
Highlights
-
JUC-610-nanosheet exhibits highly efficient oxygen reduction reaction (ORR) catalytic activity in alkaline electrolyte with half-wave potential of 0.72 V versus reversible hydrogen electrode, which is one of the best covalent organic frameworks (COF)-based ORR electrocatalysts reported so far.
-
It has been confirmed by experiments and density functional theory calculations that the abundant electrophilic structure in Q3CTP-COFs induces a highly density of carbon active sites, and the unique bilayer stacking facilitates the exposure of active carbon sites and accelerates the mass diffusion during ORR.
-
JUC-610-nanosheet can also serve as a promising cathode for Zn-air batteries (power density of 156 mW cm–2 at 300 mA cm–2), which promotes the development of metal-free carbon-based electrocatalysts.
Abstract
Metal-free carbon-based materials are considered as promising oxygen reduction reaction (ORR) electrocatalysts for clean energy conversion, and their highly dense and exposed carbon active sites are crucial for efficient ORR. In this work, two unique quasi-three-dimensional cyclotriphosphazene-based covalent organic frameworks (Q3CTP-COFs) and their nanosheets were successfully synthesized and applied as ORR electrocatalysts. The abundant electrophilic structure in Q3CTP-COFs induces a high density of carbon active sites, and the unique bilayer stacking of [6 + 3] imine-linked backbone facilitates the exposure of active carbon sites and accelerates mass diffusion during ORR. In particular, bulk Q3CTP-COFs can be easily exfoliated into thin COF nanosheets (NSs) due to the weak interlayer π–π interactions. Q3CTP-COF NSs exhibit highly efficient ORR catalytic activity (half-wave potential of 0.72 V vs. RHE in alkaline electrolyte), which is one of the best COF-based ORR electrocatalysts reported so far. Furthermore, Q3CTP-COF NSs can serve as a promising cathode for Zn-air batteries (delivered power density of 156 mW cm–2 at 300 mA cm–2). This judicious design and accurate synthesis of such COFs with highly dense and exposed active sites and their nanosheets will promote the development of metal-free carbon-based electrocatalysts.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Oxygen reduction reaction (ORR) plays a significant role in clean and sustainable energy conversion, such as metal-air batteries and fuel cells [1,2,3,4]. Pt-based materials are currently considered as most efficient electrocatalysts for ORR. However, their high-cost, scarcity, and instability in operation conditions restrict the future large-scale applications of these energy conversion devices [5,6,7]. In the past decade, metal-free materials (MFMs), such as porous carbon and porous organic polymers, have exerted a great effect on the design of low-cost and efficient electrocatalysts for ORR [8,9,10,11,12,13,14,15]. Notably, covalent organic frameworks (COFs) are an emerging class of ORR electrocatalysts due to their high surface areas, tunable porous structures, structural tunability, and well-defined building units [16,17,18,19,20]. Reasonably designing and synthesizing COF materials with chemically adjustable specific blocks can facilitate the development of MFM-based ORR electrocatalysts [21,22,23]. During ORR electrocatalysis process, efficient active sites and fast kinetic mass diffusion of electrocatalysts are indispensable ingredients. Expectantly, it has been confirmed through both experiment and theory that the electronic redistribution of carbon (C) atoms in MFMs can optimize the adsorption and dissociation behaviors for reactant (O2) and intermediates (OOH*, O*, and OH*) [24,25,26], inducing ORR catalytic activity. For example, Dai et al. [27] and Nakamura et al. [28] demonstrated that the C atoms with Lewis basicity (positive charge density) were the efficient ORR active sites. Thus, constructing electrophilic structures in COFs can induce positively charged carbon active sites. Furthermore, optimal pore structures to expose more active sites and excellent conductivity of electrocatalysts can accelerate mass diffusion and electron transport to facilitate the ORR catalytic process [29]. Hence, controllably and precisely synthesizing COFs with highly dense and exposed carbon active sites from the perspective of customizing structures is of great potential to develop highly active metal-free ORR electrocatalysts [30].
Among COF materials, three-dimensional (3D) COFs exhibit hierarchical pore structures to expose numerous open sites [31] profitably, but the structural instability and poor conductivity limit their practical applications as electrocatalysis. Two-dimensional (2D) COFs with excellent interlayer regulation have significant advantages in stable structures and electron transport properties [32, 33] but are restricted by mass diffusion due to their 2D stacking [34, 35]. In view of this situation, it may be possible to construct efficient 2D COF electrocatalysts with quasi-3D (Q-3D) structures and channel features through clever structural design. However, the emergence of structural units centered on the Q-3D cyclotriphosphazene structure makes our hypothesis plausible. The distinctive structure of Q-3D COF makes its 2D planar structure regularly distort, thus creating a new vertical multi-channel to enhance the pore permeability and the mass diffusion [36]. Moreover, the weak interlayered π–π interactions of Q-3D COFs can be easily exfoliated into nanosheets (NSs) [37]. Due to large number of active sites, faster ion diffusion, and high conductivity of thin layers, COF NSs was expected to further improve the performance of electrocatalysts [38,39,40,41]. Therefore, the active carbon sites and mass diffusion requirements of ORR electrocatalysts initiate us to judiciously design and accurately synthesize the novel Q-3D COF NSs with highly exposed carbon active sites for efficient ORR.
Herein, we report the design and synthesis of two unique Q-3D cyclotriphosphazene-based COFs (Q3CTP-COFs, namely JUC-610 and JUC-611, JUC = Jilin University China) with 2D stacking structures and their exfoliated nanosheet (JUC-610-CON) as efficient ORR electrocatalysts. The electrophilic structures of N–P–O blocks in 6-node hexa(4-formyl-phenoxy) cyclotriphosphazene (CTP-6-CHO) and pyridinic-N in 2,4,6-Tris(4-aminophenyl) triazine (TAPT) induce the highly dense C (positively charged) ORR active sites of JUC-610. The novel [6 + 3] imine-linked frameworks [42, 43] exhibit unique bilayer stacking to expose more active sites and promote mass diffusion during ORR. Furthermore, the weak π–π stacking effect between layers of Q3CTP-COFs makes possible to exfoliate ultra-thin nanosheets (~ 4 nm) easily, which can act as an ORR electrocatalyst with half-wave potential of 0.72 V versus RHE in alkaline electrolyte and the cathode for Zn-air batteries (ZABs) with the power density of 156 mW cm–2 at 300 mA cm–2, which surpasses those of almost all reported COF materials [20,21,22,23]. This work represents a newly synthesized COF architecture for efficient ORR and demonstrates its promising potential in metal-air batteries.
2 Experiment and Characterization
2.1 Experiment and Structural Determination of Q3CTP-COFs
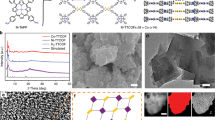
As shown in Scheme 1 and experimental procedures, Q3CTP-COFs were synthesized by condensing CTP-6-CHO with TAPT or 2,4,6-Tris(4-aminophenyl)benzene (TAPB) in the mixed solution of o-DCB/n-BuOH under 120 °C for 72 h with the yields of 75% and 72%, respectively. The powder X-ray diffraction (PXRD) patterns suggested that the crystal structures of Q3CTP-COFs well matched with simulated by using the Materials Studio software package (version 7.0) [44] based on the bilayer hcb net [45] (Fig. 1a, b). After geometrical energy minimization by using the force field to optimize the geometry of the molecular building blocks, the unit cell parameters (a = b = 24.6620 Å, c = 5.9759 Å and α = β = 90°, γ = 120° for JUC-610; a = b = 25.6008 Å, c = 6.4398 Å and α = β = 90°, γ = 120° for JUC-611, respectively) were obtained. Based on P3 space group (No. 143), Bragg peaks at 2θ = 4.14°, 7.18°, and 10.95° for JUC-610 were correspond to the (100), (110) and (120) planes, and peaks at 2θ = 4.02°, 6.97° and 10.65° for JUC-611 were correspond to the (100), (110), and (120) planes, respectively. The refinement results yielded unit cell parameters nearly equivalent to the predictions (Rwp = 1.65% and Rp = 1.30% for JUC-610; Rwp = 2.09% and Rp = 1.54% for JUC-611). A comparison between experimental and calculated PXRD curves (AA and AB stacking models) revealed that both COFs crystallized in AA stacking mode (Figs. S1–S2 and Tables S1–S4).

Schematic illustration of constructing JUC-610, JUC-611 (in the mixed solution of o-DCB/n-BuOH under 120 °C for 72 h with the yields of 75% and 72% respectively), and JUC-610-CON

a, b Powder XRD patterns of JUC-610 and JUC-611. c, d N2 adsorption–desorption isotherms at 77 K and pore-size distribution (inset) based on the NLDFT model to the adsorption data for JUC-610 and JUC-611. e, f FT-IR spectra of JUC-610 and JUC-611
2.2 Characterization of Q3CTP-COFs
Nitrogen adsorption–desorption analysis under 77 K of both prepared COFs showed a sharp uptake at a low pressure of P/P0 < 0.05, which is a typical characteristic of microporous material [46,47,48] (Fig. 1c, d). The pore size distribution was calculated on the basis of non-local density functional theory (NLDFT), resulting in a value of 14.0 Å for JUC-610 and 15.1 Å for JUC-611 respectively, which is in good agreement with those of the proposed models (15.6 Å for JUC-610 and 15.8 Å for JUC-611). The Brunauer–Emmett–Teller (BET) equation revealed their surface areas, 475 m2 g−1 for JUC-610 and 694 m2 g−1 for JUC-611 (Figs. S4–S5). The Fourier transform infrared (FT-IR) spectra peak around 1622 cm−1 for JUC-610 and 1624 cm−1 for JUC-611 indicate the formation of C=N (Figs. 1e, f and S6–S7). Interestingly, the FT-IR spectra of CTP-6-CHO, Q3CTP-COFs showed that the P=N/P–O–Ar bonds on the phosphazene ring had barely noticeable different shifts with the peaks of P=N bond at 1207, 1158, and 885 cm−1 and P–O–Ar bond at 959 cm−1 (Fig. 1e, f). Furthermore, the solid-state 13C cross-polarization magic-angle-spinning (CP/MAS) NMR spectra verified that the presence of carbons from the C=N bonds at 162 ppm for JUC-610 and 161 ppm for JUC-611 (Figs. S8–S9). According to the thermogravimetric analysis (TGA), these prepared COFs showed excellent thermal stability and were thermally stable up to ~ 400 °C under nitrogen (Figs. S10–S11). The morphologies of prepared COFs were observed by scanning electron microscopy (SEM) and transmission electron microscope (TEM), in which Q3CTP-COFs showed isometric crystals (Figs. S12–S13). The powder X-ray diffraction (PXRD) patterns revealed that COFs were stable after immersing in acetone and THF solvents and alkaline aqueous solutions for 24 h (Figs. S14–S16). The FT-IR spectra of COFs showed that the C=N bonds still existed after treatment under 6 M KOH for 24 h (Figs. S17–S19). Interestingly, we elaborately selected CTP-6-CHO with the structure of six cross-side arms outside the central plane as the node module of COFs. The CTP-6-CHO has a unique stereoscopic structure in which the O–P–O plane (β plane) is perpendicular to the aromatic ring plane of N3P3 (α plane, see Experimental Procedures) [37, 49]. In addition, the TEM images with elemental mappings verified that C, N, O, and P atoms were homogeneously distributed in Q3CTP-COFs (Fig. 2d-f).

a AFM image of JUC-610-CON, and b the corresponding height curves for the selective areas in a. c Lateral size distribution histogram of JUC-610-CON, and Gaussian fit curve is colored in red. d TEM image of JUC-610-CON. e, f TEM and the related elemental mapping images of carbon, nitrogen, oxygen, and phosphorus for JUC-610 and JUC-611
2.3 Experiment and Characterization of JUC-610-CON
In addition, given consisting of unique rhombus pores, large interlayer spacings, and the uniquely flexible structure of CTP-6-CHO units lead to the weakened interlayer conjugation and possess the larger c distance (Table S1) of JUC-610, and the bulk JUC-610 can be easily exfoliated into ultrathin NSs (denoted as JUC-610-CON) in large quantities (see Supporting Information) only 3 h by using sonication method. The ultrathin nature of JUC-610-CON was uncovered by TEM and AFM technologies. Tyndall effect was observed when a green laser went through the solution of JUC-610-CON (Fig. S20), confirming its colloidal nature. The atomic force microscopy (AFM) image revealed that the thickness of the obtained JUC-610-CON was ~ 4 nm, corresponding to ~ 8 layers and a flake size of ~ 800 nm (Fig. 2a–c and Table S7). Meanwhile, JUC-610-CON exhibited thin nanosheets by TEM and SEM (Figs. 2d and S21). To determine the crystal structure of JUC-610-CON, XRD and nitrogen adsorption–desorption analyses were performed. The results indicate the crystallinity and porosity of JUC-610-CON are identical to that of the bulk JUC-610, but the intensities of the first XRD peak (100) and N2 adsorption isotherm are decreased (Figs. S22–S23).
3 Results and Discussion
3.1 ORR Catalytic Activity
To survey the ORR catalytic activity of prepared COFs and their nanosheets, the rotating disk electrode (RDE) experiments were performed in O2-saturated 0.1 M KOH electrolyte. In addition, JUC-612 was also synthesized by condensing 1,3,5-tri(1,3-Diformyl biphenyl)benzene (TBPB-6-CHO) and TAPT as a control sample for ORR performance (Figs. S24–S31). According to the linear sweep voltammetry (LSV) curves, JUC-610-CON and Q3CTP-COFs exhibited higher ORR performance than that of JUC-612, revealing that the existence of the Q-3D structures and abundant heteroatoms significantly induces positively charged carbon active sites and improves the ORR active sites (Figs. 3a, S32–S36, and Scheme 2). The ORR catalytic activity of JUC-610 was superior to that of JUC-611, which indicates that N atoms in TAPT blocks also play an important role in improving the ORR performance. In addition, the half-wave potential of JUC-610-CON and the bulk JUC-610 were 0.72 and 0.69 V versus RHE, respectively (Fig. S37), suggesting that ultrathin nanosheet structure exposes more active sites and facilitates mass diffusion during ORR process, which can be comparable to previously reported COF materials and most of metal-based ORR electrocatalysts (Fig. 3f and Tables S8–S9). The Tafel slope of JUC-610-CON (61.95 mV dec−1) is lower than those of JUC-610 (65.8 mV dec−1), JUC-611 (70.9 mV dec−1) and JUC-612 (79.7 mV dec−1), indicating the superior ORR kinetics of JUC-610-CON (Fig. 3b). The electrochemically active surface areas (ECSA) of prepared samples were conducted by electrochemical double-layer capacitance (Cdl) (Figs. S38–S41). The Cdl of JUC-610-CON (19.2 mF cm−2) is larger than those of JUC-610 (17.8 mF cm−2), JUC-611 (7.6 mF cm−2), and JUC-612 (7.6 mF cm−2, Fig. 3c). To further explore the intrinsic activity of the prepared COFs, the turnover frequency (TOF) was carried out at 0.7 V versus RHE, indicating that C atoms in adjacent N–O–P atoms are active sites, and the highly dense carbon active sites accelerate mass diffusion during ORR process. As shown in Fig. 3d, the TOF value of JUC-610-CON is 0.0035 s−1, which has higher active site utilization efficiency than Q3CTP-COFs and JUC-612. The mass activity of JUC-610-CON (3.43 A g−1) is also larger than those of JUC-610 (2.89 A g−1), JUC-611 (1.68 A g−1), and JUC-612 (0.46 A g−1). These results thus reveal that the JUC-610-CON exhibits the most efficient ORR catalytic performance due to ultrathin nanosheet structure and the highly dense carbon active sites. Moreover, all the electron transfer numbers (n) of JUC-610-CON, JUC-610, JUC-611, and JUC-612 derived from Koutecky–Levich (K–L) plots at 0.2 V versus RHE (Fig. 3e) were closed to 4 (3.82, 3.43, 3.42, and 3.67 respectively).

a LSV curves of JUC-610-CON, JUC-610, JUC-611, and JUC-612 at 1600 rpm in O2-saturated 0.1 M KOH electrolyte. b Comparison of Tafel plots, c estimated electrochemical bilayer capacitance (Cdl). d TOF and mass activity, and e K–L plots for JUC-610-CON, JUC-610, JUC-611, and JUC-612. f Plot of onset potential against half-wave potential in metal-free organic porous materials without pyrolysis of ORR reactions. g Discharge curves of JUC-610-CON-based ZAB at different current densities (25, 100, 200, and 300 mA cm−2). h The stability of JUC-610-CON-based ZAB at 5 mA cm−2 in ambient air conditions, and photographs of red “COF” LED panel powered by two ZABs in series (inset)

Proposed ORR processes on JUC-610-CON
Subsequently, a ZAB was assembled using the JUC-610-CON as the air cathode due to its excellent ORR activity in alkaline solutions. The current density of JUC-610-CON-based ZAB was about 87.2 mA cm−2 at 1.0 V, and the maximum peak power density at 0.60 V was about 0.15 W cm−2 (Fig. S42). In addition, the JUC-610-CON-based ZAB also exhibited potential plateaus of 1.16, 0.95, 0.76, and 0.64 V at discharge current densities of 25, 100, 200, and 300 mA cm−2, respectively (Fig. 3g). Such ZAB could operate for 200 h with negligible voltage loss by regular replacement of electrolyte (6 M KOH) and zinc plate (Fig. 3h). Two JUC-610-CON-based ZABs in series were able to light a 2 V rated “COF” LED (Fig. 3h inset), which indicates this material is very promising as electrode material in metal-air batteries.
3.2 DFT Calculations
To reveal the location of carbon active sites for ORR in our samples, the DFT calculations were performed (Fig. 4). All calculations were carried out with the Gaussian 09 package and Vienna Ab-initio Simulation Package (VASP). The natural population analysis (NPA) was performed on the theoretical level of B3LYP/6-311G (d, p) using the NBO program. The electrostatic potential was considered in Gaussian 09 to describe the charge distribution of two chemical systems [50, 51]. The average electrostatic potential of O atoms in CTP-6-CHO and N atoms in TAPT were smaller than those of their adjacent C atoms, indicating the relative electrophilicity of the N and O atoms. The NPA charge results illustrated that both C atoms adjacent to the electrophilic O atom in CTP-6-CHO and the electrophilic N atom in TAPT showed positive charge, which are 0.31 and 0.47, respectively (Fig. 4a, b). Moreover, for these active carbon sites in CTP-6-CHO and TAPT, the 3D charge densities of three reactions of ORR exhibited obvious redistribution of electrons between the intermediate and substrate structure, verifying the favorable adsorption of positively charged C atoms to the ORR intermediate.

a, b NPA charges and the corresponding surface electrostatic potential maps of CTP-6-CHO and TAPT calculated by DFT. c, d Side view of the 3D charge densities for the three ORR reaction steps of C site for CTP-6-CHO and TAPT. Gray, purple, red, blue and light pink balls represent C, P, O, N, and H atoms, respectively
4 Conclusions
In summary, we have precisely and controllably synthesized the unique Q3CTP-COFs and their nanosheets using CTP-based blocks, which can act as efficient ORR electrocatalysts for Zn-air batteries. It was confirmed that the electrophilic structures in such Q3CTP-COFs induced abundant positively charged carbon ORR active sites to facilitate O2 adsorption and reduction, which has been supported by DFT calculations. The unique bilayer stacking structures of Q3CTP-COFs promote the exposure of active carbon sites to accelerate ORR process and the mass (ions, O2 and intermediate) diffusion efficiency during ORR. Furthermore, Q3CTP-COFs could be easily exploited into nanosheets, which improves their ORR catalytic activity (half-wave potential of 0.72 V vs. RHE in alkaline electrolyte) and can be applied for promising cathodes for Zn-air batteries (delivered power density of 156 mW cm–2 at 300 mA cm–2). Thus, this work provides a new way to fabricate metal-free ORR electrocatalysts with atomically definite carbon active sites and promotes their extensive application in clean energy conversion devices.
Change history
25 January 2024
ESM missed to be uploaded and is now uploaded to the publication.
Abbreviations
- ORR:
-
Oxygen reduction reaction
- COFs:
-
Covalent organic frameworks
- Q-3D:
-
Quasi-three-dimensional
- CTP:
-
Cyclotriphosphazene
- NSs:
-
Nanosheets
- MFMs:
-
Metal-free materials
- 3D:
-
Three-dimensional
- 2D:
-
Two-dimensional
- JUC:
-
Jilin University China
- LSV:
-
Linear sweep voltammetry
- ECSA:
-
Electrochemically active surface areas
- ZABs:
-
Zn-air batteries
- Cdl :
-
Double-layer capacitance
- TOF:
-
Turnover frequency
- K–L:
-
Koutecky–Levich
References
A. Kumar, Y. Zhang, W. Liu, X. Sun, The chemistry, recent advancements and activity descriptors for macrocycles based electrocatalysts in oxygen reduction reaction. Coord. Chem. Rev. 402, 213047 (2020). https://doi.org/10.1016/j.ccr.2019.213047
A. Kumar, V.K. Vashistha, D.K. Das, Recent development on metal phthalocyanines based materials for energy conversion and storage applications. Coord. Chem. Rev. 431, 213678 (2021). https://doi.org/10.1016/j.ccr.2020.213678
Y. Wang, A. Kumar, M. Ma, Y. Jia, Y. Wang et al., Hierarchical peony-like FeCo-NC with conductive network and highly active sites as efficient electrocatalyst for rechargeable Zn-air battery. Nano Res. 13, 1090 (2020). https://doi.org/10.1007/s12274-020-2751-7
C. Li, N.C.S. Selvam, J. Fang, Shape-controlled synthesis of platinum-based nanocrystals and their electrocatalytic applications in fuel cells. Nano-Micro Lett. 15, 83 (2023). https://doi.org/10.1007/s40820-023-01060-2
C. Zhang, X. Shen, Y. Pan, Z. Peng, A review of Pt-based electrocatalysts for oxygen reduction reaction. Front. Energy 11, 268 (2017). https://doi.org/10.1007/s11708-017-0466-6
H. Cruz-Martinez, H. Rojas-Chavez, P.T. Matadamas-Ortiz, J.C. Ortiz-Herrera, E. Lopez-Chavez et al., Current progress of Pt-based ORR electrocatalysts for PEMFCs: an integrated view combining theory and experiment. Mater. Today Phys. 19, 100406 (2021). https://doi.org/10.1016/j.mtphys.2021.100406
C. Wang, Q. Zhang, B. Yan, B. You, J. Zheng et al., Facet engineering of advanced electrocatalysts toward hydrogen/oxygen evolution reactions. Nano-Micro Lett. 15, 52 (2023). https://doi.org/10.1007/s40820-023-01024-6
Y. Jia, L. Zhang, L. Zhuang, H. Liu, X. Yan et al., Identification of active sites for acidic oxygen reduction on carbon catalysts with and without nitrogen doping. Nat. Catal. 2, 688 (2019). https://doi.org/10.1038/s41929-019-0297-4
Q. Xu, Y. Tang, X. Zhang, Y. Oshima, Q. Chen et al., Template conversion of covalent organic frameworks into 2d conducting nanocarbons for catalyzing oxygen reduction reaction. Adv. Mater. 30, 1706330 (2018). https://doi.org/10.1002/adma.201706330
J. Wu, Z. Yang, X. Li, Q. Sun, C. Jin et al., Phosphorus-doped porous carbons as efficient electrocatalysts for oxygen reduction. J. Mater. Chem. A 1, 9889 (2013). https://doi.org/10.1039/c3ta11849e
D. Li, Y. Jia, G. Chang, J. Chen, H. Liu et al., A defect-driven metal-free electrocatalyst for oxygen reduction in acidic electrolyte. Chem 4, 2345 (2018). https://doi.org/10.1016/j.chempr.2018.07.005
Y. Jia, K. Jiang, H. Wang, X. Yao, The role of defect sites in nanomaterials for electrocatalytic energy conversion. Chem 5, 1371 (2019). https://doi.org/10.1038/s41929-019-0297-4
J. Zhang, G. Chen, K. Muellen, X. Feng, Carbon-rich nanomaterials: fascinating hydrogen and oxygen electrocatalysts. Adv. Mater. 30, 1800528 (2018). https://doi.org/10.1002/adma.201800528
Y. Song, P. Ruan, C. Mao, Y. Chang, L. Wang et al., Metal–organic frameworks functionalized separators for robust aqueous zinc-ion batteries. Nano-Micro Lett. 14, 218 (2022). https://doi.org/10.1007/s40820-023-01024-6
Y. Xue, Y. Guo, Q. Zhang, Z. Xie, J. Wei et al., MOF-derived Co and Fe species loaded on N-doped carbon networks as efficient oxygen electrocatalysts for Zn-air batteries. Nano-Micro Lett. 14, 162 (2022). https://doi.org/10.1007/s40820-022-00890-w
K. Geng, T. He, R. Liu, S. Dalapati, K.T. Tan et al., Covalent organic frameworks: design, synthesis, and functions. Chem. Rev. 120, 8814 (2020). https://doi.org/10.1021/acs.chemrev.9b00550
X. Feng, X. Ding, D. Jiang, Covalent organic frameworks. Chem. Soc. Rev. 41, 6010 (2012). https://doi.org/10.1039/c2cs35157a
X. Guan, F. Chen, Q. Fang, S. Qiu, Design and applications of three dimensional covalent organic frameworks. Chem. Soc. Rev. 49, 1357 (2020). https://doi.org/10.1039/c9cs00911f
Y. Yusran, X. Guan, H. Li, Q. Fang, S. Qiu, Postsynthetic functionalization of covalent organic frameworks. National Sci. Rev. 7, 170 (2020). https://doi.org/10.1093/nsr/nwz122
D. Li, C. Li, L. Zhang, H. Li, L. Zhu et al., Metal-free thiophene-sulfur covalent organic frameworks: precise and controllable synthesis of catalytic active sites for oxygen reduction. J. Am. Chem. Soc. 142, 8104 (2020). https://doi.org/10.1021/jacs.0c02225
S. Roy, S. Mari, M.K. Sai, S.C. Sarma, S. Sarkar et al., Highly efficient bifunctional oxygen reduction/evolution activity of a non-precious nanocomposite derived from a tetrazine-COF. Nanoscale 12, 22718 (2020). https://doi.org/10.1039/d0nr05337f
J.H. Park, C.H. Lee, J.-M. Ju, J.-H. Lee, J. Seol et al., Bifunctional covalent organic framework-derived electrocatalysts with modulated p-band centers for rechargeable Zn-Air batteries. Adv. Funct. Mater. 31, 2101727 (2021). https://doi.org/10.1002/adfm.202101727
Y. Yue, T. Wang, X. Wu, P. Yang, Y. Ma et al., Two-dimensional porphyrin covalent organic frameworks with tunable catalytic active sites for the oxygen reduction reaction. Chem. Commun. 57, 12619 (2021). https://doi.org/10.1039/d1cc04928c
B. Yang, J. Miao, F. Hung, J. Chen, H.B. Tao et al., Identification of catalytic sites for oxygen reduction and oxygen evolution in N-doped graphene materials: development of highly efficient metal-free bifunctional electrocatalyst. Sci. Adv. 2, 1501122 (2016). https://doi.org/10.1126/sciadv.1501122
J. Liang, Y. Jiao, M. Jaroniec, S.Z. Qiao, Sulfur and nitrogen dual-doped mesoporous graphene electrocatalyst for oxygen reduction with synergistically enhanced performance. Angew. Chem. Int. Ed. 51, 11496 (2012). https://doi.org/10.1002/anie.201206720
Y. Su, Z. Yao, F. Zhang, H. Wang, Z. Mics et al., Sulfur-enriched conjugated polymer nanosheet derived sulfur and nitrogen Co-doped porous carbon nanosheets as electrocatalysts for oxygen reduction reaction and zinc-air battery. Adv. Funct. Mater. 26, 5893 (2016). https://doi.org/10.1002/adfm.201602158
K. Gong, F. Du, Z. Xia, M. Durstock, L. Dai, Nitrogen-doped carbon nanotube arrays with high electrocatalytic activity for oxygen reduction. Science 323, 760 (2009). https://doi.org/10.1126/science.1168049
D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo et al., Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts. Science 351, 361 (2016). https://doi.org/10.1126/science.aad0832
Q. Wu, Y. Jia, Q. Liu, X. Mao, Q. Guo et al., Ultra-dense carbon defects as highly active sites for oxygen reduction catalysis. Chem 8, 2715 (2022). https://doi.org/10.1016/j.chempr.2022.06.013
D. Li, X. Long, Y. Wu, H. Hou, X. Wang et al., Hierarchically porous and defective carbon fiber cathode for efficient Zn-air batteries and microbial fuel cells. Adv. Fiber. Mater. 4, 795 (2022). https://doi.org/10.1007/s42765-022-00139-6
B. Gui, H. Ding, Y. Cheng, A. Mal, C. Wang, Structural design and determination of 3D covalent organic frameworks. Trends Chem. 5, 437 (2022). https://doi.org/10.1016/j.trechm.2022.01.002
C. Yang, S. Tao, N. Huang, X. Zhang, J. Duan et al., Heteroatom-doped carbon electrocatalysts derived from nanoporous two-dimensional covalent organic frameworks for oxygen reduction and hydrogen evolution. ACS Appl. Nano Mater. 3, 5481 (2020). https://doi.org/10.1021/acsanm.0c00786
X.-T. Wang, X.-F. Lin, D.-S. Yu, Metal-containing covalent organic framework: a new type of photo/electrocatalyst. Rare Met. 41, 1160 (2022). https://doi.org/10.1007/s12598-021-01873-3
Y. Liu, J. Ren, Y. Wang, X. Zhu, X. Guan et al., A stable luminescent covalent organic framework nanosheet for sensitive molecular recognition. CCS Chem 4, 1 (2022). https://doi.org/10.31635/ccschem.022.202202352
X. Ren, C. Li, W. Kang, L. He, T. Na et al., Enormously promoted photocatalytic activity by using a near-single layer COFs. CCS Chem. 7, 2429 (2021). https://doi.org/10.31635/ccschem.021.202101090
X. Guo, Y. Li, M. Zhang, K. Cao, Y. Tian et al., Colyliform crystalline 2d covalent organic frameworks (COFs) with quasi-3d topologies for rapid I(2)adsorption. Angew. Chem. Int. Ed. 59, 22697 (2020). https://doi.org/10.1002/anie.202010829
X. Li, S. Xu, K. Leng, S.W. Chee, X. Zhao et al., Partitioning the interlayer space of covalent organic frameworks by embedding pseudorotaxanes in their backbones. Nat. Chem. 12, 1115 (2020). https://doi.org/10.1038/s41557-020-00562-5
H.S. Sasmal, A.K. Mahato, P. Majumder, R. Banerjee, Landscaping covalent organic framework nanomorphologies. J. Am. Chem. Soc. 144, 11482 (2022). https://doi.org/10.1021/jacs.2c02301
S. Mohata, K. Dey, S. Bhunia, N. Thomas, E.B. Gowd et al., Dual nanomechanics in anisotropic porous covalent organic framework janus-type thin films. J. Am. Chem. Soc. 144, 400 (2022). https://doi.org/10.1021/jacs.1c10263
A.K. Mahato, S. Bag, H.S. Sasmal, K. Dey et al., Crystallizing sub 10 nm covalent organic framework thin films via interfacial–residual concomitance. J. Am. Chem. Soc. 143, 20916 (2021). https://doi.org/10.1021/jacs.1c09740
K. Dey, S. Mohata, R. Banerjee, Covalent organic frameworks and supramolecular nano-synthesis. ACS Nano 15, 12723 (2021). https://doi.org/10.1021/acsnano.1c05194
Y. Ding, J. Gao, Q. Wang, Y. Zhang, G. Song et al., Construction of covalent organic framework for catalysis: Pd/COF-LZU1 in suzuki–miyaura coupling reaction. J. Am. Chem. Soc. 133, 19816 (2021). https://doi.org/10.1021/ja206846p
F.J. Uribe-Romo, J.R. Hunt, H. Furukawa, C. Klock, M. O’Keeffe et al., A crystalline imine-linked 3-d porous covalent organic framework. J. Am. Chem. Soc. 131, 4570 (2009). https://doi.org/10.1021/ja8096256
Materials Studio v.7.0 (Accelrys Inc, 2013). http://www.3dsbiovia.com/products/collaborative-science/biovia-materials-studio
M.O. Keeffe, M.A. Peskov, S.J. Ramsden, O.M. Yaghi, The reticular chemistry structure resource (RCSR) database of, and symbols for, crystal nets. Acc. Chem. Res. 41, 1782 (2008). https://doi.org/10.1021/ar800124u
Q. Fang, J. Wang, S. Gu, R.B. Kaspar, Z. Zhuang et al., 3D porous crystalline polyimide covalent organic frameworks for drug delivery. J. Am. Chem. Soc. 137, 8352 (2015). https://doi.org/10.1021/jacs.5b04147
C. Ji, K. Su, W. Wang, J. Chang et al., Tunable cage based three-dimensional covalent organic frameworks. CCS Chem. 9, 3095 (2021). https://doi.org/10.31635/ccschem.021.202101453
Y. Liu, C. Wu, Q. Sun, F. Hu, Q. Pan et al., Spirobifluorene-based three-dimensional covalent organic frameworks with rigid topological channels as efficient heterogeneous catalyst. CCS Chem. 4, 2418 (2020). https://doi.org/10.31635/ccschem.020.202000493
T.-M. Geng, F.-Q. Wang, X.-C. Fang, H. Xu, Dual functional N,O,P containing covalent organic frameworks for adsorbing iodine and fluorescence sensing to p-nitrophenol and iodine. Microporous Mesoporous Mater. 317, 111001 (2021). https://doi.org/10.1016/j.micromeso.2021.111001
R. Ditchfield, W.J. Hehre, J.A. Pople, An extended gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 54, 724 (1971). https://doi.org/10.1063/1.1674902
W.J. Hehre, R. Ditchfield, J.A. Pople, Self—consistent molecular orbital methods. XII. Further extensions of gaussian—type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 56(5), 2257–2261 (1972). https://doi.org/10.1063/1.1677527
Acknowledgements
This work was supported by National Key R&D Program of China (2022YFB3704900 and 2021YFF0500500), National Natural Science Foundation of China (22025504, 21621001, and 22105082), the SINOPEC Research Institute of Petroleum Processing, "111" project (BP0719036 and B17020), China Postdoctoral Science Foundation (2020TQ0118 and 2020M681034), the program for JLU Science and Technology Innovative Research Team, and the Taishan Scholars Program (No. tsqn202211124).
Funding
Open access funding provided by Shanghai Jiao Tong University.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, J., Li, C., Wang, X. et al. Quasi-Three-Dimensional Cyclotriphosphazene-Based Covalent Organic Framework Nanosheet for Efficient Oxygen Reduction. Nano-Micro Lett. 15, 159 (2023). https://doi.org/10.1007/s40820-023-01111-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40820-023-01111-8