects) [144,145,146,147,148]. In this section, the relationship between the Cu surface and the performance of ECO2RR is discussed, with the aim of comprehending the origin of the improvement observed in C2+ production over Cu-based materials. The discussion is approached from a materials viewpoint, and the Cu-based materials for ECO2RR are categorized into three groups: Cu metal/oxides, Cu alloys, and Cu-based single-atom catalysts (Cu SACs). Table 3 provides a summary of the use of Cu-based catalysts in ECO2RR to C2+ compounds.
3.2.1 Cu Metal/Oxides
The copper surface morphology and geometry have significant effects on the type of products generated during ECO2RR. The faradic efficiencies (FE) for the formation of methane and C2 products (ethylene and ethanol) on polycrystalline Cu surfaces at − 5 mA cm−2 in 0.1 M KHCO3 are approximately 29% and 37%, respectively. The presence of heterogeneous catalytic sites on the polycrystalline Cu plane may account for the insufficient selectivity [135]. Significant improvements were found by using single-crystal Cu(100) and cleaved Cu(100) surfaces with high-indexed planes. For example, Cu(S)-[4(100) × (111)] surface showed the formation of ethanol and ethylene with a total FE of about 57% [149]. It is suggested that the atomic steps and a square arrangement of Cu atoms in the (100) terraces facilitate the coupling of CHxO intermediates, thereby contributing to the generation of more C2+ products. Even though Cu(100) single crystals are the optimal for ethylene production, they still produce a considerable quantity of methane. However, the use of Cu cubes with (100) facets, having an edge length of approximately 100 nm, can further enhance ethylene selectivity by almost completely suppressing methane formation [150].
In a study by Buonsanti et al., the influence of size and shape of Cu nanocrystals (NCs) on the activity and selectivity in ECO2RR was investigated [151]. By using colloidal chemistry approach, Cu NC spheres with two distinct sizes (7.5 and 27 nm) and Cu NC cubes with three different sizes (24, 44, and 63 nm) were synthesised (Fig. 15a–e). The X-ray diffraction patterns for Cu NC cubes and spheres were compared, revealing that the Cu cubes were dominated by {100} facets, as indicated by the more pronounced (200) peak compared to the bulk fcc Cu reference (Fig. 15f). The results showed that, while smaller nanoparticles with the same morphology demonstrated greater activity, the cube-shaped nanoparticles displayed greater intrinsic activity compared to the spheres (Fig. 15g). A noteworthy observation was the nonlinear trend in selectivity, as Cu cube nanoparticles with a side length of 44 nm exhibited 80% selectivity toward carbon products, of which 50% was identified as ethylene. The superior activity observed in the Cu NC cubes (44 nm) can be attributed to the optimal proportion of edge sites to (100) plane sites (Nedge/N100 = 0.025), as evidenced by the statistical analysis of the surface atom density (Fig. 13h), which emphasizes the crucial role of edge atoms in the active sites that selectively drive ECO2RR and ethylene production in these Cu nanocrystal cubes.

TEM images of Cu cubes with an average edge length of a 24 nm, b 44 nm, c 63 nm, TEM images of Cu spheres with an average diameter of d 7.5 nm, e 27 nm, f XRD patterns of the Cu cubes and Cu spheres, g current density at − 1.1 VRHE plotted versus the size of the Cu NC catalysts, and h density of adsorption sites in Cu NC cubes (left axis) and trend Nedge/N100 and N100/Nedge (right axis) versus the edge length. Reproduced with permission [151]. Copyright 2016, Wiley–VCH
In another study, researchers explored the effect of copper nanowire (Cu NW) morphology on ECO2RR toward C2+ hydrocarbons [152]. It was observed that the selectivity for C2+ hydrocarbons, on Cu NWs array electrodes could be finely adjusted by systematically modifying the Cu NW length and density, which could be controlled by varying the reaction times (Fig. 16a–d). Their findings indicated a gradual increase in the formation of C2H4 as the length and thickness of Cu nanowires increased. As shown in Fig. 15e, at a length of 8.1 mm for the Cu NWs, the FE for C2H4 formation reached 17.4%, while the production of H2 is simultaneously reduced.

a–d SEM images of Cu(OH)2 nanowires with synthesis time of 1, 3, 5, and 8 min, respectively, e FE for different products on Cu nanowire arrays with different lengths at − 1.1 VRHE in CO2-saturated 0.1 M KHCO3 electrolytes. Reproduced with permission [152]. Copyright 2016, Wiley–VCH. f-i TEM images of as-etched Cu NCs subjected to different etching periods of 4 h, 8 h, 12 h, and 24 h, respectively. Reproduced with permission [153]. Copyright 2016, American Chemical Society
Nanomaterials with controlled morphology play a pivotal role in both assessing the effect of different facets on the ECO2RR and designing catalysts with superior performance and selectivity. In this regard, Yin and colleagues [153] reported the synthesis of cubic Cu NPs through chemical etching, spanning across different time intervals, leading to the formation of various shapes as illustrated in Fig. 16f–i. Notably, when the nanocrystals were etched for a duration exceeding 12 h, they exhibited a rhombic dodecahedral morphology, prominently exposing high-energy (110) facets. When compared to Cu nanocubes, rhombic dodecahedral Cu NPs exhibit a positive onset potential of − 1.1 VRHE. Additionally, at − 1.4 VRHE, the current density for the rhombic dodecahedral structure is approximately three times higher than that of Cu nanocubes. This observation indicates that the Cu crystal structure with enriched (110) facets outperforms the one with (100) facets in terms of catalytic activity as depicted in Fig. 16j. The presence of high-energy (110) facets in the rhombic dodecahedral Cu structure leads to increased selectivity for the formation of C2+ products, including C2H4, C2H6, and C3H8 when compared to the original (100) facet of cubic Cu. These findings suggest that the (110) facet of rhombic dodecahedral Cu is particularly conducive to the formation of C2+ hydrocarbons in ECO2RR.
In addition to considering structural parameters including particle size/shape and reactive facets, it has been suggested that the surface state of Cu is an important factor influencing the activity and product selectivity in ECO2RR. Recently, it was demonstrated that the interaction between the surfaces made of Cu+ and Cu0 restrains the C1 pathways while promoting CO–CO dimerization owing to the attraction between carbon atoms with opposite charges stimulated by the interface between Cu+ and Cu0 [154]. According to a recent study, oxide-derived copper catalysts, activated using oxygen plasma, showed a noteworthy ethylene FE of 60% at − 0.9 VRHE. This high efficiency was related to the existence of Cu+ species. Interestingly, through the in situ hard X-ray absorption spectroscopy (hXAS) analysis, it was observed that stable Cu+ species were detected at notably negative potentials of about − 1.0 VRHE during the ECO2RR [155].
In a study conducted by Hwang et al., it was found that the initial stages of ECO2RR caused fragmentation of Cu2O NPs, leading to improved C2H4 selectivity and activity [156]. The researchers utilized a cysteamine immobilization agent to synthesize Cu2O nanoparticles by direct growth on carbon black, which initially measured at a size of 20 nm, but were found to be broken down into smaller sizes (2–4 nm) with denser arrangement and deformed crystal planes after 10 h of ECO2RR. These morphological changes over reaction time led to higher activity, with a C2H4 FE of 57.3% achieved at − 1.1 VRHE in 0.1 M KHCO3. In situ/operando XANES analysis revealed a shift in oxidation state from Cu+ to Cu0, suggesting that Cu0 was the dominant state during the electrochemical CO2 reduction, even in Cu catalysts derived from the oxide material. Upon fragmentation of the initial Cu NPs, they underwent re-oxidation to Cu+ states without air exposure, whereas large Cu NPs retained their Cu0 state when subjected to open-circuit potential conditions, suggesting that the morphological alterations observed in fragmented Cu2O nanoparticles enable greater oxygen access and contribute to improved CO2 reduction efficiency.
In another study, Sargent et al. reported the electro-redeposition of Cu (ERD Cu) from a Cu2(OH)3Cl sol–gel precursor to control the morphology and oxidation state of Cu in order to inhibit CH4 formation and enhance C2H4 production [150]. Various structural morphologies appeared as a function of applied potential, including rounded nanostructures (at − 0.7 VRHE), sharper needles (at − 1.0 VRHE), sharper nanowhiskers (at − 1.2 VRHE), and dendrites with rounded tips (at − 1.4 VRHE). The ERD Cu catalyst was found to exhibit a 54% FE for C2+ products (ethylene, acetate, and ethanol) and an 18% FE for C1 products (carbon monoxide, methane, and formate) at − 1.2 VRHE. Additionally, the C2H4 partial current density was high for ERD Cu in both the H-cell (22 mA cm−2 at − 1.2 VRHE) and flow cell (161 mA cm−2 at − 1.0 VRHE) configurations accompanied by notable inhibition of CH4 with a C2H4/CH4 ratio of 200. Notably, the high local pH induced by ERD Cu is an important factor in the suppression of CH4. This is because ERD Cu generated remarkably high current densities, with a recorded 60 mA cm−2 in H-cell and 450 mA cm−2 in flow cell, resulting in a rapid consumption of protons that led to the elevated local pH [157, 158]. At high pH levels, CO dimerization pathway is most likely to occur because the CO hydrogenation step is kinetically limited. At very negative potentials (less than − 1.0 VRHE), in situ soft X-ray absorption (sXAS) spectroscopy indicated the existence of Cu+ species. For over 1 h of operation, it was up to 23% of the ERD Cu electrocatalyst consisted of Cu+ species, even when subjected to a negative applied bias as low as − 1.2 VRHE, indicating a crucial role of Cu2(OH)3Cl sol–gel in stabilizing Cu+ species at higher negative applied potentials. According to DFT calculation, it was observed that the ERD Cu(211) model, comprising of 25% Cu+ species, closely resembling the optimal Cu+ quantity identified through sXAS analysis, demonstrated the minimum Gibbs free energy of formation of the OCCOH* intermediate (1.13 eV). This value was 0.76 eV lower than that of the Cu(211) model. Moreover, the ERD Cu(211) surface showed the highest CO binding, as evidenced by a binding energy of − 1.45 eV, indicating that the Cu+ species has a significant impact on the stabilization of the OCCOH* intermediate, thereby promoting the formation of C2 products over C1 products. Overall, the Cu+ site plays a crucial role in enhancing the selectivity toward C2+ products by promoting the C–C coupling reaction through strengthened *CO adsorption and stabilized C2 intermediates. Nevertheless, the low stability of Cu+ species at negative potentials during the ECO2RR remains a major impediment to its large-scale implementation. To address this challenge, metal doping is suggested as effective way to boost the stability of Cu+ species [159,160,161,162,163].
Recently, the study conducted by Han et al. applied DFT calculations and multiple characterization tests to investigate the impact of atomic dopants on the efficiency of Cu-based catalysts or CO2 electroreduction to C2+ products [164]. Starting with DFT calculations, they explored the effectiveness of doping various single-atom metals (Ag, Pd, Ni, Zn, Sn, Ce, Sm, and Gd) of Cu2O and identified single-atom Gd-doped Cu2O as a highly promising choice for ECO2RR to C2+ products. This was attributed to its ability to enhance the CO2 conversion to *CO, suppress HER, demonstrated lower thermodynamic limiting potentials for ECO2RR, and exhibited moderate *CO binding energy. The characterization results of as-prepared Gd/CuOx confirmed that has the ability to stabilize Cu+ species and induce tensile strain in the catalyst. The Gd/CuOx catalyst exhibited superior CO2 conversion to C2+ product, with a Faradaic efficiency of 81.4% and a partial current density of C2+ product reaching 444.3 mA cm−2 at − 0.8 VRHE. Both DFT calculations and experiments revealed that the stability of the key intermediate O*CCO was enhanced by Gd doping. Additionally, the energy barrier for the CO dimerization to O*CCO step is significantly affected by tensile strain, ultimately contributing to the superior performance of the Gd/CuOx catalyst in ECO2RR to C2+ products.
3.2.2 Cu Alloys
The introduction of a secondary metal to single Cu crystal can alter its chemical composition, leading to improved activity/selectivity by adjusting the binding strength of key intermediates on the catalyst surface. Thus, Cu alloys have garnered significant attention in this regard. Based on the previous experiments, Qiao et al. [165] introduced a classification system for the secondary metal of Cu alloys based on their relative affinities toward H and oxygen O, as illustrated in Fig. 17a. They proceeded to examine the distinct catalytic mechanisms and resultant product variations associated with each of these four affinity categories separately. For ECO2RR, metals that exhibit a higher affinity for O but a lower affinity for H compared to Cu, such as Sn, In, Hg, and Pb, tend to result in the formation of a weakly bound *COOH intermediate after the initial reaction step. Consequently, the primary product of CO2 reduction on these metal surfaces is HCOOH. There is even evidence to suggest that the reaction may proceed through an O-bound *OCHO intermediate on these metals. Conversely, metals with lower affinities for both O and H than Cu, including Zn, Ag, and Au, exhibit a stronger binding affinity for *COOH compared to *CO. Consequently, CO is desorbed as the primary product, with the formation of *COOH serving as the potential determining step [166]. On the other hand, metals that possess both a stronger affinity for O and H than Cu, which include Co, Ni, Fe, and Pt, generally tend to favor the competitive HER with a minor amounts of hydrocarbons and alcohols being detected on the surfaces of these metal catalysts.

a Classification of various metals into four groups in relation to Cu alloys based on O and H affinities from the previous studies. Reproduced with permission [165]. Copyright 2018, Elsevier. b Reaction products of electrocatalytic CO2 reduction on different Cu alloys
It was found that when group 1 metals including Au, Zn, and Ag (denoted as M1) are the predominant component in Cu–M1 systems, the primary product is CO, and the FE is generally better than that of the parent metals. This outcome is likely due to the increased O affinity of Cu, which stabilizes the *COOH intermediate, while the weak binding ability of M1 metals for *CO facilitates the desorption of CO. However, in cases where M1 provides a moderate contribution to the active sites, C2+ products are generally produced with a high FE due to the synergistic effect between Cu and M1. Moreover, the weak binding of H also suppresses HER, enhancing the overall selectivity for ECO2RR (as illustrated in Fig. 17b). For group 2 metals (M2), the trend suggests a preference for 2e− reduction products in Cu-M2 systems, likely due to the weak H binding, which inhibits pathways beyond CO. When M2 metals dominate the active sites, HCOOH becomes the major reduction product. This is attributed to the increased availability of O-binding sites, potentially leading to better stabilization of the *OCHO intermediate and a favorable pathway to HCOOH formation. It is noteworthy that there is limited research on the coupling of group 3 metals (M3, specifically Pd) with Cu, and the intermetallic arrangement of these materials significantly influences product selectivity. When the metallic arrangement is more regular, CO becomes the major product and is produced with a high FE. There are fewer examples of group 4 metals including Pt and Ni (M4) being coupled with Cu in the literature. Although M4 metals with a strong affinity for CO should be avoided, these materials can produce CH4 and CH3OH with relatively high FE, indicating their potential for specific applications in ECO2RR [165].
As mentioned above, the synergetic effect between Cu and group 1 metals (e.g., Au, Ag, or Zn) has been reported to improve the production of C2+ compounds [160, 167,168,169,170,171]. Specifically, the *CO dimerization process (which generates *OCCO) on bimetallic Cu surfaces could be facilitated owing to the enhanced reduction of CO2 to CO via Au, Ag, or Zn sites. For instance, Kuhl et al. reported that the selectivity toward C2+ products of Au–Cu alloys was significantly higher than that of a monometallic Cu catalyst at − 0.7 VRHE using 0.1 M KHCO3 as electrolyte [160]. In a study by Du et al., it was found that of C2H4 FE increased from 15 to 33% for a Cu and a Cu4Zn1 alloy catalyst, respectively [168]. Meanwhile, Buonsanti et al. explored Ag–Cu alloys and determined that, when operating at − 1.1 VRHE, using 0.1 M KHCO3 as electrolyte, the C2H4 FE increased from 13 to 37% with Ag1–Cu1.1 alloy catalyst, as compared to the pure Cu catalyst [169]. Based on these studies, the guest metals (Au, Ag, and Zn) can boost the CO concentration near the catalyst surface, making it easier for Cu sites to convert it into C2+ products. The disposition of the guest metals on the surface of copper has the potential to alter the electronic states of the catalyst and to modify the interatomic distance between surface atoms and influence the reaction kinetics of the adsorbed intermediates on the surface. These factors may ultimately affect the chemical binding strengths of reaction, thereby modulating the reaction toward pathways that are more advantageous [171, 172]. For example, recent experiments by Timoshenko et al. using in situ EXAFS demonstrated how the composition and structure affect the product distribution of CO2RR. The researchers observed that Cu-ZnNPs with shorter interatomic distances have a greater affinity for producing CH4, whereas NPs with longer Cu–Zn distances are more likely to generate CO, which is then reduced to C2+ compounds [172].
Related to the high levels of C2+ selectivity at high activity challenges (current density << − 200 mA cm−2, applied potentials >> − 1.0 VRHE) on Cu alloy-based catalysts, Gewirth et al. demonstrated the enhanced selectivity of ECO2RR toward ethylene and ethanol with bimetallic CuAg catalyst synthesized by using additive-controlled electrodeposition method [173]. In an alkaline flow electrolyzer, the CuAg catalyst that has a nanoporous structure and a small amount of Ag (< 10%) showed high ethylene selectivity (~ 60%) and ethanol production (~ 25%) at a relatively low applied potential of − 0.7 VRHE and a high current density of − 300 mA cm−2. According to the findings from in situ Raman analysis and control experiments, it can be inferred that the improved selectivity toward ethylene and ethanol can be ascribed to the presence of an optimal amount of Ag responsible for stabilizing the Cu2O overlayer and to the higher flux of CO generated by the active promoter Ag that generated more CO intermediates during the ECO2RR process. Recently, Shen et al. reported the use of Ag–Cu bimetallic surface alloys (AgxCu100−x) prepared by a modified polyol method followed by electrochemical reduction as illustrated in Fig. 18a [174]. The chemical compositions of AgxCu100−x alloys were rationally adjusted from Cu-rich to Ag-rich. Interestingly, different products including CO, C1 hydrocarbons/alcohols (C1-H/A) and C2 hydrocarbons/alcohols were selectively obtained over AgxCu100−x alloys by modulating the surface chemical compositions and applied potentials. The findings indicated that the AgxCu100−x alloys catalysts, having Cu-rich surface ranging from 14 to 34 at%, exhibited good selectivity toward C2-H/A products (C2H4 and C2H5OH) when subjected to applied potentials within the range of − 0.85 to − 1.0 VRHE, as illustrated in Fig. 18b and c. For example, Ag16Cu84 catalyst showed a high FEC2-H/A of 60.3% at − 0.90 VRHE. Meanwhile, the increase in surface Ag compositions (Ag: 27–55 at%) in AgxCu100−x alloys resulted in an enhanced formation of C1-H/A (namely, CH4 and CH3OH) with applied potentials shifted from − 1.00 to − 1.10 VRHE. Specifically, the nearly-balanced Ag/Cu surface Ag43Cu57 displayed a high FEC1-H/A reaching 41.4% at − 1.10 VRHE. As expected, the Ag-rich surface alloys (Ag: > 74 at%) selectively convert CO2 to CO at applied potentials between − 0.90 and − 1.15 VRHE. As example, the Ag83Cu17 catalyst exhibited a high FECO of 74.0% at − 1.10 VRHE. The findings from DFT calculations show that the high-energy levels of d-band centers (Ed) on the surface of Cu-rich AgxCu100−x result in a strong binding affinity for *CO and allow for coupling of *CO into *COCO over a wide potential range (− 0.80 to − 1.20 VRHE), leading to a yield of C2H/A with high selectivity. For Ag/Cu balanced AgxCu100−x alloys (Fig. 18e), a moderate binding strength of *CO was determined by the downshifted Ed (− 3.24 to − 3.80 eV), which renders the hydrogenation of *CO to *CHO as predominate reaction, leading to selective formation of C1H/A products at high potential (more negative than − 1.0 VRHE). However, in the Ag-rich AgxCu100−x alloys (Ag: > 60 at%), the further decreased Ed (< − 4.08 eV) resulting in weaker binding of *CO intermediates on the surface (Fig. 18f), which facilitate the desorption of *CO intermediates from the surface to generate CO with high selectivity across a wide range of potentials (− 0.80 to − 1.20 VRHE). This study presents a potential avenue for controlling the selectivity of bimetallic alloys to produce desired value-added chemicals.

a Schematic illustration of the synthetic processes of AgxCu100−x alloys, b 3D colormap surface plot, c corresponding colortour maps for FE of different products vs. surface compositions and applied cathodic potentials, mechanism illustrations of selective production on AgxCu100−x alloys, d C2H/A over surface Cu-rich, e C1H/A over surface Ag/Cu nearly-balanced, and f CO over surface Ag-rich. Reproduced with permission [174]. Copyright 2023, Wiley–VCH
3.2.3 Cu SACs
Catalysts consisting of a single atom (SAs) with active metal centers distributed at the atomic level have been found to exhibit improved activity and tunable selectivity in ECO2RR. This can be attributed mainly to the outstanding effectiveness in atom utilization, distinctive electronic structure, and unsaturated coordination surroundings of the metallic centers [175,176,177]. Considering that the CO2RR occurs over solid–liquid–gas boundaries, maximizing the surface area is crucial in order to attain optimal electron and proton transfer across these interfaces. This can be achieved by reducing the CO2RR to a single-atom scale and utilizing substrates that are highly porous and stable. Cu-SACs are of great interest among the SACs due to their remarkable ability to produce high C2+ products with high selectivity. N-doped conductive carbon supports with N-chelated metal sites are frequently used examples for ECO2RR. For instance, Guan et al. reported the use of a nitrogen coordination strategy for dispersing SAs Cu catalysts on nitrogen-doped carbon [178]. The incorporation of nitrogen promoted the dispersion and interaction of atomic copper species onto carbon frameworks doped with nitrogen via Cu–Nx coordinations. The study revealed that when the Cu concentration reached 4.9%mol, the close proximity of Cu–Nx species was sufficient to facilitate C–C coupling, leading to the generation of C2H4. Conversely, when the Cu concentration was below 2.4%mol, the gap between Cu–Nx species was significant, favoring the production of CH4. According to DFT calculations, the generation of C2H4 was facilitated by coupling two *CO intermediates on adjacent Cu–N2 sites, whereas isolated Cu–N2 and Cu–N4 sites as well as neighboring Cu–N4 sites promoted the production of CH4. Despite the promising performance of Cu-SACs, there is still a need for more improvement in terms of C2+ product selectivity and Faradaic efficiency. This is mainly due to the existence of ambiguous catalytic sites and the adverse electronic properties of Cu when coordinated with N.
According to reports, Cu-SACs have the ability to facilitate the crucial C–C coupling reaction, which is necessary for C2+ products formation. Nonetheless, it was proven that these catalysts undergo in situ transformation into metallic agglomerations during operation [179,180,181]. The utilization of a Cu SACs coordinated with nitrogen has demonstrated remarkable ethanol selectivity in ECO2RR. Operando X-ray absorption spectroscopy (XAS) has indicated that isolated Cu species underwent reversible conversion to Cu NPs, which functioned as the authentic active sites during operation [180, 181]. The stability of Cu single sites during ECO2RR and their ability to catalyze C–C coupling without undergoing initial structural changes that lead to Cu agglomeration is uncertain. In line with this, Zeng et al. reported a Cu coordination polymer (Cu(OH)BTA) with a stable single-site and periodic neighboring Cu centers, as illustrated in Fig. 19a [182]. The Cu(OH)BTA catalysts have been found to efficiently promote the conversion of CO2 to C2+ products, including ethylene, ethanol, acetate, and n-propanol, at a low overpotential with ethylene has been identified as the predominant product. At a potential of − 0.87 V, the total FE of C2+ was 73%, and a maximum FE of 57% for ethylene was achieved with a partial current density of 285 mA cm−2. The FE for ethanol, acetate, and n-propanol reached 11%, 4%, and 1%, respectively (Fig. 19b). Cu(OH)BTA demonstrated a 1.5-fold enhancement in ethylene selectivity in comparison with its metallic analogue. Operando XAS and in situ Raman as well as infrared spectroscopies (Fig. 19c–f) revealed that Cu(OH)BTA retained its structural stability and did not undergo any dynamic transformation during ECO2RR. Cyclic voltammetry (CV) studies were conducted in order to gain additional insight into the stability origins of the Cu(OH)BTA catalyst. The findings indicate that Cu(OH)BTA exhibits instability when subjected to Ar atmosphere during redox reactions involving Cu2+/Cu1+/Cu0, and such instability was observed only after 10 scans. In contrast, the use of 1 M KOH as electrolyte in the same flow cell under a CO2 atmosphere did not result in any redox process. The researchers postulated that the ECO2RR had the ability to establish a favorable local environment that acted as a protective shield for the Cu(OH)BTA molecule against the harsh alkaline environment, thereby maintaining its structural integrity. The results obtained from DFT calculations indicate that the polymer’s neighboring Cu atoms provide dual-Cu sites that are appropriately spaced. These sites facilitate the formation of an energetically suitable *OCCHO intermediate following a rate-determining step of CO hydrogenation. The energy barrier for C–C coupling was low, measuring 0.82 V, as illustrated in Fig. 19g and h.

a The proposed model of Cu(OH)BTA with isolated Cu atoms, b FEs of all products at different applied potentials over Cu(OH)BTA, c operando Cu K-edge XANES, d EXAFS of Cu(OH)BTA under CO2 atmosphere at applied potentials ranging from − 0.2 to − 1.2 V, e in situ Raman spectra of Cu(OH)BTA under CO2 atmosphere at the applied potential of − 0.6 to − 1.2 V, f in situ ATR-SEIRAS spectra of Cu(OH)BTA in CO2-saturated 0.1 M KHCO3 electrolyte at the applied potential of − 0.2 to − 1.0 V, g Gibbs free energy diagram of *CO hydrogenation on Cu(OH)BTA slab and Cu(111) slab, and h Gibbs free energy diagram of ECO2RR to C2+ and CH4 pathway on Cu(OH)BTA slab. Reproduced with permission [182]. Copyright 2023, Nature
It is noteworthy to mention that the effective conversion of CO2 into C3 products using Cu-based electrocatalysts is rarely reported. It is necessary to conduct an in-depth investigation of the reaction pathways of Cu-based electrocatalysts and their resultant products in order to gain a comprehensive understanding of the electrocatalytic CO2 conversion to C3 compounds. Chen et al. have recently reported that Cu SAs anchored in N-doped porous carbon (Cu-SA/NPC) catalysts can convert CO2 to ethanol, acetic acid, and acetone products at a low overpotential. Interestingly, acetone was found to be the major product, with a production rate of 336.1 μg h−1 and a Faradaic efficiency of 36.7%. The authors performed an investigation into the impact of copper distribution and local coordination environment of Cu SAs in order to gain insight into the ECO2RR to C3 oxygenates [183]. The DFT calculations and the experimental results revealed that the high ability of Cu-SA/NPC catalyst for producing acetone from CO2 reduction was ascribed to the crucial role of Cu-pyrrolic-N4 active sites in stabilizing the intermediates associated with acetone production and promoting C–C coupling reactions owing to the synergistic effect in Cu–N configuration.
4 Summary and Outlook
This review provides an overview of the latest research advancements in Cu-based materials and their role in photocatalytic and electrocatalytic CO2 reduction to C2+ products. The primary emphasis is placed on understanding the structure–activity relationships. The economic advantages of C2+ products surpass those of C1 products, thereby underscoring the significance of developing efficient catalysts. The inherent advantages of Cu-based materials make them a promising option for CO2 reduction processes. However, there is a need to improve maximum selectivity and intrinsic activity for C–C bond formation, which remains a challenge in C2+ chemical production.
Various strategies have been employed to enhance the effectiveness and selectivity of PCO2RR toward C2+ products, including crystal phase and morphology optimization, metal doping, defect engineering, heterostructure fabrication, and bimetallic synergies. The aforementioned improvements are related to the semiconductor’s ability to effectively separate and transfer photo-induced charge carriers to the active site with minimal recombination, concentrate electrons on the active site, increase CO2 adsorption capacity, and facilitate C–C coupling reactions for the formation of C2+ products. The C–C coupling process in PCO2RR involves stabilizing intermediates and accumulating electrons to reduce *CO intermediates into C2+ products. In order to enhance the reduction capability of the photocatalyst and extend its charge lifetime, it is imperative to adequately inhibit charge recombination and precisely adjust the conduction band’s position toward a significantly negative direction.
Improving the proportion of Cu species can enhance the efficiency of Cu-based materials in PCO2RR to C2+ compounds. Nevertheless, the influence of various ratios of copper valences on its catalytic activity has not been exhaustively studied due to difficulties in precisely controlling Cu0, Cu+, and Cu2+ ratios in Cu-based catalysts. Therefore, modifying the structural properties of these catalysts to enhance their PCO2RR performance is a preferred research direction. Strategies such as pursuing controlled morphology and particle size with abundant efficient active sites as well as inducing surface defects can significantly improve the catalyst’s performance. As example, engineering the defects or oxygen/metal vacancies on the surface of copper catalyst may create more efficient active sites and allows for a continuous supply of Cu+ species to be available in sufficient amounts. Presently, the available methods for characterization are limited to the assessment of the microstructure and local coordination environment of the catalyst before and after the reaction. Therefore, it is necessary to employ sophisticated in situ/operando characterization methodologies to effectively to dynamically monitor the evolution of copper species on Cu-based materials under reaction conditions. Copper single-atom catalysts (Cu-SACs) exhibit great potential for effective photoconversion of CO2 to C2+ products. However, despite their promising performance, they have not been widely investigated or examined. Hence, the development of Cu-SACs that enable C–C coupling for the production of higher hydrocarbon products is necessary. Optimization of the host material, incorporation of various dopants for anchoring Cu-SAs, or modification of the photoreaction parameters may potentially facilitate the aforementioned outcome.
Despite the significant progress in PCO2RR to C2+ products, the selectivity for C2+ products over C1 products, as well as the suppression of HER, remain inadequate for practical applications. This is due to various factors such as charge recombination and inefficient reaction rates. In addition, it is a commendable goal to produce C3+ products which possess higher energy and value compared to C2 products. Despite the considerable differences in charge supply and density from electrochemistry, and the presence of a high kinetic barrier, empirical findings demonstrate the possibility of achieving CO2 reduction C3+ products through photocatalysis, providing that near theoretical efficiency is attained.
Reaction conditions can have a significant impact on product selectivity. When compared to vapor-phase CO2 reduction conditions, aqueous-phase CO2 reduction reactions offer advantages in terms of charge and mass transfer processes, which are conducive to CO2 reduction. However, the aqueous phase also tends to result in more pronounced competitive HER, which can lead to changes in product selectivity. Furthermore, the use of a suitable hole sacrificial agent can enhance the separation of photogenerated holes and electrons by eliminating the holes. This allows a greater number of photogenerated electrons to participate in photoreduction reactions, offering a better opportunity to produce C2+ products. Furthermore, to thoroughly investigate product selectivity, it is imperative to employ a robust product detection system that can both qualitatively and quantitatively measure all possible products, even those found at extremely low concentrations. Developing such a detection system is of paramount importance, as it greatly contributes to achieving more accurate studies on reaction mechanisms and product selectivity.
For ECO2RR to C2+ products, most of the electrocatalysts are made of Cu as the base due to its unique ability to facilitate the C–C coupling process. Nevertheless, attaining high FE at low overpotential for the production of C2+ chemicals, including ethylene and ethanol, remains a challenging task. Typically, these products are generated at higher potentials due to the necessity of surpassing the kinetic barriers of CO2 intermediates associated with the progression toward the C2+ pathway. Various research studies have been conducted to optimize the properties of Cu-based catalysts for attaining high performance in ECO2RR. Aspects such as morphology, surface composition, size, facet, defect density, and oxidation state are considered while designing these catalysts. The efficiency and selectivity of ECO2RR on Cu catalysts depend largely on various factors such as the composition of the electrocatalyst, local pH, and applied potential. The improvement of catalytic activity/selectivity in Cu-based electrocatalysts can be attributed to several factors, which results in complexity in identifying the catalytic center driving the ECO2RR performance. Although the supply of stable Cu0 and oxidized Cu (Cu+ or Cu2+) is essential during catalysis, other factors such as particle size, surface area, facet, defect, and morphological effects also play a significant role in achieving excellent ECO2RR performance. Thus, understanding the relationship between the Cu surface and CO2RR activity is necessary to design effective catalysts for ECO2RR in the future.
Moreover, incorporating secondary metals such as Pt, Ag, Au, or Zn into the single Cu crystal or Cu SAs catalyst has shown to be an effective strategy, as these alloys or bimetallic species have shown higher selectivity and stability toward C2+ products during ECO2RR. The role of guest metals in altering the catalyst’s electronic states, reaction kinetics, and binding energies of surface adsorbed intermediates depends on their arrangement on the Cu surface. The combination of metallic copper with secondary metals such as Au, Ag, and Zn may produce interfaces that change the electronic structure and facilitate tandem catalytic reactions, so enhancing the metals' intrinsic activity. The segregation of bimetallic phases may potentially enhance the transfer of CO from the guest metal sites responsible for CO production to the Cu sites, thereby leading to an increase in the concentration of *CO on the active Cu sites, which could lead to higher C–C coupling rates. Tailoring Cu-based electrocatalysts with support materials like carbon or MOFs not only preserves the catalyst structure but also provides beneficial synergistic effects that improve catalytic efficiency.
Furthermore, the utilization of DFT calculation is imperative in investigating the correlation between the microstructure of Cu-based catalysts and their catalytic performance, given the rapid progressions in computer technology and artificial intelligence. Additionally, the implementation of machine learning techniques can facilitate the exploration of prospective Cu-based catalysts for the purpose of CO2 reduction. The utilization of in situ and operando characterization techniques is crucial in recognizing the CO2 reduction mechanism and identifying the intermediates produced during the reaction. Regarding this matter, the use of in situ infrared and Raman spectroscopy, alongside in situ and operando XAS and APXPS, has been found to offer reliable data, thereby enabling the suggestion of more precise reaction mechanisms. The combined use of in situ and operando analysis is urged for the investigation of the complex and ongoing controversial mechanism of CO2 reduction reaction.
The advancement of technologies such as flow cells, GDE, and MEA has led to significant enhancements in reducing overpotential, improving Faradaic efficiency, increasing current density, and ensuring the stability of the ECO2RR. As a result, the integrated approach of combining catalyst design with operational technology represents a promising avenue for achieving the production of C2+ products at a low economic cost. It is anticipated that practical applications of ECO2RR will be realized in the industrial sector in the near future. Finally, this review aims to stimulate novel approaches in the design of Cu-based materials that can improve the efficiency of PCO2RR and ECO2RR in producing C2+ products.
References
J. Xie, R. Jin, A. Li, Y. Bi, Q. Ruan et al., Highly selective oxidation of methane to methanol at ambient conditions by titanium dioxide-supported iron species. Nat. Catal. 1, 889–896 (2018). https://doi.org/10.1038/s41929-018-0170-x
S. Yao, X. Zhang, W. Zhou, R. Gao, W. Xu et al., Atomic-layered Au clusters on α-MoC as catalysts for the low-temperature water-gas shift reaction. Science 357, 389–393 (2017). https://doi.org/10.1126/science.aah4321
E. Gong, S. Ali, C.B. Hiragond, H.S. Kim, N.S. Powar et al., Solar fuels: research and development strategies to accelerate photocatalytic CO2 conversion into hydrocarbon fuels. Energy Environ. Sci. 15, 880–937 (2022). https://doi.org/10.1039/D1EE02714J
S.I. Seneviratne, M.G. Donat, A.J. Pitman, R. Knutti, R.L. Wilby, Allowable CO2 emissions based on regional and impact-related climate targets. Nature 529, 477–483 (2016). https://doi.org/10.1038/nature16542
M. Forkel, N. Carvalhais, C. Rödenbeck, R. Keeling, M. Heimann et al., Enhanced seasonal CO2 exchange caused by amplified plant productivity in northern ecosystems. Science 351, 696–699 (2016). https://doi.org/10.1126/science.aac4971
Z.-J. Wang, H. Song, H. Liu, J. Ye, Coupling of solar energy and thermal energy for carbon dioxide reduction: status and prospects. Angew. Chem. Int. Ed. 59, 8016–8035 (2020). https://doi.org/10.1002/anie.201907443
G.H. Han, J. Bang, G. Park, S. Choe, Y.J. Jang et al., Recent advances in electrochemical, photochemical, and photoelectrochemical reduction of CO2 to C2+ products. Small 19, 2205765 (2023). https://doi.org/10.1002/smll.202205765
X. Li, J. Wang, X. Lv, Y. Yang, Y. Xu et al., Hetero-interfaces on Cu electrode for enhanced electrochemical conversion of CO2 to multi-carbon products. Nano-Micro Lett. 14, 134 (2022). https://doi.org/10.1007/s40820-022-00879-5
C. Wang, Z. Sun, Y. Zheng, Y.H. Hu, Recent progress in visible light photocatalytic conversion of carbon dioxide. J. Mater. Chem. A 7, 865–887 (2019). https://doi.org/10.1039/C8TA09865D
Y. Gao, K. Qian, B. Xu, Z. Li, J. Zheng et al., Recent advances in visible-light-driven conversion of CO2 by photocatalysts into fuels or value-added chemicals. Carbon Resour. Convers. 3, 46–59 (2020). https://doi.org/10.1016/j.crcon.2020.02.003
J. Albero, Y. Peng, H. García, Photocatalytic CO2 reduction to C2+ products. ACS Catal. 10, 5734–5749 (2020). https://doi.org/10.1021/acscatal.0c00478
S. Nitopi, E. Bertheussen, S.B. Scott, X. Liu, A.K. Engstfeld et al., Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 119, 7610–7672 (2019). https://doi.org/10.1021/acs.chemrev.8b00705
M. Jouny, W. Luc, F. Jiao, General techno-economic analysis of CO2 electrolysis systems. Ind. Eng. Chem. Res. 57, 2165–2177 (2018). https://doi.org/10.1021/acs.iecr.7b03514
Q. Lu, F. Jiao, Electrochemical CO2 reduction: electrocatalyst, reaction mechanism, and process engineering. Nano Energy 29, 439–456 (2016). https://doi.org/10.1016/j.nanoen.2016.04.009
E. Vahidzadeh, S. Zeng, A.P. Manuel, S. Riddell, P. Kumar et al., Asymmetric multipole plasmon-mediated catalysis shifts the product selectivity of CO2 photoreduction toward C2+ products. ACS Appl. Mater. Interfaces 13, 7248–7258 (2021). https://doi.org/10.1021/acsami.0c21067
X. Wu, Y. Li, G. Zhang, H. Chen, J. Li et al., Photocatalytic CO2 conversion of M0.33WO3 directly from the air with high selectivity: insight into full spectrum-induced reaction mechanism. J. Am. Chem. Soc. 141, 5267–5274 (2019). https://doi.org/10.1021/jacs.8b12928
X. Li, J. Wang, X. Lv, Y. Yang, Y. Xu et al., Hetero-interfaces on Cu electrode for enhanced electrochemical conversion of CO2 to multi-carbon products. Nanomicro Lett. 14, 134 (2022). https://doi.org/10.1007/s40820-022-00879-5
V.S.S. Mosali, X. Zhang, Y. Liang, L. Li, G. Puxty et al., CdS-enhanced ethanol selectivity in electrocatalytic CO2 reduction at sulfide-derived Cu–Cd. Chemsuschem 14, 2924–2934 (2021). https://doi.org/10.1002/cssc.202100903
D. Zang, Q. Li, G. Dai, M. Zeng, Y. Huang et al., Interface engineering of Mo8/Cu heterostructures toward highly selective electrochemical reduction of carbon dioxide into acetate. Appl. Catal. B Environ. 281, 119426 (2021). https://doi.org/10.1016/j.apcatb.2020.119426
M.G. Méndez-Medrano, E. Kowalska, B. Ohtani, D. Bahena Uribe, C. Colbeau-Justin et al., Heterojunction of CuO nanoclusters with TiO2 for photo-oxidation of organic compounds and for hydrogen production. J. Chem. Phys. 153, 034705 (2020). https://doi.org/10.1063/5.0015277
Y. Song, M. Ichimura, H2O2 treatment of electrochemically deposited Cu2O thin films for enhancing optical absorption. Int. J. Photoenergy 2013, 738063 (2013). https://doi.org/10.1155/2013/738063
C.-F. Li, R.-T. Guo, Z.-R. Zhang, T. Wu, W.-G. Pan, Converting CO2 into value-added products by Cu2O-based catalysts: from photocatalysis, electrocatalysis to photoelectrocatalysis. Small 19, 2207875 (2023). https://doi.org/10.1002/smll.202207875
W. Wang, L. Wang, W. Su, Y. Xing, Photocatalytic CO2 reduction over copper-based materials: a review. J. CO2 Util. 61, 102056 (2022). https://doi.org/10.1016/j.jcou.2022.102056
D.D. Zhu, J.L. Liu, S.Z. Qiao, Recent advances in inorganic heterogeneous electrocatalysts for reduction of carbon dioxide. Adv. Mater. 28, 3423–3452 (2016). https://doi.org/10.1002/adma.201504766
Y. Lum, T. Cheng, W.A. Goddard 3rd., J.W. Ager, Electrochemical CO reduction builds solvent water into oxygenate products. J. Am. Chem. Soc. 140, 9337–9340 (2018). https://doi.org/10.1021/jacs.8b03986
Y.Y. Birdja, E. Pérez-Gallent, M.C. Figueiredo, A.J. Göttle, F. Calle-Vallejo et al., Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 4, 732–745 (2019). https://doi.org/10.1038/s41560-019-0450-y
W. Wang, C. Deng, S. Xie, Y. Li, W. Zhang et al., Photocatalytic C–C coupling from carbon dioxide reduction on copper oxide with mixed-valence copper(I)/copper(II). J. Am. Chem. Soc. 143, 2984–2993 (2021). https://doi.org/10.1021/jacs.1c00206
F. Chang, M. Xiao, R. Miao, Y. Liu, M. Ren et al., Copper-based catalysts for electrochemical carbon dioxide reduction to multicarbon products. Electrochem. Energy Rev. 5, 4 (2022). https://doi.org/10.1007/s41918-022-00139-5
M.K. Birhanu, M.-C. Tsai, A.W. Kahsay, C.-T. Chen, T.S. Zeleke et al., Copper and copper-based bimetallic catalysts for carbon dioxide electroreduction. Adv. Mater. Interfaces 5, 1800919 (2018). https://doi.org/10.1002/admi.201800919
A. Bagger, W. Ju, A.S. Varela, P. Strasser, J. Rossmeisl, Electrochemical CO2 reduction: a classification problem. ChemPhysChem 18, 3266–3273 (2017). https://doi.org/10.1002/cphc.201700736
O. Zoubir, L. Atourki, H. Ait Ahsaine, A. BaQais, Current state of copper-based bimetallic materials for electrochemical CO2 reduction: a review. RSC Adv. 12, 30056–30075 (2022). https://doi.org/10.1039/D2RA05385C
W. Fu, Z. Liu, T. Wang, J. Liang, S. Duan et al., Promoting C2+ production from electrochemical CO2 reduction on shape-controlled cuprous oxide nanocrystals with high-index facets. ACS Sustain. Chem. Eng. 8, 15223–15229 (2020). https://doi.org/10.1021/acssuschemeng.0c04873
B. Liu, X. Yao, Z. Zhang, C. Li, J. Zhang et al., Synthesis of Cu2O nanostructures with tunable crystal facets for electrochemical CO2 reduction to alcohols. ACS Appl. Mater. Interfaces 13, 39165–39177 (2021). https://doi.org/10.1021/acsami.1c03850
X. Yuan, S. Chen, D. Cheng, L. Li, W. Zhu et al., Controllable Cu0-Cu+ sites for electrocatalytic reduction of carbon dioxide. Angew. Chem. Int. Ed. Engl. 60, 15344–15347 (2021). https://doi.org/10.1002/anie.202105118
K. Yang, Z. Yang, C. Zhang, Y. Gu, J. Wei et al., Recent advances in CdS-based photocatalysts for CO2 photocatalytic conversion. Chem. Eng. J. 418, 129344 (2021). https://doi.org/10.1016/j.cej.2021.129344
M.Q. Yang, M. Gao, M. Hong, G.W. Ho, Visible-to-NIR photon harvesting: progressive engineering of catalysts for solar-powered environmental purification and fuel production. Adv. Mater. 30, e1802894 (2018). https://doi.org/10.1002/adma.201802894
X. Zhang, F. Han, B. Shi, S. Farsinezhad, G.P. Dechaine et al., Photocatalytic conversion of diluted CO2 into light hydrocarbons using periodically modulated multiwalled nanotube arrays. Angew. Chem. Int. Ed. 51, 12732–12735 (2012). https://doi.org/10.1002/anie.201205619
S. Yan, J. Wang, H. Gao, N. Wang, H. Yu et al., An ion-exchange phase transformation to ZnGa2O4 nanocube towards efficient solar fuel synthesis. Adv. Funct. Mater. 23, 758–763 (2013). https://doi.org/10.1002/adfm.201202042
L. Chen, M. Sun, J. Meng, B. Chu, X. Xie et al., Control of charge transfer direction by spatial engineering of redox active centers for boosting photocatalytic CO2 reduction. Appl. Surf. Sci. 637, 157948 (2023). https://doi.org/10.1016/j.apsusc.2023.157948
S. Verma, X. Lu, S. Ma, R.I. Masel, P.J.A. Kenis, The effect of electrolyte composition on the electroreduction of CO2 to CO on Ag based gas diffusion electrodes. Phys. Chem. Chem. Phys. 18, 7075–7084 (2016). https://doi.org/10.1039/c5cp05665a
C.M. Gabardo, A. Seifitokaldani, J.P. Edwards, C.-T. Dinh, T. Burdyny et al., Combined high alkalinity and pressurization enable efficient CO2 electroreduction to CO. Energy Environ. Sci. 11, 2531–2539 (2018). https://doi.org/10.1039/C8EE01684D
J. Wicks, M.L. Jue, V.A. Beck, J.S. Oakdale, N.A. Dudukovic et al., 3D-printable fluoropolymer gas diffusion layers for CO2 electroreduction. Adv. Mater. 33, e2003855 (2021). https://doi.org/10.1002/adma.202003855
F.P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum et al., CO2 electrolysis to multicarbon products at activities greater than 1 A cm–2. Science 367, 661–666 (2020). https://doi.org/10.1126/science.aay4217
G. Díaz-Sainz, M. Alvarez-Guerra, A. Irabien, Continuous electroreduction of CO2 towards formate in gas-phase operation at high current densities with an anion exchange membrane. J. CO2 Util. 56, 101822 (2022). https://doi.org/10.1016/j.jcou.2021.101822
W. Lee, Y.E. Kim, M.H. Youn, S.K. Jeong, K.T. Park, Catholyte-free electrocatalytic CO2 reduction to formate. Angew. Chem. Int. Ed. 57, 6883–6887 (2018). https://doi.org/10.1002/anie.201803501
H.-K. Lim, H. Shin, W.A. Goddard 3rd., Y.J. Hwang, B.K. Min et al., Embedding covalency into metal catalysts for efficient electrochemical conversion of CO2. J. Am. Chem. Soc. 136, 11355–11361 (2014). https://doi.org/10.1021/ja503782w
K.A. Adegoke, R.O. Adegoke, A.O. Ibrahim, S.A. Adegoke, O.S. Bello, Electrocatalytic conversion of CO2 to hydrocarbon and alcohol products: realities and prospects of Cu-based materials. Sustain. Mater. Technol. 25, e00200 (2020). https://doi.org/10.1016/j.susmat.2020.e00200
U.J. Etim, C. Zhang, Z. Zhong, Impacts of the catalyst structures on CO2 activation on catalyst surfaces. Nanomaterials 11, 3265 (2021). https://doi.org/10.3390/nano11123265
X. Chang, T. Wang, J. Gong, CO2 photo-reduction: insights into CO2 activation and reaction on surfaces of photocatalysts. Energy Environ. Sci. 9, 2177–2196 (2016). https://doi.org/10.1039/C6EE00383D
Megha, K. Mondal, A. Banerjee, T.K. Ghanty, Adsorption and activation of CO2 on Zrn (n = 2–7) clusters. Phys. Chem. Chem. Phys. 22, 16877–16886 (2020). https://doi.org/10.1039/d0cp02505d
S.N. Habisreutinger, L. Schmidt-Mende, J.K. Stolarczyk, Photocatalytic reduction of CO2 on TiO2 and other semiconductors. Angew. Chem. Int. Ed. 52, 7372–7408 (2013). https://doi.org/10.1002/anie.201207199
J. Li, S.U. Abbas, H. Wang, Z. Zhang, W. Hu, Recent advances in interface engineering for electrocatalytic CO2 reduction reaction. Nano-Micro Lett. 13, 216 (2021). https://doi.org/10.1007/s40820-021-00738-9
W. Tu, Y. Zhou, Z. Zou, Photocatalytic conversion of CO2 into renewable hydrocarbon fuels: state-of-the-art accomplishment, challenges, and prospects. Adv. Mater. 26, 4607–4626 (2014). https://doi.org/10.1002/adma.201400087
J. Fu, K. Jiang, X. Qiu, J. Yu, M. Liu, Product selectivity of photocatalytic CO2 reduction reactions. Mater. Today 32, 222–243 (2020). https://doi.org/10.1016/j.mattod.2019.06.009
S. Samanta, R. Srivastava, Catalytic conversion of CO2 to chemicals and fuels. Mater. Adv. 1, 1506–1545 (2020). https://doi.org/10.1039/D0MA00293C
Megha, A. Banerjee, T.K. Ghanty, Role of metcar on the adsorption and activation of carbon dioxide: a DFT study. Phys. Chem. Chem. Phys. 23, 5559–5570 (2021). https://doi.org/10.1039/d0cp05756h
J. Jia, C. Qian, Y. Dong, Y.F. Li, H. Wang et al., Heterogeneous catalytic hydrogenation of CO2 by metal oxides: defect engineering–perfecting imperfection. Chem. Soc. Rev. 46, 4631–4644 (2017). https://doi.org/10.1039/C7CS00026J
A.K. Mishra, A. Roldan, N.H. de Leeuw, CuO surfaces and CO2 activation: a dispersion-corrected DFT+U study. J. Phys. Chem. C 120, 2198–2214 (2016). https://doi.org/10.1021/acs.jpcc.5b10431
J. Lee, D.C. Sorescu, X. Deng, Electron-induced dissociation of CO2 on TiO2(110). J. Am. Chem. Soc. 133, 10066–10069 (2011). https://doi.org/10.1021/ja204077e
W. Chu, Q. Zheng, O.V. Prezhdo, J. Zhao, CO2 photoreduction on metal oxide surface is driven by transient capture of hot electrons: Ab initio quantum dynamics simulation. J. Am. Chem. Soc. 142, 3214–3221 (2020). https://doi.org/10.1021/jacs.9b13280
S. Huygh, A. Bogaerts, E.C. Neyts, How oxygen vacancies activate CO2 dissociation on TiO2 anatase (001). J. Phys. Chem. C 120, 21659–21669 (2016). https://doi.org/10.1021/acs.jpcc.6b07459
Z. Cheng, B.J. Sherman, C.S. Lo, Carbon dioxide activation and dissociation on ceria (110): a density functional theory study. J. Chem. Phys. 138, 014702 (2013). https://doi.org/10.1063/1.4773248
P. Gao, S. Li, X. Bu, S. Dang, Z. Liu et al., Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 9, 1019–1024 (2017). https://doi.org/10.1038/nchem.2794
L. Liu, W. Fan, X. Zhao, H. Sun, P. Li et al., Surface dependence of CO2 adsorption on Zn2GeO4. Langmuir 28, 10415–10424 (2012). https://doi.org/10.1021/la301679h
H. Chen, T. Fan, Y. Ji, CO2 reduction mechanism on the Cu2O(110) surface: a first-principles study. ChemPhysChem 24, e202300047 (2023). https://doi.org/10.1002/cphc.202300047
L.I. Bendavid, E.A. Carter, CO2 adsorption on Cu2O(111): a DFT+U and DFT-D study. J. Phys. Chem. C 117, 26048–26059 (2013). https://doi.org/10.1021/jp407468t
Z. Gu, N. Yang, P. Han, M. Kuang, B. Mei et al., Oxygen vacancy tuning toward efficient electrocatalytic CO2 reduction to C2H4. Small Methods 3, 1800449 (2019). https://doi.org/10.1002/smtd.201800449
C. Hiragond, S. Ali, S. Sorcar, Su-Il In, Hierarchical nanostructured photocatalysts for CO2 photoreduction. Catalysts 9, 370 (2019). https://doi.org/10.3390/catal9040370
H. Molavi, A. Eskandari, A. Shojaei, S.A. Mousavi, Enhancing CO2/N2 adsorption selectivity via post-synthetic modification of NH2-UiO-66(Zr). Microporous Mesoporous Mater. 257, 193–201 (2018). https://doi.org/10.1016/j.micromeso.2017.08.043
X. Ma, L. Li, R. Chen, C. Wang, H. Li et al., Heteroatom-doped nanoporous carbon derived from MOF-5 for CO2 capture. Appl. Surf. Sci. 435, 494–502 (2018). https://doi.org/10.1016/j.apsusc.2017.11.069
R. Zhao, P. Ding, P. Wei, L. Zhang, Q. Liu et al., Recent progress in electrocatalytic methanation of CO2 at ambient conditions. Adv. Funct. Mater. 31, 2009449 (2021). https://doi.org/10.1002/adfm.202009449
J.T. Feaster, C. Shi, E.R. Cave, T. Hatsukade, D.N. Abram et al., Understanding selectivity for the electrochemical reduction of carbon dioxide to formic acid and carbon monoxide on metal electrodes. ACS Catal. 7, 4822–4827 (2017). https://doi.org/10.1021/acscatal.7b00687
J. Santatiwongchai, K. Faungnawakij, P. Hirunsit, Comprehensive mechanism of CO2 electroreduction toward ethylene and ethanol: the solvent effect from explicit water–Cu(100) interface models. ACS Catal. 11, 9688–9701 (2021). https://doi.org/10.1021/acscatal.1c01486
A.J. Garza, A.T. Bell, M. Head-Gordon, Mechanism of CO2 reduction at copper surfaces: pathways to C2 products. ACS Catal. 8, 1490–1499 (2018). https://doi.org/10.1021/acscatal.7b03477
Z. Sun, T. Ma, H. Tao, Q. Fan, B. Han, Fundamentals and challenges of electrochemical CO2 reduction using two-dimensional materials. Chem 3, 560–587 (2017). https://doi.org/10.1016/j.chempr.2017.09.009
S. Pablo-García, F.L.P. Veenstra, L.R.L. Ting, R. García-Muelas, F. Dattila et al., Mechanistic routes toward C3 products in copper-catalysed CO2 electroreduction. Catal. Sci. Technol. 12, 409–417 (2022). https://doi.org/10.1039/d1cy01423d
G.M. Tomboc, S. Choi, T. Kwon, Y.J. Hwang, K. Lee, Potential link between Cu surface and selective CO2 electroreduction: perspective on future electrocatalyst designs. Adv. Mater. 32, e1908398 (2020). https://doi.org/10.1002/adma.201908398
N.J. Azhari, N. Nurdini, S. Mardiana, T. Ilmi, A.T.N. Fajar et al., Zeolite-based catalyst for direct conversion of CO2 to C2+ hydrocarbon: a review. J. CO2 Util. 59, 101969 (2022). https://doi.org/10.1016/j.jcou.2022.101969
L. Liao, G. Xie, X. Xie, N. Zhang, Advances in modulating the activity and selectivity of photocatalytic CO2 reduction to multicarbon products. J. Phys. Chem. C 127, 2766–2781 (2023). https://doi.org/10.1021/acs.jpcc.2c08963
M.E. Aguirre, R. Zhou, A.J. Eugene, M.I. Guzman, M.A. Grela, Cu2O/TiO2 heterostructures for CO2 reduction through a direct Z-scheme: protecting Cu2O from photocorrosion. Appl. Catal. B Environ. 217, 485–493 (2017). https://doi.org/10.1016/j.apcatb.2017.05.058
S. Rej, M. Bisetto, A. Naldoni, P. Fornasiero, Well-defined Cu2O photocatalysts for solar fuels and chemicals. J. Mater. Chem. A 9, 5915–5951 (2021). https://doi.org/10.1039/D0TA10181H
L. Yu, G. Li, X. Zhang, X. Ba, G. Shi et al., Enhanced activity and stability of carbon-decorated cuprous oxide mesoporous nanorods for CO2 reduction in artificial photosynthesis. ACS Catal. 6, 6444–6454 (2016). https://doi.org/10.1021/acscatal.6b01455
H. Huo, D. Liu, H. Feng, Z. Tian, X. Liu et al., Double-shelled Cu2O/MnOx mesoporous hollow structure for CO2 photoreduction with enhanced stability and activity. Nanoscale 12, 13912–13917 (2020). https://doi.org/10.1039/D0NR02944K
L. Cui, L. Hu, Q. Shen, X. Liu, H. Jia et al., Three-dimensional porous Cu2O with dendrite for efficient photocatalytic reduction of CO2 under visible light. Appl. Surf. Sci. 581, 152343 (2022). https://doi.org/10.1016/j.apsusc.2021.152343
S. Wang, X. Bai, Q. Li, Y. Ouyang, L. Shi et al., Selective visible-light driven highly efficient photocatalytic reduction of CO2 to C2H5OH by two-dimensional Cu2S monolayers. Nanoscale Horiz. 6, 661–668 (2021). https://doi.org/10.1039/d1nh00196e
H. Park, H.-H. Ou, A.J. Colussi, M.R. Hoffmann, Artificial photosynthesis of C1–C3 hydrocarbons from water and CO2 on titanate nanotubes decorated with nanoparticle elemental copper and CdS quantum dots. J. Phys. Chem. A 119, 4658–4666 (2015). https://doi.org/10.1021/jp511329d
P. Li, L. Liu, W. An, H. Wang, H. Guo et al., Ultrathin porous g-C3N4 nanosheets modified with AuCu alloy nanoparticles and C–C coupling photothermal catalytic reduction of CO2 to ethanol. Appl. Catal. B Environ. 266, 118618 (2020). https://doi.org/10.1016/j.apcatb.2020.118618
Y. Yu, X.-A. Dong, P. Chen, Q. Geng, H. Wang et al., Synergistic effect of Cu single atoms and Au–Cu alloy nanoparticles on TiO2 for efficient CO2 photoreduction. ACS Nano 15, 14453–14464 (2021). https://doi.org/10.1021/acsnano.1c03961
S. Sorcar, Y. Hwang, J. Lee, H. Kim, K.M. Grimes et al., CO2, water, and sunlight to hydrocarbon fuels: a sustained sunlight to fuel (Joule-to-Joule) photoconversion efficiency of 1%. Energy Environ. Sci. 12, 2685–2696 (2019). https://doi.org/10.1039/C9EE00734B
Y. Zhang, B. Xia, J. Ran, K. Davey, S.Z. Qiao, Atomic-level reactive sites for semiconductor-based photocatalytic CO2 reduction. Adv. Energy Mater. 10, 1903879 (2020). https://doi.org/10.1002/aenm.201903879
J. Zhang, S. Yuan, J. Lin, Cd1–xZnxS nanorod solid solutions with sulfur vacancies as effective electron traps for highly efficient photocatalytic hydrogen evolution. J. Phys. Chem. C 125, 25600–25607 (2021). https://doi.org/10.1021/acs.jpcc.1c08457
P. Xia, M. Antonietti, B. Zhu, T. Heil, J. Yu et al., Designing defective crystalline carbon nitride to enable selective CO2 photoreduction in the gas phase. Adv. Funct. Mater. 29, 1900093 (2019). https://doi.org/10.1002/adfm.201900093
J. Wang, C. Yang, L. Mao, X. Cai, Z. Geng et al., Regulating the metallic Cu–Ga bond by S vacancy for improved photocatalytic CO2 reduction to C2H4. Adv. Funct. Mater. 33, 2213901 (2023). https://doi.org/10.1002/adfm.202213901
C. Gao, J. Low, R. Long, T. Kong, J. Zhu et al., Heterogeneous single-atom photocatalysts: fundamentals and applications. Chem. Rev. 120, 12175–12216 (2020). https://doi.org/10.1021/acs.chemrev.9b00840
M.B. Gawande, K. Ariga, Y. Yamauchi, Single-atom catalysts. Small 17, 2101584 (2021). https://doi.org/10.1002/smll.202101584
Y. Xia, M. Sayed, L. Zhang, B. Cheng, J. Yu, Single-atom heterogeneous photocatalysts. Chem. Catal. 1, 1173–1214 (2021). https://doi.org/10.1016/j.checat.2021.08.009
C.B. Hiragond, N.S. Powar, J. Lee, S.-I. In, Single-atom catalysts (SACs) for photocatalytic CO2 reduction with H2O: activity, product selectivity, stability, and surface chemistry. Small 18, 2201428 (2022). https://doi.org/10.1002/smll.202201428
A. Zhang, M. Zhou, S. Liu, M. Chai, S. Jiang, Synthesis of single-atom catalysts through top-down atomization approaches. Front. Catal. 1, 754167 (2021). https://doi.org/10.3389/fctls.2021.754167
X. Tan, H. Li, S. Yang, Single-atom catalysts-enabled reductive upgrading of CO2. ChemCatChem 13, 4859–4877 (2021). https://doi.org/10.1002/cctc.202100966
W. Xie, K. Li, X.-H. Liu, X. Zhang, H. Huang, P-mediated Cu–N4 sites in carbon nitride realizing CO2 photoreduction to C2 H4 with selectivity modulation. Adv. Mater. 35, e2208132 (2023). https://doi.org/10.1002/adma.202208132
G. Wang, C.-T. He, R. Huang, J. Mao, D. Wang et al., Photoinduction of Cu single atoms decorated on UiO-66-NH2 for enhanced photocatalytic reduction of CO2 to liquid fuels. J. Am. Chem. Soc. 142, 19339–19345 (2020). https://doi.org/10.1021/jacs.0c09599
R. Xu, D.-H. Si, S.-S. Zhao, Q.-J. Wu, X.-S. Wang et al., Tandem photocatalysis of CO2 to C2H4 via a synergistic rhenium-(I) bipyridine/copper-porphyrinic triazine framework. J. Am. Chem. Soc. 145, 8261–8270 (2023). https://doi.org/10.1021/jacs.3c02370
N. Li, B. Wang, Y. Si, F. Xue, J. Zhou et al., Toward high-value hydrocarbon generation by photocatalytic reduction of CO2 in water vapor. ACS Catal. 9, 5590–5602 (2019). https://doi.org/10.1021/acscatal.9b00223
H. Shi, H. Wang, Y. Zhou, J. Li, P. Zhai et al., Atomically dispersed indium-copper dual-metal active sites promoting C–C coupling for CO2 photoreduction to ethanol. Angew. Chem. Int. Ed. 61, e202208904 (2022). https://doi.org/10.1002/anie.202208904
T. Wang, L. Chen, C. Chen, M. Huang, Y. Huang et al., Engineering catalytic interfaces in Cuδ+/CeO2–TiO2 photocatalysts for synergistically boosting CO2 reduction to ethylene. ACS Nano 16, 2306–2318 (2022). https://doi.org/10.1021/acsnano.1c08505
D. Zeng, H. Wang, X. Zhu, H. Cao, W. Wang et al., Photocatalytic conversion of CO2 to acetic acid by CuPt/WO3: chloride enhanced C–C coupling mechanism. Appl. Catal. B Environ. 323, 122177 (2023). https://doi.org/10.1016/j.apcatb.2022.122177
Y. Shen, C. Ren, L. Zheng, X. Xu, R. Long et al., Room-temperature photosynthesis of propane from CO2 with Cu single atoms on vacancy-rich TiO2. Nat. Commun. 14, 1117 (2023). https://doi.org/10.1038/s41467-023-36778-5
J.J. Plata, J. Graciani, J. Evans, J.A. Rodriguez, J.F. Sanz, Cu deposited on CeOx-modified TiO2(110): synergistic effects at the metal–oxide interface and the mechanism of the WGS reaction. ACS Catal. 6, 4608–4615 (2016). https://doi.org/10.1021/acscatal.6b00948
J. Yang, Y. Huang, H. Qi, C. Zeng, Q. Jiang et al., Modulating the strong metal-support interaction of single-atom catalysts via vicinal structure decoration. Nat. Commun. 13, 4244 (2022). https://doi.org/10.1038/s41467-022-31966-1
T. Pu, W. Zhang, M. Zhu, Engineering heterogeneous catalysis with strong metal–support interactions: characterization, theory and manipulation. Angew. Chem. Int. Ed. 62, e202212278 (2023). https://doi.org/10.1002/anie.202212278
S. Yu, P.K. Jain, Plasmonic photosynthesis of C1–C3 hydrocarbons from carbon dioxide assisted by an ionic liquid. Nat. Commun. 10, 2022 (2019). https://doi.org/10.1038/s41467-019-10084-5
H. Zhang, X. Chang, J.G. Chen, W.A. Goddard 3rd., B. Xu et al., Computational and experimental demonstrations of one-pot tandem catalysis for electrochemical carbon dioxide reduction to methane. Nat. Commun. 10, 3340 (2019). https://doi.org/10.1038/s41467-019-11292-9
D.-L. Meng, M.-D. Zhang, D.-H. Si, M.-J. Mao, Y. Hou et al., Highly selective tandem electroreduction of CO2 to ethylene over atomically isolated nickel-nitrogen site/copper nanoparticle catalysts. Angew. Chem. Int. Ed. 60, 25485–25492 (2021). https://doi.org/10.1002/anie.202111136
T. Zhang, J.C. Bui, Z. Li, A.T. Bell, A.Z. Weber et al., Highly selective and productive reduction of carbon dioxide to multicarbon products via in situ CO management using segmented tandem electrodes. Nat. Catal. 5, 202–211 (2022). https://doi.org/10.1038/s41929-022-00751-0
F. Zhang, Y.-H. Li, M.-Y. Qi, Z.-R. Tang, Y.-J. Xu, Boosting the activity and stability of Ag-Cu2O/ZnO nanorods for photocatalytic CO2 reduction. Appl. Catal. B Environ. 268, 118380 (2020). https://doi.org/10.1016/j.apcatb.2019.118380
Z. Jiang, W. Wan, H. Li, S. Yuan, H. Zhao, P.K. Wong, A hierarchical Z-scheme α-Fe2O3/g-C3N4 hybrid for enhanced photocatalytic CO2 reduction. Adv. Mater. 30(10), 1706108 (2018). https://doi.org/10.1002/adma.201706108
B. Wang, J. Zhao, H. Chen, Y.-X. Weng, H. Tang et al., Unique Z-scheme carbonized polymer dots/Bi4O5Br2 hybrids for efficiently boosting photocatalytic CO2 reduction. Appl. Catal. B Environ. 293, 120182 (2021). https://doi.org/10.1016/j.apcatb.2021.120182
C.-Y. Wu, C.-J. Lee, Y.-H. Yu, H.-W. Tsao, Y.-H. Su et al., Efficacious CO2 photoconversion to C2 and C3 hydrocarbons on upright SnS–SnS2 heterojunction nanosheet frameworks. ACS Appl. Mater. Interfaces 13, 4984–4992 (2021). https://doi.org/10.1021/acsami.0c18420
I. Shown, H.C. Hsu, Y.C. Chang, C.H. Lin, P.K. Roy et al., Highly efficient visible light photocatalytic reduction of CO2 to hydrocarbon fuels by Cu-nanoparticle decorated graphene oxide. Nano Lett. 14, 6097–6103 (2014). https://doi.org/10.1021/nl503609v
H. Ge, B. Zhang, H. Liang, M. Zhang, K. Fang et al., Photocatalytic conversion of CO2 into light olefins over TiO2 nanotube confined Cu clusters with high ratio of Cu+. Appl. Catal. B Environ. 263, 118133 (2020). https://doi.org/10.1016/j.apcatb.2019.118133
L. Zhang, T. Liu, T. Liu, S. Hussain, Q. Li et al., Improving photocatalytic performance of defective titania for carbon dioxide photoreduction by Cu cocatalyst with SCN- ion modification. Chem. Eng. J. 463, 142358 (2023). https://doi.org/10.1016/j.cej.2023.142358
A.E. Nogueira, G.T.S.T. Silva, J.A. Oliveira, O.F. Lopes, J.A. Torres et al., CuO decoration controls Nb2O5 photocatalyst selectivity in CO2 reduction. ACS Appl. Energy Mater. 3, 7629–7636 (2020). https://doi.org/10.1021/acsaem.0c01047
H. Du, X. Gao, Q. Ma, X. Yang, T.-S. Zhao, Cu/PCN metal-semiconductor heterojunction by thermal reduction for photoreaction of CO2-aerated H2O to CH3OH and C2H5OH. ACS Omega 7, 16817–16826 (2022). https://doi.org/10.1021/acsomega.2c01827
H. Du, Q. Ma, X. Gao, T.-S. Zhao, Cu/ZnV2O4 heterojunction interface promoted methanol and ethanol generation from CO2 and H2O under UV–vis light irradiation. ACS Omega 7, 7278–7286 (2022). https://doi.org/10.1021/acsomega.1c07108
S. Xie, Y. Li, B. Sheng, W. Zhang, W. Wang et al., Self-reconstruction of paddle-wheel copper-node to facilitate the photocatalytic CO2 reduction to ethane. Appl. Catal. B Environ. 310, 121320 (2022). https://doi.org/10.1016/j.apcatb.2022.121320
N. Li, X. Liu, J. Zhou, W. Chen, M. Liu, Encapsulating CuO quantum dots in MIL-125(Ti) coupled with g-C3N4 for efficient photocatalytic CO2 reduction. Chem. Eng. J. 399, 125782 (2020). https://doi.org/10.1016/j.cej.2020.125782
X. Yu, V.V. Ordomsky, A.Y. Khodakov, Selective deposition of cobalt and copper oxides on BiVO4 facets for enhancement of CO2 photocatalytic reduction to hydrocarbons. ChemCatChem 12, 740–749 (2020). https://doi.org/10.1002/cctc.201901115
J. Cai, Y. Xiao, Y. Tursun, A. Abulizi, Z-type heterojunction of Cu2O-modified layered BiOI composites with superior photocatalytic performance for CO2 reduction. Mater. Sci. Semicond. Process. 149, 106891 (2022). https://doi.org/10.1016/j.mssp.2022.106891
D. Kim, C.S. Kley, Y. Li, P. Yang, Copper nanoparticle ensembles for selective electroreduction of CO2 to C2–C3 products. Proc. Natl. Acad. Sci. U.S.A. 114, 10560–10565 (2017). https://doi.org/10.1073/pnas.1711493114
B. Yang, L. Chen, S. Xue, H. Sun, K. Feng et al., Electrocatalytic CO2 reduction to alcohols by modulating the molecular geometry and Cu coordination in bicentric copper complexes. Nat. Commun. 13, 5122 (2022). https://doi.org/10.1038/s41467-022-32740-z
C.W. Li, M.W. Kanan, CO2 reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films. J. Am. Chem. Soc. 134, 7231–7234 (2012). https://doi.org/10.1021/ja3010978
Y. Song, R. Peng, D.K. Hensley, P.V. Bonnesen, L. Liang et al., High-selectivity electrochemical conversion of CO2 to ethanol using a copper nanoparticle/N-doped graphene electrode. ChemistrySelect 1, 6055–6061 (2016). https://doi.org/10.1002/slct.201601169
C. Reller, R. Krause, E. Volkova, B. Schmid, S. Neubauer et al., Selective electroreduction of CO2 toward ethylene on nano dendritic copper catalysts at high current density. Adv. Energy Mater. 7, 1602114 (2017). https://doi.org/10.1002/aenm.201602114
D. Ren, Y. Deng, A.D. Handoko, C.S. Chen, S. Malkhandi et al., Selective electrochemical reduction of carbon dioxide to ethylene and ethanol on copper(I) oxide catalysts. ACS Catal. 5, 2814–2821 (2015). https://doi.org/10.1021/cs502128q
K.P. Kuhl, E.R. Cave, D.N. Abram, T.F. Jaramillo, New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 5, 7050–7059 (2012). https://doi.org/10.1039/C2EE21234J
K.L. Bae, J. Kim, C.K. Lim, K.M. Nam, H. Song, Colloidal zinc oxide-copper(I) oxide nanocatalysts for selective aqueous photocatalytic carbon dioxide conversion into methane. Nat. Commun. 8, 1156 (2017). https://doi.org/10.1038/s41467-017-01165-4
A.S. Varela, M. Kroschel, T. Reier, P. Strasser, Controlling the selectivity of CO2 electroreduction on copper: the effect of the electrolyte concentration and the importance of the local pH. Catal. Today 260, 8–13 (2016). https://doi.org/10.1016/j.cattod.2015.06.009
C.T. Dinh, T. Burdyny, M.G. Kibria, A. Seifitokaldani, C.M. Gabardo et al., CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 360, 783–787 (2018). https://doi.org/10.1126/science.aas9100
K.D. Yang, W.R. Ko, J.H. Lee, S.J. Kim, H. Lee et al., Morphology-directed selective production of ethylene or ethane from CO2 on a Cu mesopore electrode. Angew. Chem. Int. Ed. Engl. 56, 796–800 (2017). https://doi.org/10.1002/anie.201610432
A.S. Varela, W. Ju, T. Reier, P. Strasser, Tuning the catalytic activity and selectivity of Cu for CO2 electroreduction in the presence of halides. ACS Catal. 6, 2136–2144 (2016). https://doi.org/10.1021/acscatal.5b02550
Y. Lum, B. Yue, P. Lobaccaro, A.T. Bell, J.W. Ager, Optimizing C–C coupling on oxide-derived copper catalysts for electrochemical CO2 reduction. J. Phys. Chem. C 121, 14191–14203 (2017). https://doi.org/10.1021/acs.jpcc.7b03673
W. Luo, X. Nie, M.J. Janik, A. Asthagiri, Facet dependence of CO2 reduction paths on Cu electrodes. ACS Catal. 6, 219–229 (2016). https://doi.org/10.1021/acscatal.5b01967
T. Cheng, H. Xiao, W.A. Goddard 3rd., Full atomistic reaction mechanism with kinetics for CO reduction on Cu(100) from ab initio molecular dynamics free-energy calculations at 298 K. Proc. Natl. Acad. Sci. U.S.A. 114, 1795–1800 (2017). https://doi.org/10.1073/pnas.1612106114
W. Tang, A.A. Peterson, A.S. Varela, Z.P. Jovanov, L. Bech et al., The importance of surface morphology in controlling the selectivity of polycrystalline copper for CO2 electroreduction. Phys. Chem. Chem. Phys. 14, 76–81 (2012). https://doi.org/10.1039/c1cp22700a
A. Eilert, F. Cavalca, F.S. Roberts, J. Osterwalder, C. Liu et al., Subsurface oxygen in oxide-derived copper electrocatalysts for carbon dioxide reduction. J. Phys. Chem. Lett. 8, 285–290 (2017). https://doi.org/10.1021/acs.jpclett.6b02273
R. Yang, J. Duan, P. Dong, Q. Wen, M. Wu et al., In situ halogen-ion leaching regulates multiple sites on tandem catalysts for efficient CO2 electroreduction to C2+ products. Angew. Chem. Int. Ed. 61, e202116706 (2022). https://doi.org/10.1002/anie.202116706
B. Deng, M. Huang, K. Li, X. Zhao, Q. Geng et al., Crystal plane is not the key factor for CO2-to-methane electrosynthesis on reconstructed Cu2O microparticles. Angew. Chem. Int. Ed. 61, 202114080 (2021). https://doi.org/10.1002/anie.202114080
P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M.B. Ross, O.S. Bushuyev et al., Catalyst electro-redeposition controls morphology and oxidation state for selective carbon dioxide reduction. Nat. Catal. 1, 103–110 (2018). https://doi.org/10.1038/s41929-017-0018-9
Y. Hori, I. Takahashi, O. Koga, N. Hoshi, Electrochemical reduction of carbon dioxide at various series of copper single crystal electrodes. J. Mol. Catal. A Chem. 199, 39–47 (2003). https://doi.org/10.1016/S1381-1169(03)00016-5
F.S. Roberts, K.P. Kuhl, A. Nilsson, High selectivity for ethylene from carbon dioxide reduction over copper nanocube electrocatalysts. Angew. Chem. Int. Ed. 54, 5179–5182 (2015). https://doi.org/10.1002/anie.201412214
A. Loiudice, P. Lobaccaro, E.A. Kamali, T. Thao, B.H. Huang et al., Tailoring copper nanocrystals towards C2 products in electrochemical CO2 reduction. Angew. Chem. Int. Ed. 128, 5883–5886 (2016). https://doi.org/10.1002/ange.201601582
M. Ma, K. Djanashvili, W.A. Smith, Controllable hydrocarbon formation from the electrochemical reduction of CO2 over Cu nanowire arrays. Angew. Chem. Int. Ed. 55, 6680–6684 (2016). https://doi.org/10.1002/anie.201601282
Z. Wang, G. Yang, Z. Zhang, M. Jin, Y. Yin, Selectivity on etching: creation of high-energy facets on copper nanocrystals for CO2 electrochemical reduction. ACS Nano 10, 4559–4564 (2016). https://doi.org/10.1021/acsnano.6b00602
H. Xiao, W.A. Goddard 3rd., T. Cheng, Y. Liu, Cu metal embedded in oxidized matrix catalyst to promote CO2 activation and CO dimerization for electrochemical reduction of CO2. Proc. Natl. Acad. Sci. U.S.A. 114, 6685–6688 (2017). https://doi.org/10.1073/pnas.1702405114
H. Mistry, A.S. Varela, C.S. Bonifacio, I. Zegkinoglou, I. Sinev et al., Highly selective plasma-activated copper catalysts for carbon dioxide reduction to ethylene. Nat. Commun. 7, 12123 (2016). https://doi.org/10.1038/ncomms12123
H. Jung, S.Y. Lee, C.W. Lee, M.K. Cho, D.H. Won et al., Electrochemical fragmentation of Cu2O nanoparticles enhancing selective C–C coupling from CO2 reduction reaction. J. Am. Chem. Soc. 141, 4624–4633 (2019). https://doi.org/10.1021/jacs.8b11237
H. Xiao, T. Cheng, W.A. Goddard 3rd., Atomistic mechanisms underlying selectivities in C(1) and C(2) products from electrochemical reduction of CO on Cu(111). J. Am. Chem. Soc. 139, 130–136 (2017). https://doi.org/10.1021/jacs.6b06846
T. Burdyny, P.J. Graham, Y. Pang, C.-T. Dinh, M. Liu et al., Nanomorphology-enhanced gas-evolution intensifies CO2 reduction electrochemistry. ACS Sustain. Chem. Eng. 5, 4031–4040 (2017). https://doi.org/10.1021/acssuschemeng.7b00023
P. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng et al., Boosting electrocatalytic CO2-to-ethanol production via asymmetric C–C coupling. Nat. Commun. 13, 3754 (2022). https://doi.org/10.1038/s41467-022-31427-9
C.G. Morales-Guio, E.R. Cave, S. Nitopi, J.T. Feaster, L. Wang et al., Improved CO2 reduction activity towards C2+ alcohols on a tandem gold on copper electrocatalyst. Nat. Catal. 1, 764–771 (2018). https://doi.org/10.1038/s41929-018-0139-9
W. Zhang, P. He, C. Wang, T. Ding, T. Chen et al., Operando evidence of Cu+ stabilization via a single-atom modifier for CO2 electroreduction. J. Mater. Chem. A 8, 25970–25977 (2020). https://doi.org/10.1039/D0TA08369K
W. Guo, S. Liu, X. Tan, R. Wu, X. Yan et al., Highly efficient CO2 electroreduction to methanol through atomically dispersed Sn coupled with defective CuO catalysts. Angew. Chem. Int. Ed. 60, 21979–21987 (2021). https://doi.org/10.1002/anie.202108635
W. Li, L. Li, Q. Xia, S. Hong, L. Wang et al., Lowering C−C coupling barriers for efficient electrochemical CO2 reduction to C2H4 by jointly engineering single Bi atoms and oxygen vacancies on CuO. Appl. Catal. B Environ. 318, 121823 (2022). https://doi.org/10.1016/j.apcatb.2022.121823
J. Feng, L. Wu, S. Liu, L. Xu, X. Song et al., Improving CO2-to-C2+ product electroreduction efficiency via atomic lanthanide dopant-induced tensile-strained CuOx catalysts. J. Am. Chem. Soc. 145, 9857–9866 (2023). https://doi.org/10.1021/jacs.3c02428
A. Vasileff, C. Xu, Y. Jiao, Y. Zheng, S.-Z. Qiao, Surface and interface engineering in copper-based bimetallic materials for selective CO2 electroreduction. Chem 4, 1809–1831 (2018). https://doi.org/10.1016/j.chempr.2018.05.001
Z. Yang, F.E. Oropeza, K.H.L. Zhang, P-block metal-based (Sn, In, Bi, Pb) electrocatalysts for selective reduction of CO2 to formate. APL Mater. 8, 060901 (2020). https://doi.org/10.1063/5.0004194
D. Ren, B.S.-H. Ang, B.S. Yeo, Tuning the selectivity of carbon dioxide electroreduction toward ethanol on oxide-derived CuxZn catalysts. ACS Catal. 6, 8239–8247 (2016). https://doi.org/10.1021/acscatal.6b02162
Y. Feng, Z. Li, H. Liu, C. Dong, J. Wang et al., Laser-prepared CuZn alloy catalyst for selective electrochemical reduction of CO2 to ethylene. Langmuir 34, 13544–13549 (2018). https://doi.org/10.1021/acs.langmuir.8b02837
J. Huang, M. Mensi, E. Oveisi, V. Mantella, R. Buonsanti, Structural sensitivities in bimetallic catalysts for electrochemical CO2 reduction revealed by Ag–Cu nanodimers. J. Am. Chem. Soc. 141, 2490–2499 (2019). https://doi.org/10.1021/jacs.8b12381
T. Zhang, Z. Li, J. Zhang, J. Wu, Enhance CO2-to-C2+ products yield through spatial management of CO transport in Cu/ZnO tandem electrodes. J. Catal. 387, 163–169 (2020). https://doi.org/10.1016/j.jcat.2020.05.002
A.H.M. da Silva, S.J. Raaijman, C.S. Santana, J.M. Assaf, J.F. Gomes et al., Electrocatalytic CO2 reduction to C2+ products on Cu and CuxZny electrodes: effects of chemical composition and surface morphology. J. Electroanal. Chem. 880, 114750 (2021). https://doi.org/10.1016/j.jelechem.2020.114750
J. Timoshenko, H.S. Jeon, I. Sinev, F.T. Haase, A. Herzog et al., Linking the evolution of catalytic properties and structural changes in copper–zinc nanocatalysts using operando EXAFS and neural-networks. Chem. Sci. 11, 3727–3736 (2020). https://doi.org/10.1039/D0SC00382D
T.T.H. Hoang, S. Verma, S. Ma, T.T. Fister, J. Timoshenko et al., Nanoporous copper–silver alloys by additive-controlled electrodeposition for the selective electroreduction of CO2 to ethylene and ethanol. J. Am. Chem. Soc. 140, 5791–5797 (2018). https://doi.org/10.1021/jacs.8b01868
D. Wei, Y. Wang, C.-L. Dong, Z. Zhang, X. Wang et al., Decrypting the controlled product selectivity over Ag–Cu bimetallic surface alloys for electrochemical CO2 reduction. Angew. Chem. Int. Ed. 62, e202217369 (2023). https://doi.org/10.1002/anie.202217369
Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu et al., Design of single-atom co-N5 catalytic site: a robust electrocatalyst for CO2 reduction with nearly 100% CO selectivity and remarkable stability. J. Am. Chem. Soc. 140, 4218–4221 (2018). https://doi.org/10.1021/jacs.8b00814
H. Zhang, J. Li, S. Xi, Y. Du, X. Hai et al., A graphene-supported single-atom FeN5 catalytic site for efficient electrochemical CO2 reduction. Angew. Chem. Int. Ed. 58, 14871–14876 (2019). https://doi.org/10.1002/anie.201906079
Y. Cai, J. Fu, Y. Zhou, Y.-C. Chang, Q. Min et al., Insights on forming N, O-coordinated Cu single-atom catalysts for electrochemical reduction CO2 to methane. Nat. Commun. 12, 586 (2021). https://doi.org/10.1038/s41467-020-20769-x
A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang et al., Boosting CO2 electroreduction to CH4 via tuning neighboring single-copper sites. ACS Energy Lett. 5, 1044–1053 (2020). https://doi.org/10.1021/acsenergylett.0c00018
D.-H. Nam, O.S. Bushuyev, J. Li, P. De Luna, A. Seifitokaldani et al., Metal-organic frameworks mediate Cu coordination for selective CO2 electroreduction. J. Am. Chem. Soc. 140, 11378–11386 (2018). https://doi.org/10.1021/jacs.8b06407
D. Karapinar, N.T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley et al., Electroreduction of CO2 on single-site copper-nitrogen-doped carbon material: selective formation of ethanol and reversible restructuration of the metal sites. Angew. Chem. Int. Ed. 58, 15098–15103 (2019). https://doi.org/10.1002/anie.201907994
H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu et al., Highly selective electrocatalytic CO2 reduction to ethanol by metallic clusters dynamically formed from atomically dispersed copper. Nat. Energy 5, 623–632 (2020). https://doi.org/10.1038/s41560-020-0666-x
Y. Liang, J. Zhao, Y. Yang, S.-F. Hung, J. Li et al., Stabilizing copper sites in coordination polymers toward efficient electrochemical C–C coupling. Nat. Commun. 14, 474 (2023). https://doi.org/10.1038/s41467-023-35993-4
K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan et al., Selective electroreduction of CO2 to acetone by single copper atoms anchored on N-doped porous carbon. Nat. Commun. 11, 2455 (2020). https://doi.org/10.1038/s41467-020-16381-8
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22178149), Jiangsu Distinguished Professor Program, Natural Science Foundation of Jiangsu Province for Outstanding Youth Scientists (BK20211599), Key R and D Project of Zhenjiang City (CQ2022001), Scientific Research Startup Foundation of Jiangsu University (Nos. 202096 and 22JDG020), Open Project Program of the State Key Laboratory of Photocatalysis on Energy and Environment of Fuzhou University (SKLPEE-KF202310), and the Opening Project of Structural Optimization and Application of Functional Molecules Key Laboratory of Sichuan Province (2023GNFZ-01).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The author declares no interest conflict. He has no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rhimi, B., Zhou, M., Yan, Z. et al. Cu-Based Materials for Enhanced C2+ Product Selectivity in Photo-/Electro-Catalytic CO2 Reduction: Challenges and Prospects. Nano-Micro Lett. 16, 64 (2024). https://doi.org/10.1007/s40820-023-01276-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40820-023-01276-2