
Highlights
-
A MgH2/TiO2 heterostructure with nano MgH2 assembled on oxygen vacancy-rich 2D TiO2 nanosheets was successfully fabricated via a simple solvothermal strategy.
-
The MgH2/TiO2 heterostructure shows rapid desorption kinetics, low dehydrogenation temperature, and excellent cycling stability.
-
In situ HRTEM observations and ex situ XPS analyses reveal that multi-valance of Ti species, presence of abundant oxygen vacancies, formation of catalytic Mg-Ti oxides, and confinement of TiO2 nanosheets, contribute to the high stability and kinetically accelerated hydrogen sorption performances of Mg.
Abstract
MgH2 has attracted intensive interests as one of the most promising hydrogen storage materials. Nevertheless, the high desorption temperature, sluggish kinetics, and rapid capacity decay hamper its commercial application. Herein, 2D TiO2 nanosheets with abundant oxygen vacancies are used to fabricate a flower-like MgH2/TiO2 heterostructure with enhanced hydrogen storage performances. Particularly, the onset hydrogen desorption temperature of the MgH2/TiO2 heterostructure is lowered down to 180 °C (295 °C for blank MgH2). The initial desorption rate of MgH2/TiO2 reaches 2.116 wt% min−1 at 300 °C, 35 times of the blank MgH2 under the same conditions. Moreover, the capacity retention is as high as 98.5% after 100 cycles at 300 °C, remarkably higher than those of the previously reported MgH2-TiO2 composites. Both in situ HRTEM observations and ex situ XPS analyses confirm that the synergistic effects from multi-valance of Ti species, accelerated electron transportation caused by oxygen vacancies, formation of catalytic Mg-Ti oxides, and stabilized MgH2 NPs confined by TiO2 nanosheets contribute to the high stability and kinetically accelerated hydrogen storage performances of the composite. The strategy of using 2D substrates with abundant defects to support nano-sized energy storage materials to build heterostructure is therefore promising for the design of high-performance energy materials.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
With the ever-increasing global energy demands and environmental concerns, clean and renewable energy is urgently demanded worldwide. Hydrogen has been considered as one of the most promising candidates to alternate fossil fuels, due to its intrinsic clean nature, environmental-friendliness, and high gravimetric energy density (142 MJ kg−1) [1,2,3,4]. However, an effective strategy to store hydrogen is essential to realizing the "hydrogen economy" [5, 6]. Compared with hydrogen storage in pressurized tanks or cryogenic containers, solid-state hydrogen storage methods potentially have the advantages of high safety, low cost, and exceptional hydrogen storage capacity [7]. Among various solid-state hydrogen storage materials, magnesium hydride (MgH2) has attracted tremendous attention, due to its high gravimetric (~ 7.6 wt% H2) and volumetric (~ 110 kg m−3 H2) hydrogen storage density, excellent reversibility, low cost, and non-toxicity [8, 9]. Unfortunately, the industrial application of MgH2 has been seriously hampered by its high operating temperature and slow de/re-hydrogenation kinetics. Particularly, a high temperature of at least 280 °C is required at 1 bar for desorption of coarse-grained MgH2, which far exceeds the working temperature of the proton exchange membrane fuel cell (PEMFC). Consequently, it is essential to improve the hydrogen storage performances of MgH2 for possible applications in the hydrogen energy field.
In the last few decades, extensive researches have been executed toward fabricating novel Mg-based hydrogen storage materials with enhanced performances, such as adding catalysts [10,11,12,13], alloying [14,15,16], and reducing the particle size of MgH2 [17,18,19]. From the extrinsic perspective, the use of additives to weaken the Mg-H bonding and promote the diffusion rate of hydrogen atoms, thus reducing the dehydrogenation temperature and accelerating the desorption rates, is regarded as an efficient strategy for enhancing the hydrogen desorption properties of MgH2. These additives include transition metals (TM) [20, 21], transition metal oxides [22, 23], nitrides [24, 25], halides [26, 27], and hydrides [28, 29]. Among them, transition metals (Ti, V, Nb, Ni, Co) and their compounds (TiO2, Nb2O5, V2O3) strike more attentions and have been proven to be the most effective additives to ameliorate the hydrogen sorption performances of MgH2, since transition metals with multiple valences favor the electron transfer during the de/re-hydrogenation processes [26]. The Fermi level of transition metals is located around s-type orbitals, which is a necessary condition to promote hydrogen dissociation and recombination [30]. Cui et al. [21] systematically investigated the catalytic mechanism of TM on the MgH2 by the synthesis of core (Mg)-shell (TM) structure. The results show that Mg-Ti composites, which can release hydrogen even under 200 °C, exhibit optimal desorption performances, owing to the lower electronegativity of Ti. Notably, it has been proved that TiO2 possesses a superior catalytic effect than metallic Ti [31]. Furthermore, TiO2 has many advantages, such as high natural abundance, low cost, and nontoxicity. Though the addition of catalysts through simple ball milling method can indeed ameliorate the kinetics of MgH2 to some extent, the improvement is restricted by the inevitable self-aggregation and growth of the additives during cycling, as well as the low density of exposed active sites.
On the other hand, from an intrinsic point of view, nano-sized MgH2, which owes the shortened diffusion path for hydrogen atoms and increased active sites for re/de-hydrogenation, has been successfully synthesized through various nanotechnologies, giving rise to improved hydrogen storage performances. For instance, the nano-confinement method has been proved to be a facile strategy to simultaneously improve the thermodynamics and kinetics of MgH2 while reducing the capacity loss as little as possible. Nano-sized MgH2 confined into porous carbon has demonstrated a noticeable decrease in its initial desorption temperature (~ 90 °C) compared with its bulk counterpart [32]. Yu et al. [33] synthesized Ni nanocrystal-decorated MgH2 nanoparticles anchored on graphene sheets that exhibit excellent hydrogen sorption properties. The composites absorb 5.4 wt% H2 within 10 min at 200 °C, and identical capacity can be released at the same temperature within 150 min. It is noted, however, that neither graphene nor porous carbon possesses good catalytic activities, and additional catalysts are needed to further improve hydrogen storage performances of MgH2. Although huge achievements have been achieved, it is still far from enough to enhance the hydrogen storage performances of MgH2 by solely depending on any methods mentioned above. In this respect, it is essential to fabricate MgH2 with extraordinary hydrogen storage performances by taking full advantage of both the intrinsic (nano-size) and the extrinsic (catalyst addition) strategy.
Recently, graphene-like two-dimensional (2D) transition metal oxides have been extensively studied due to their potential in energy-related applications with high surface area and unique electronic/thermal properties [34,35,36,37]. However, preparing hydrogen storage composites composed of graphene-like 2D TiO2 nanosheets (TiO2 NS) and MgH2 by a simple ball milling method would cause irreversible agglomeration of TiO2 NS, leading to seriously reduced catalytic active sites and deterioration of various spectacular intrinsic properties of 2D materials [38]. Thus, the idea of fabricating MgH2 (0D)/TiO2 (2D) heterostructure using graphene-like 2D TiO2 NS is triggered as it can not only facilitate the formation of nano-sized MgH2 through the confinement effect of TiO2 NS but also possess catalytic effect for MgH2. More importantly, the construction of MgH2/TiO2 heterostructure can effectively inhibit the self-stacking of TiO2 layers, leading to increased surface catalytic sites for MgH2. Additionally, defect engineering has also aroused more attention to adjusting the band structure and providing more catalytic active sites [39]. Among them, oxygen vacancy-rich TiO2 has been proved to remarkably promote electron transportation, enhance electrical conductivity, and provide more active sites for the capture and diffusion of hydrogen [40, 41].
In light of the above consideration, herein, taking full advantage of the intrinsic (nano-size) and extrinsic (catalyst addition) effects, we report a first attempt to design a novel self-assembled MgH2/TiO2 heterostructure with remarkably enhanced hydrogen sorption properties through facile solvothermal approach, where 2D TiO2 NS exhibit both nano-confinement and catalytic effects. Surprisingly, we found that the oxygen vacancies were introduced into TiO2 NS in the process of solvothermal treatment. Owing to the presence of oxygen vacancies and unique heterostructure, the obtained MgH2/TiO2 heterostructure exhibits impressive hydrogen storage performances with high stability and accelerated kinetics. Specifically, the 60MgH2/TiO2 heterostructure presents low onset desorption temperature (180 °C) and superior long-term cycling stability with a capacity retention of 98.5% after 100 cycles at 300 °C, remarkably higher than that of the state-of-the-art TiO2 catalyzed MgH2 systems.
2 Experimental Section
2.1 Synthesis of MgH2/TiO2 Heterostructure
2.1.1 Synthesis of Graphene-like 2D TiO2 Nanosheets
TiO2 nanosheets were synthesized by a surfactant self-assembly method [34]. Firstly, 1.48 mL titanium isopropoxide (TTIP, 99.9%, Macklin) was added to 2.1 mL concentrated HCl under vigorous stirring (solution A). Meanwhile, 0.4 g poly(ethylene oxide)-block-poly(propylene oxide)-block-poly(ethylene oxide) triblock copolymer (P123, Mn = 5,800) was dissolved in 8 mL ethanol (solution B) by ultrasonication. Afterward, solution B was added dropwise into solution A and kept stirring for 30 min at room temperature. Then, 5 mL of the above resultants with 40 mL ethylene glycol was then transferred into a 100 mL stainless steel autoclave and heated at 150 °C for 20 h. Finally, the products were centrifuged and washed with ethanol, followed by freeze-drying to obtain 2D TiO2 NS. Moreover, to improve the crystallinity and remove the impurities in TiO2 NS, the pristine TiO2 NS were annealed at different temperatures (300, 400, and 500 °C) for 2 h in the air (denoted as TiO2-T, T represents the annealing temperature).
2.1.2 Synthesis of the MgH2/TiO2 Heterostructure
MgH2/TiO2 heterostructure was prepared by a bottom-up solvothermal strategy of in situ growth of MgH2 nanoparticles on TiO2 substrates. Di-n-butyl-magnesium (MgBu2, 1 M solution in hexane, Sigma-Aldrich) and cyclohexane (C6H12, 99.5%, extra Dry, Sigma-Aldrich) were used as received states without further treatment. As provided in Table S1, different loading capacities of MgH2 on TiO2 NS of 40, 50, 60, 70, and 80 wt% were synthesized by changing the ratio between MgBu2 and TiO2 NS, which were denoted as 40MgH2/TiO2, 50MgH2/TiO2, 60MgH2/TiO2, 70MgH2/TiO2, and 80MgH2/TiO2, respectively. In a typical synthesis of 60MgH2/TiO2, 30 mg as-prepared TiO2-300 NS and 1.8 mL MgBu2 were firstly dispersed in 40 mL cyclohexane under sonication. The mixture was then transferred into a 100 mL custom-designed autoclave, filled with 4.5 MPa H2 pressure, and heated at 200 °C for 12 h under vigorous magnetic stirring [19, 42]. Finally, the resultants (named 60MgH2/TiO2 heterostructure) were collected by centrifugation, washed with cyclohexane several times, and dried at 80 °C overnight under dynamic vacuum. Moreover, blank MgH2 without TiO2 NS were also prepared under the same conditions for comparison.
2.2 Characterization and Measurements
The phase composition was characterized by using X-ray diffraction (XRD, Mini Flex 600) with Cu Kα radiation operated at 40 kV and 15 mA. The samples were prepared in an argon-filled glovebox and sealed in a custom-designed holder covered by the Scotch tape to avoid the possible oxidation during the XRD tests. The microstructure and morphological analyses were performed by scanning electron microscope (SEM, MIRA3 LHM) and transmission electron microscope (TEM, FEI Talos F200X G2). For TEM observations, the preparation of samples was carried out in the glovebox. The samples were dispersed in cyclohexane, sonicated, dropped on a copper grid, and rapidly transferred to the equipment. The preparation conditions of samples for SEM tests were similar to those for the TEM observation, except that the samples were dispersed in cyclohexane and dropped onto the silicon wafer. The in situ high-resolution transmission electron microscope (HRTEM) observation was applied to explore the microstructural evolution during the decomposition of the MgH2/TiO2 heterostructure under an irradiation beam current of 3 nA [43]. The valence state and chemical bonding nature of constituent elements of MgH2/TiO2 composites were analyzed by X-ray photoelectron spectroscopy (XPS, Kratos AXIS Ultra DLD). For XPS analyses, a special air-proof transfer vessel was used to transfer the samples from glovebox to the equipment. Electron paramagnetic resonance (EPR) measurement was performed at room temperature using a Bruker spectrometer (EMXplus-9.5/12). Solid-state 1H magic-angle spinning nuclear magnetic resonance (MAS NMR) was carried out on a Bruker AVANCE NEO 600 spectrometer. Brunner–Emmet–Teller (BET) surface areas of all samples were measured by the N2 isothermal ad/de-sorption tests at 77 K on Autosorb-IQ3 apparatus. Differential scanning calorimetry (DSC, NETZSCH STA 449 F3) measurements were performed at different heating rates (3, 5, and 10 °C min−1) under a flowing Ar atmosphere to analyze the desorption kinetic properties. The DSC data were further processed by the Kissinger method to figure out the desorption apparent activation energy (Ea) according to the following equation:
where β represents the heating rate, Tp refers to the peak temperature, R is the universal gas constant (R = 8.314 J mol−1 K−1), and C is a constant.
The isothermal ab/de-sorption tests and temperature program desorption (TPD) measurements of samples were carried out by a Sievert-type pressure-composition-temperature apparatus (PCT, Shanghai Institute of Microsystem and Information Technology). For the isothermal ab/de-sorption tests, samples were loaded into the vessel in the glove box and were heated to the preset temperature at a heating rate of 10 °C min−1. The initial hydrogen pressures for isothermal ab/de-sorption were 3 and 0.001 MPa, respectively. Besides, the hydrogenated samples were heated from room temperature to 350 °C with a heating rate of 2 °C min−1 to perform TPD measurements. The hydrogen pressure/duration/temperature parameters for cycling tests are set as 3 MPa/15 min/300 °C for re-hydrogenation and 0.001 MPa/15 min/300 °C for dehydrogenation. The hydrogen storage capacity was calculated in the weight percent of the entire composites including the TiO2 scaffold.
3 Results and Discussion
3.1 Synthesis and Characterizations of the MgH2/TiO2 Heterostructure
The preparation procedure of the MgH2/TiO2 heterostructures is schematically illustrated in Scheme 1, including the synthesis of 2D TiO2 NS and subsequent impregnation of MgBu2 as well as the bottom-up self-assembly of MgH2 nanoparticles anchored on TiO2 NS. In a typical surfactant self-assembly synthesis process of TiO2 NS, P123 and ethylene glycol (EG) acted as co-surfactants which played the role of structure-directing agents to form inverse lamellar micelles. The precursor oligomers (TTIP) were then self-assembled into lamellar structures with surfactant molecules, inducing the formation of layered inorganic oligomer agglomerates. Afterward, solvothermal treatment was carried out to promote the organization and crystallization of 2D TiO2 NS. Subsequently, the TiO2 NS were ultrasonically dispersed into the C6H12 solution followed by the dropwise addition of MgBu2. Finally, the MgH2/TiO2 heterostructure was obtained in the above dispersed suspension by hydrogenation of MgBu2 on TiO2 NS through a facile solvothermal method.

Synthesis process illustration of the MgH2/TiO2 heterostructure
The typical morphology and structural features of the ultrathin 2D TiO2 NS were observed using SEM and TEM. As shown in Fig. 1a–c, the pristine TiO2 NS exhibit a morphology of 2D corrugated ultrathin nanosheets with their edges roll up owing to the surface tension, which is also observed in graphene as a general phenomenon. The sizes of the TiO2 NS vary from 150 to 200 nm as seen from the TEM images. The thickness of the prepared TiO2 NS was further characterized using AFM and TEM (Fig. S1). The obtained TiO2 NS have a thickness of around 3.7 nm, corresponding to 5–6 monolayers, which confirms the formation of ultrathin nanosheets. In addition, the sheet-like structure of TiO2 can be clearly distinguished in the HAADF and BF images (Fig. 1d), which may play a morphology-directing role in the solution-phase synthesis of nanostructured MgH2. The EDS elemental mapping images demonstrate the homogeneous distribution of Ti and O elements (Fig. 1d).

a Typical SEM image of TiO2 NS. b, c Typical TEM images at different scales. d Representative HAADF and BF images of TiO2 NS as well as corresponding EDS elemental mapping results. e XRD patterns of TiO2 NS. f, g N2 ad/de-sorption isotherms and corresponding specific surface areas and pore-size distribution of pristine TiO2 NS, commercial TiO2, and TiO2 NS treated at different annealing temperatures. h-j BF, HRTEM, and SAED images of as-synthesized 60MgH2/TiO2 heterostructure. k HAADF images of as-synthesized 60MgH2/TiO2 heterostructure and corresponding EDS elemental mapping results
Furthermore, the influence of the annealing temperature on the morphology of TiO2 NS was also taken into account (Fig. S2). It is worth emphasizing that TiO2-300 and TiO2-400 exhibit a similar sheet-like structure as the pristine TiO2 NS, except for the slight granulation after annealing at 400 °C. However, the crystallite size and crystallinity of TiO2-500 increase obviously and the 2D nanostructures can hardly be observed. This phenomenon coincides well with the previous report [44]. Moreover, we also explored the stability of TiO2 NS in the hydrogen atmosphere and it can be seen that the sheet-like structure of TiO2 is still preserved after treatment in H2 at 300 °C for 2 h (Fig. S2). Figure 1e shows the XRD patterns of pristine TiO2, TiO2-300, TiO2-400, and TiO2-500. According to the XRD patterns, pristine TiO2 NS are mainly composed of the anatase phase (JCPDS No. 21–1272), as well as a small amount of rutile phase (JCPDS No. 21–1276). After calcination treatment, the intensities of the diffraction peaks are much sharper and stronger, manifesting an increase in crystallinity, coinciding well with the SEM and TEM results (Fig. S2). Additionally, the diffraction peaks of rutile phase disappear and crystalline diffraction peaks assigned to anatase TiO2 crystal can be identified when the annealing temperature increases up to 400 and 500 °C. The specific surface area and pore size distribution of TiO2 NS were further investigated (Fig. 1f–g). Compared with commercial TiO2, the specific surface area of TiO2 NS is increased by 14–100 times, attributed to the special 2D sheet-like structures. The TiO2 NS possess a specific surface area of 431.32 m2 g−1, which is almost 100 times higher than that of the commercial TiO2 (~ 4 m2 g−1). Besides, it still possesses a high specific surface area of 268.31 m2 g−1 even after calcination treatment (300 °C). Thus, we selected TiO2-300 as the substrates to fabricate the MgH2/TiO2 heterostructure, and the TiO2 mentioned below all refers to TiO2-300 unless otherwise specified. The TiO2 NS with ultrahigh specific surface area will provide more nucleation sites for nanoparticles, which could be ingeniously employed as the excellent scaffold for the self-assembly of MgH2 NPs.
To fabricate MgH2/TiO2 heterostructure, MgBu2 was used as the precursor and added into TiO2 suspension followed by the solvothermal method under 4.5 MPa of hydrogen pressure. Impressively, the color of TiO2 suspension fortuitously changed from white to dark blue with the addition of MgBu2. The obvious blue color indicates the presence of oxygen vacancies caused by MgBu2 [39], which will be discussed in detail in the following section. Figure 1h shows the typical TEM images of as-synthesized 60MgH2/TiO2 heterostructure. It can be seen that TiO2 nanosheets still maintain a graphene-like structure after the self-assembly of MgH2 nanoparticles on them. Besides, 2D TiO2 nanosheets are automatically arranged into flower-like structures to support and encapsulate MgH2 nanoparticles, tremendously restricting the agglomeration of MgH2 NPs and simultaneously hampering the re-stacking of TiO2 NS in turn. According to the measurement of lattice spacing in HRTEM images (Fig. 1i), the interplanar spacings of 0.225 and 0.219 nm could be attributed to the (200) plane of MgH2 (JCPDS No. 12–0697) and (111) plane of TiO2 (JCPDS No. 21–1276), respectively. In addition, the diffraction rings in SAED pattern of 60MgH2/TiO2 (Fig. 1j) reveal the formation of polycrystalline structure. Furthermore, the 2D wrinkled sheet-like structure of TiO2 could be well distinguished from the HAADF images. EDS elemental mapping images confirm the homogeneous distribution of Mg, Ti, and O elements in the MgH2/TiO2 heterostructure (Fig. 1k).
3.2 Improved Hydrogen Storage Properties of the MgH2/TiO2 Heterostructure
The hydrogen storage performances of the MgH2/TiO2 heterostructure were further examined. Figure 2a shows the TPD profiles of blank MgH2 and the MgH2/TiO2 with different loading of MgH2 and Fig. 2b shows the comparison of the onset desorption temperature and hydrogen capacity of different samples. As the control sample, the blank MgH2 starts to desorb hydrogen at about 295 °C, which is higher than those of MgH2/TiO2 composites, indicating that the TiO2 NS indeed have a positive effect on the dehydrogenation of MgH2. Concerning the MgH2/TiO2 with different loading capacities of MgH2, the onset desorption temperatures decrease in the sequence of 50MgH2/TiO2 (260 °C), 70MgH2/TiO2 (233 °C), and 60MgH2/TiO2 (200 °C). Thus, we further explored the hydrogen storage performances of 60MgH2/TiO2, and the MgH2/TiO2 heterostructure mentioned below refers to the 60MgH2/TiO2 unless otherwise specified. In the first non-isothermal desorption process, the 60MgH2/TiO2 exhibits the initial desorption temperature of 200 °C with a total hydrogen release of 4 wt%. Notably, the 60MgH2/TiO2 composite possesses a lower onset desorption temperature of 180 °C along with a hydrogen capacity of 3.4 wt% in the second non-isothermal dehydrogenation process. The lower desorption temperature in the second cycle reveals that the catalytic activity of TiO2 NS varies upon de/ab-sorption behavior. The capacity loss after the first desorption may be attributed to the irreversible reaction between MgH2 and TiO2. It is therefore reasonable to speculate that the chemical environment of MgH2 and TiO2 in the first dehydrogenated sample should be considerably different from the as-synthesized sample, indicating the formation of catalytic species during initial hydrogen cycling, which is similar to what shown in previous reports [45,46,47,48], and will be discussed in later sections. In addition, DSC profiles (Figs. 2c and S3) show that the peak desorption temperature of commercial MgH2 is 408.6 °C under the heating rate of 3 °C min−1, while it tremendously reduces to 220.2 °C for the 60MgH2/TiO2 heterostructure. According to the fitting results of DSC by Kissinger’s equation, the apparent activation energy of 60MgH2/TiO2 is determined to be 106.7 kJ mol−1 H2, much lower than that of the commercial MgH2 (142.27 kJ mol−1 H2).

a, b TPD curves and corresponding onset desorption temperature and hydrogen capacity of blank MgH2, 50MgH2/TiO2, 60MgH2/TiO2-1st, 60MgH2/TiO2-2nd, and 70MgH2/TiO2. c DSC curves of 60MgH2/TiO2. d Isothermal dehydrogenation profiles of 60MgH2/TiO2. e Isothermal dehydrogenation curves and f initial desorption rate of 60MgH2/TiO2 and blank MgH2 at 300 °C. g Isothermal re-hydrogenation profiles of 60MgH2/TiO2. h Isothermal re-hydrogenation curves and i initial absorption rate of 60MgH2/TiO2 and blank MgH2 at 200 °C. j Comparison of the onset desorption temperature and initial desorption rates at 300 °C of MgH2/TiO2 heterostructure with those of other nano-confined MgH2 systems and TiO2 catalyzed MgH2 systems in the literature (Table S2)
In order to further explore the influence of TiO2 NS on MgH2, the isotherm de/ab-sorption measurements were carried out for the 60MgH2/TiO2 heterostructure as well as control samples (blank MgH2 and commercial MgH2). Figure 2d shows the isotherm desorption curves of the 60MgH2/TiO2. As can been seen, the 60MgH2/TiO2 releases hydrogen of 0.51, 1.56, 2.14, 2.83, and 2.43 wt% at the temperature of 180, 200, 250, 275, and 300 °C within 10 min, respectively. Moreover, even at a lower temperature of 180 °C, the composite can still desorb 1.24 wt% of hydrogen within 60 min, corresponding to 41% of the maximum hydrogen storage capacity. At 250 °C, 2.14 wt% of hydrogen can be desorbed within 10 min, while no hydrogen desorption is detected for the blank MgH2 and commercial MgH2 under identical conditions (Fig. S4). In addition, blank MgH2 needs 68 min to release 80% of its theoretical hydrogen capacity at 300 °C. In contrast, it takes only 1.8 min for the 60MgH2/TiO2 composite to reach the identical reaction extent (Fig. 2e). Impressively, the initial desorption rate is increased by 35 times for the 60MgH2/TiO2 (Fig. 2f) when compared to that of blank MgH2. This demonstrates the remarkably improved hydrogen storage performances of MgH2 through the introduction of TiO2 NS.
The isothermal re-hydrogenation curves of samples are presented in Fig. 2g-i. The 60MgH2/TiO2 could absorb hydrogen even at a temperature as low as 80 °C with a hydrogen capacity of 1.86 wt% within 60 min. Such a low hydrogenation temperature meets the target of working temperature set by DOE for light-duty vehicle applications [7]. A hydrogen capacity of 2.65 wt%, corresponding to 80% of the maximum hydrogen capacity, is achieved at 200 °C within 11.9 min for the 60MgH2/TiO2 heterostructure. However, it takes a long time of 54.8 min for the blank MgH2 to achieve the same reaction extent (Fig. 2h). Additionally, it is noteworthy that the initial absorption rate of 60MgH2/TiO2 at 200 °C is 6.5 times that of the blank MgH2 (Fig. 2i). The superior hydrogen desorption performances, such as high initial desorption rate and low desorption temperature of the 60MgH2/TiO2, remarkably exceed those of the state-of-the-art MgH2 systems reported in literature (Fig. 2j and Table S2).
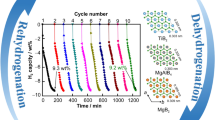
Cycling stability is also a key factor for the practical application of hydrogen storage materials. To further evaluate the stability of the 60MgH2/TiO2, long-term (100 cycles) isothermal de/re-hydrogenation was tested at a relatively high temperature of 300 °C (Fig. 3a). It can be seen that MgH2/TiO2 shows negligible degradation of the capacity even after 100 cycles (from 2.71 to 2.67 wt%) and a capacity retention as high as 98.5% is achieved (Fig. 3b), which is highly comparable with and even surpasses those of the previously reported Mg-based hydrogen storage materials catalyzed by TiO2 and other transition metal oxides. Additionally, compared to the first cycle, the MgH2/TiO2 heterostructure exhibits a faster initial desorption rate during the second cycle (Fig. S5), which is associated with the formation of the catalytic oxide mentioned above. Moreover, the MgH2/TiO2 heterostructure still maintains flower-like structures and shows no obvious agglomeration of MgH2 and restacking of TiO2 NS even after 100 cycles (Fig. 3c). However, with the absence of TiO2 NS, blank MgH2 exhibits severe growth and agglomeration after several de/re-hydrogenation cycles (Fig. S6). The high stability of the MgH2/TiO2 heterostructure is presumably ascribed to the confinement effect of 2D TiO2 NS on MgH2 and robust interfacial contact between host and support materials.

a Reversible hydrogen absorption and desorption cycling profiles of 60MgH2/TiO2 at 300 °C. b Hydrogen desorption curves of 60MgH2/TiO2 at different cycles (1st and 100th). c HAADF images of 60MgH2/TiO2 after 100 cycles and corresponding EDS elemental mapping results
Therefore, the MgH2/TiO2 heterostructure exhibits rapid hydrogen ab/de-sorption kinetics and excellent long-term cycling stability than those of blank MgH2, highlighting the crucial role of the 2D TiO2 nanosheets, which simultaneously possess catalytic effect to MgH2 and inhibit the agglomeration of MgH2.
3.3 Catalytic Mechanism of the MgH2/TiO2 Heterostructure
In order to further understand the mechanism of improved sorption performances of the MgH2/TiO2 heterostructure, we further characterized the phase components and microstructure of samples at different states. Figure 4 displays the optical photographs and microstructure of pristine TiO2 NS and MgBu2-treated TiO2 NS. As shown in Fig. 4a, the MgBu2-treated TiO2 NS exhibit in different colors ranging from blue to black, in marked contrast with the white pristine TiO2 powders. The color change of TiO2 is ascribed to the presence of oxygen vacancies, which coincides well with the previous work [39,40,41, 49]. Herein, we introduce oxygen vacancies into TiO2 NS by a facile strategy, that is, the impregnation of MgBu2 into TiO2 NS. We discover that the MgBu2 could not only act as the reducing agent to fabricate oxygen vacancy-rich blue TiO2 NS but also serve as the precursor of MgH2 to promote the self-assembly of MgH2 nanoparticles onto TiO2 NS through subsequent solvothermal approaches. Such a "one-stone-two-birds" effect enables the MgH2/TiO2 heterostructure as a promising hydrogen storage material with superior performances. Figure 4b displays the identical crystalline structure with the anatase phase in the majority and that of rutile in minority for different TiO2 samples, indicating that the impregnation of MgBu2 does not destroy the intrinsic crystal structure of the TiO2 nanosheets. Besides, XPS spectra of elemental Ti (Fig. 4c) and O (Fig. S7) are provided. The high-resolution XPS spectra of Ti 2p of the pristine TiO2 NS and TiO2-300 at 464.4 and 458.6 eV can be attributed to Ti4+ 2p1/2 and Ti4+ 2p3/2, respectively, while the peaks of TiO2 NS treated with MgBu2 slightly shift toward lower binding energy, indicating that Ti3+ is generated with the formation of oxygen vacancies. The O 1s peaks located at 531.8, 530.8, and 529.5 eV belong to adsorbed oxygen (OA), oxygen vacancy (OV), and lattice oxygen (OL) in the MgBu2-treated TiO2 NS, respectively (Fig. S7). Based on the peak area of OV to the overall peak area of O 1s, the percentage of OV on the surface of the MgBu2-treated TiO2 is determined to be 32.53%. Additionally, the concentration of oxygen vacancies on the surface of the as-synthesized MgH2/TiO2 heterostructure (33.4%) shows no significant change when compared to that on the TiO2 NS treated with MgBu2 (Fig. S7).

a Optical photographs of TiO2 NS suspension treated with different amounts of MgBu2. b XRD patterns of TiO2-300 and MgBu2-treated TiO2 NS. c High-resolution XPS spectra of Ti 2p. d EPR spectra of TiO2-300 and TiO2 NS treated with different amounts of MgBu2 ("40wt%/ 50wt%/ 60wt%/ 70wt%/ 80wt% " refers to the weight percentage of MgH2 in the MgH2/TiO2 heterostructure)
Moreover, EPR measurement was carried out to further explore the existence of oxygen vacancies in TiO2 NS treated with MgBu2. As shown in Fig. 4d, the TiO2 NS treated with MgBu2 exhibit typical EPR signals at about g = 1.972 and g = 2.002, which could be attributed to Ti3+ and oxygen vacancies in the lattice, respectively [40, 50]. All these results strongly prove that the oxygen vacancies have been successfully introduced into TiO2 nanosheets by the "one-stone-two-birds" strategy. Due to the higher electrical conductivity, numerous active sites, and enlarged lattice space derived from oxygen vacancies, the formation of oxygen vacancy-rich blue TiO2 could not only accelerate the migration of electrons and diffusion of hydrogen atoms but also provide more active sites for the capture of hydrogen and nucleation of MgH2/Mg. Thus, the MgH2/TiO2 heterostructure displays superior hydrogen ab/de-sorption kinetic properties.
In order to elucidate the catalytic mechanism, the structural stability of the MgH2/TiO2 composites during the hydrogen de/ab-sorption process was further investigated. Figure 5 shows the TEM images of dehydrogenated and re-hydrogenated 60MgH2/TiO2 heterostructure. Surprisingly, there are no significant differences in morphology among the as-synthesized (Fig. 1h–k), dehydrogenated (Fig. 5a–d), and re-hydrogenated (Fig. 5e–h) samples, further confirming the thermal stability of the MgH2/TiO2 composites. Notably, by measuring the interplanar spacing of the well-resolved lattice fringes (Fig. 5b, f), we observe a new Mg-Ti ternary oxide, which is discussed in detail below. Thus, the establishment of the flower-like heterostructure has rendered the ability to well maintain the primary structure of the TiO2 scaffold and MgH2 nanoparticles, thereby improving the long-term cycling stability of MgH2 and facilitating rapid transport of electrons and hydrogen.

BF, HRTEM, SAED, HAADF images as well as EDS elemental mapping results of dehydrogenated a-d and re-hydrogenated e–h 60MgH2/TiO2 heterostructure
We further explored the phase transformation and chemical valence state change of MgH2/TiO2 at different states. The XRD patterns of as-synthesized samples exhibit broad diffraction peaks indicating the presence of nano-sized MgH2 (Fig. 6a). No peaks from MgO (which generally appear at the peak position of ~ 42.9° and 62.3° [11]) can be detected in the XRD pattern of as-synthesized MgH2/TiO2, indicating the absence of MgO. After dehydrogenation, in addition to the main phase of Mg, two new broad peaks located at 42.7° and 62.0° appear in the XRD patterns, which are close to the peak positions of MgO. In the subsequent hydrogen absorption process, Mg phase transforms to MgH2 and those two characteristic peaks assigned to Mg2TiO4 (JCPDS No. 16–0215) show no obvious change. Additionally, as shown in Fig. S8, the phase composition of the MgH2/TiO2 heterostructure remains unchanged after 100 de/re-hydrogenation cycles by showing very similar XRD patterns. Such a Mg-Ti oxide phase may possess a catalytic effect for the de/re-hydrogenation of MgH2 [45, 47]. As is well known, the existence of MgO will deteriorate the performance of MgH2; hence, higher temperature is required for the release of hydrogen [51]. However, the MgH2/TiO2 composites perform excellent hydrogen desorption properties. To further prove that these two peaks belong to the highly catalytic Mg-Ti oxide instead of less-catalytic MgO, 1H NMR test was conducted (Figs. 6b and S9). A shorter spin–lattice relaxation time of MgH2/TiO2 (~ 1.54 s) compared with that of blank MgH2 (~ 4.50 s) indicates a faster H motion in MgH2/TiO2. In particular, the MgH2/TiO2 heterostructure exhibits a much shorter spin–lattice relaxation time of 1.49 s after the 1st re-hydrogenation than that of the as-synthesized MgH2/TiO2, indicating that new species with catalytic activity have formed during the first dehydrogenation process. Thus, it is demonstrated that two characteristic peaks centered at 42.7° and 62.0° are assigned to the ternary Mg-Ti oxide phase (MgTi2O4), having much higher catalytic activity than MgO. It can be deduced that the Mg-Ti oxide is formed by the chemical reaction at the interface between MgH2 and TiO2 NS during the first hydrogen desorption process. The similar phenomenon, for which Ti/Nb oxide could react with MgH2 and result in the formation of a new ternary oxide, has also been reported in the previous work [48]. Compared to Mg/MgH2, the broader diffraction peaks of the ternary Mg-Ti oxide phase manifest its smaller crystallite size than that of Mg/MgH2.

a XRD patterns of the as-synthesized, dehydrogenated, and re-hydrogenated 60MgH2/TiO2 heterostructure. b NMR spin lattice relaxation times of 60MgH2/TiO2, commercial MgH2, and blank MgH2. c In situ HRTEM images showing the microstructure evolution of the hydrogenated 60MgH2/TiO2 during the hydrogen desorption process. d High-resolution Ti 2p spectra of 60MgH2/TiO2 at different states. e Schematic diagram showing the hydrogenation and dehydrogenation mechanisms of MgH2/TiO2 heterostructure
To further clarify the role of the ternary Mg-Ti oxide in the dehydrogenation process, in situ HRTEM observations were carried out by real-time recording of the structure and composition evolution of hydrogenated MgH2/TiO2 during the hydrogen desorption process (Fig. 6c). Additional images with more details are shown in Fig. S10. Before electron beam irradiation (at 0 min), MgH2 (101) and TiO2 nanosheets could be identified according to the lattice fringe spacings. After irradiation under a beam current of 3 nA for 1.5 min, a portion of MgH2 react with TiO2, generating a thin Mg-Ti oxide layer. After 2.5 min of irradiation, the MgH2 near Mg-Ti oxide begins to release hydrogen to form Mg. The observed area releases hydrogen completely after 3 min. It can be seen that, unlike the neat MgO layer, the in-built ternary Mg-Ti oxide catalyst could act as a pathway to facilitate hydrogen diffusion, resulting in improved reaction kinetics. Through the pathways provided by Mg-Ti oxide, the hydrogen can diffuse into/out of Mg/MgH2, possibly by a formation of metastable titanium hydride (TiHx) species [19, 52], which coincides well with the XPS chemical state shifts discussed later. In addition, Mg-Ti oxide at the MgH2/TiO2 interfaces could also strongly anchor MgH2 nanoparticles, which significantly inhibits the growth and agglomeration of MgH2 particles during cycling. Moreover, the high ductility and flexibility of the Mg-Ti oxide [53] can significantly buffer the volume change of MgH2 particles during de/re-hydrogenation cycles, preventing the detachment of MgH2 from the TiO2 substrate and thereby ensuring the excellent cyclic stability.
Moreover, XPS analyses were employed to explore the evolution of the bonding state of Ti in the process of de/re-hydrogenation and the results are shown in Fig. 6d. XPS survey spectrum indicates the coexistence of Mg, O, and Ti elements (Fig. S11), well coincident with the EDS mapping results. As shown in the high-resolution Ti 2p XPS spectra, the signals of Ti4+ species (458.8 eV for 2p3/2 and 464.5 eV for 2p1/2) can be detected in all samples at different states, indicating that TiO2 nanosheets still exist in the MgH2/TiO2 heterostructure during hydrogen ab/de-sorption process. Additionally, the typical doublets located at 456.8 and 462.5 eV of Ti3+ for as-synthesized MgH2/TiO2 composites further confirm the successful introduction of oxygen vacancies. Moreover, it can be seen that the Ti 2p signals of Ti2+ appear in the dehydrogenated samples and Ti2+ peaks shift to higher binding energies in the re-hydrogenated samples. The presence of Ti2+ peaks testifies the formation of low-valence Ti-H active species. The slight change of the peak position of Ti2+ demonstrates that Ti-H active species could act as the intermediates to promote the transportation of electrons and hydrogen [19], further proving that the Mg-Ti oxide acts as a pathway to promote hydrogen diffusion. It is well known that the electronegativity of Ti (1.54) is between Mg (1.31) and H (2.2) [21, 54]. Variable valence of Ti could also facilitate the electron transfer between Mg2+ and H−, providing a favorable condition for weakening the Mg-H bond. In addition, numerous multi-phase interfaces composed of MgH2 and multivalence Ti promote the diffusion of hydrogen and electron and provide more nucleation sites for MgH2/Mg. Thus, the existence of multivalence Ti-based catalysts accounts for the significantly enhanced hydrogen storage performances of the MgH2/TiO2 heterostructure.
Based on the experimental results and analyses, it is concluded that the superior hydrogen storage performances of the MgH2/TiO2 heterostructure can be attributed to the intrinsic and extrinsic synergistic effects, as illustrated in Fig. 6e: (1) Graphene-like 2D TiO2 nanosheets with a high specific surface area provide the unimpeded channels for the rapid diffusion of hydrogen and more nucleation sites for MgH2/Mg, thereby improving the kinetics of de/re-hydrogenation. (2) Nano-sized MgH2 are uniformly confined on TiO2 nanosheets to form a flower-like heterostructure, which significantly suppresses the growth and agglomeration of MgH2 during the hydrogen ab/de-sorption processes, ensuring the excellent cycling stability. Meanwhile, the re-stacking of TiO2 nanosheets can be hampered in turn. (3) Abundant oxygen vacancies introduced by the "one-stone-two-birds" strategy significantly enhance the electrical conductivity of TiO2 NS and provide additional active sites for transportation of electrons and hydrogen, leading to greatly enhanced hydrogen sorption kinetics. (4) The ternary Mg-Ti oxide catalyst located at the interface of MgH2 and TiO2 acts as the pathway to facilitate hydrogen diffusion, resulting in improved hydrogen storage properties. In addition, Mg-Ti oxide possesses better ductility and flexibility than pure MgO. Thus, due to the stable interfacial bonding strength between oxide layers and the MgH2 nanoparticles, Mg-Ti oxide may exhibit a long-term confinement effect on MgH2 nanoparticles and significantly buffer successive expansion and contraction of MgH2 particles during de/re-hydrogenation cycles, improving reversible cycling performance. (5) Multi-valence Ti-based catalysts favor the electron transfer between Mg2+ and H−, weakening the Mg-H bond. Additionally, numerous multi-phase interfaces composed of Mg2+ and multi-valence Ti containing species could provide more diffusion pathways for hydrogen and more nucleation sites for MgH2/Mg.
4 Conclusion
In the present work, a flower-like MgH2/TiO2 heterostructure has been successfully synthesized using oxygen vacancy-rich 2D TiO2 NS as the functional scaffold. Benefitting from the intrinsic (nano-sized MgH2) and extrinsic (catalytic activity of Mg-Ti oxide at interfaces) effects as well as the incorporation of oxygen vacancies, the prepared MgH2/TiO2 heterostructure exhibits superior hydrogen storage performances. Specifically, the composite shows excellent desorption performances with a low onset desorption temperature down to 180 °C and a rapid initial dehydrogenation rate at 300 °C (2.116 wt% min−1), surpassing those of the state-of-the-art MgH2 systems catalyzed by TiO2. Meanwhile, the MgH2/TiO2 heterostructure displays an excellent cycling stability with a capacity retention of 98.5% after 100 cycles at 300 °C. This work offers a promising approach for applying graphene analogues to the construction of advanced hydrogen storage materials with 0D/2D heterostructures.
References
L. Schlapbach, A. Zuttel, Hydrogen-storage materials for mobile applications. Nature 414(6861), 353–358 (2001). https://doi.org/10.1038/35104634
D. Pukazhselvan, V. Kumar, S.K. Singh, High capacity hydrogen storage: basic aspects, new developments and milestones. Nano Energy 1(4), 566–589 (2012). https://doi.org/10.1016/j.nanoen.2012.05.004
P. Nicola, M. Andrea, M. Vittorio, P. Luca, Interfaces within biphasic nanoparticles give a boost to magnesium-based hydrogen storage. Nano Energy 72, 104654 (2020). https://doi.org/10.1016/j.nanoen.2020.104654
X. Yu, Z. Tang, D. Sun, L. Ouyang, M. Zhu, Recent advances and remaining challenges of nanostructured materials for hydrogen storage applications. Prog. Mater. Sci. 88, 1–48 (2017). https://doi.org/10.1016/j.pmatsci.2017.03.001
N.M. Dowell, N. Sunny, N. Brandon, H. Herzog, A.Y. Ku et al., The hydrogen economy: a pragmatic path forward. Joule 5(10), 2524–2529 (2021). https://doi.org/10.1016/j.joule.2021.09.014
M. Spek, C. Banet, C. Bauer, P. Gabrielli, W. Goldthorpe et al., Perspective on the hydrogen economy as a pathway to reach net-zero CO2 emissions in Europe. Energy Environ. Sci. 15(3), 1034–1077 (2022). https://doi.org/10.1039/D1EE02118D
C. Weidenthaler, M. Felderhoff, Solid-state hydrogen storage for mobile applications: Quo Vadis? Energy Environ. Sci. 4(7), 2495–2502 (2011). https://doi.org/10.1039/c0ee00771d
H. Shao, G. Xin, J. Zheng, X. Li, E. Akiba, Nanotechnology in Mg-based materials for hydrogen storage. Nano Energy 1(4), 590–601 (2012). https://doi.org/10.1016/j.nanoen.2012.05.005
P. Larsson, C.M. Araujo, J.A. Larsson, P. Jena, R. Ahuja, Role of catalysts in dehydrogenation of MgH2 nanoclusters. PNAS 105, 8227–8231 (2008). https://doi.org/10.1073/.0711743105
M. Liu, X. Xiao, S. Zhao, M. Chen, J. Mao et al., Facile synthesis of Co/Pd supported by few-walled carbon nanotubes as an efficient bidirectional catalyst for improving the low temperature hydrogen storage properties of magnesium hydride. J. Mater. Chem. A 7(10), 5277–5287 (2019). https://doi.org/10.1039/C8TA12431K
K. Xian, M. Wu, M. Gao, S. Wang, Z. Li et al., A unique nanoflake-shape bimetallic Ti–Nb oxide of superior catalytic effect for hydrogen storage of MgH2. Small (2022). https://doi.org/10.1002/smll.202107013
X. Lu, L. Zhang, H. Yu, Z. Lu, J. He et al., Achieving superior hydrogen storage properties of MgH2 by the effect of TiFe and carbon nanotubes. Chem. Eng. J. 422, 130101 (2021). https://doi.org/10.1016/j.cej.2021.130101
Z. Lan, H. Fu, R. Zhao, H. Liu, W. Zhou et al., Roles of in situ-formed NbN and Nb2O5 from N-doped Nb2C MXene in regulating the re/hydrogenation and cycling performance of magnesium hydride. Chem. Eng. J. 431, 133985 (2022). https://doi.org/10.1016/j.cej.2021.133985
J. Mao, T. Huang, S. Panda, J. Zou, W. Ding, Direct observations of diffusion controlled microstructure transition in Mg-In/Mg-Ag ultrafine particles with enhanced hydrogen storage and hydrolysis properties. Chem. Eng. J. 418, 129301 (2021). https://doi.org/10.1016/j.cej.2021.129301
X. Ding, R. Chen, X. Chen, H. Fang, Q. Wang et al., A novel method towards improving the hydrogen storage properties of hypoeutectic Mg-Ni alloy via ultrasonic treatment. J. Magnes. Alloy. (2021). https://doi.org/10.1016/j.jma.2021.06.003
F. Guo, T. Zhang, L. Shi, L. Song, Hydrogen absorption/desorption cycling performance of Mg-based alloys with in-situ formed Mg2Ni and LaHx (x = 2, 3) nanocrystallines. J. Magnes. Alloy. (2021). https://doi.org/10.1016/j.jma.2021.06.013
K.J. Jeon, H.R. Moon, A.M. Ruminski, B. Jiang, C. Kisielowski et al., Air-stable magnesium nanocomposites provide rapid and high-capacity hydrogen storage without using heavy-metal catalysts. Nat. Mater. 10(4), 286–290 (2011). https://doi.org/10.1038/nmat2978
M. Liu, S. Zhao, X. Xiao, M. Chen, C. Sun et al., Novel 1D carbon nanotubes uniformly wrapped nanoscale MgH2 for efficient hydrogen storage cycling performances with extreme high gravimetric and volumetric capacities. Nano Energy 61, 540–549 (2019). https://doi.org/10.1016/j.nanoen.2019.04.094
W. Zhu, L. Ren, C. Lu, H. Xu, F. Sun et al., Nanoconfined and in situ catalyzed MgH2 self-assembled on 3D Ti3C2 MXene folded nanosheets with enhanced hydrogen sorption performances. ACS Nano 15(11), 18494–18504 (2021). https://doi.org/10.1021/acsnano.1c08343
C. Lu, J. Zou, X. Zeng, W. Ding, Hydrogen storage properties of core-shell structured Mg@TM (TM = Co, V) composites. Int. J. Hydrog. Energy 42(22), 15246–15255 (2017). https://doi.org/10.1016/j.ijhydene.2017.04.063
J. Cui, J. Liu, H. Wang, L. Ouyang, D. Sun et al., Mg-TM (TM: Ti, Nb, V Co, Mo or Ni) core-shell like nanostructures: synthesis, hydrogen storage performance and catalytic mechanism. J. Mater. Chem. A 2(25), 9645–9655 (2014). https://doi.org/10.1039/c4ta00221k
K. Wang, X. Zhang, Y. Liu, Z. Ren, X. Zhang et al., Graphene-induced growth of N-doped niobium pentaoxide nanorods with high catalytic activity for hydrogen storage in MgH2. Chem. Eng. J. 406, 126831 (2021). https://doi.org/10.1016/j.cej.2020.126831
S. Milošević, S. Kurko, L. Pasquini, L. Matović, R. Vujasin et al., Fast hydrogen sorption from MgH2-VO2(B) composite materials. J. Power Sources 307, 481–488 (2016). https://doi.org/10.1016/j.jpowsour.2015.12.108
K. Wang, X. Zhang, Z. Ren, X. Zhang, J. Hu et al., Nitrogen-stimulated superior catalytic activity of niobium oxide for fast full hydrogenation of magnesium at ambient temperature. Energy Stor. Mater. 23, 79–87 (2019). https://doi.org/10.1016/j.ensm.2019.05.029
Q. Zhang, Y. Wang, L. Zang, X. Chang, L. Jiao et al., Core-shell Ni3N@Nitrogen-doped carbon: synthesis and application in MgH2. J. Alloys Compd. 703, 381–388 (2017). https://doi.org/10.1016/j.jallcom.2017.01.224
C. Jie, W. Hui, L. Jiangwen, O. Liuzhang, Z. Qingan et al., Remarkable enhancement in dehydrogenation of MgH2 by a nano-coating of multi-valence Ti-based catalysts. J. Mater. Chem. A 1(18), 5603–5611 (2013). https://doi.org/10.1039/c3ta01332d
J. Ankur, A. Shivani, K. Sanjay, Y. Shotaro, M. Hiroki et al., How does TiF4 affect the decomposition of MgH2 and its complex variants? - an XPS investigation. J. Mater. Chem. A 5(30), 15543 (2017). https://doi.org/10.1039/c7ta03081a
Y. Zhang, X. Zhuang, Y. Zhu, N. Wan, L. Li et al., Synergistic effects of TiH2 and Pd on hydrogen desorption performances of MgH2. Int. J. Hydrog. Energy 40(46), 16338–16346 (2015). https://doi.org/10.1016/j.ijhydene.2015.09.029
L. Mykhaylo, D. Roman, A.Y. Volodymyr, E. Jon, G. Jonathan et al., An outstanding effect of graphite in nano-MgH2-TiH2 on hydrogen storage performance. J. Mater. Chem. A 6, 10740–10754 (2018). https://doi.org/10.1039/c8ta02969e
R.A. Pavel, C. Fermin, L. Michel, Hydrides of early transition metals as catalysts and grain growth inhibitors for enhanced reversible hydrogen storage in nanostructured magnesium. J. Mater. Chem. A 7, 23064–23075 (2019). https://doi.org/10.1039/c9ta05440e
S.K. Verma, A. Bhatnagar, V. Shukla, P.K. Soni, A.P. Pandey et al., Multiple improvements of hydrogen sorption and their mechanism for MgH2 catalyzed through TiH2@Gr. Int. J. Hydrog. Energy 45(38), 19516–19530 (2020). https://doi.org/10.1016/j.ijhydene.2020.05.031
K. Wang, G. Wu, H. Cao, H. Li, X. Zhao, Improved reversible dehydrogenation properties of MgH2 by the synergetic effects of graphene oxide-based porous carbon and TiCl3. Int. J. Hydrog. Energy 43(15), 7440–7446 (2018). https://doi.org/10.1016/j.ijhydene.2018.02.195
G. Xia, Y. Tan, X. Chen, D. Sun, Z. Guo et al., Monodisperse magnesium hydride nanoparticles uniformly self-assembled on graphene. Adv. Mater. 27(39), 5981–5988 (2015). https://doi.org/10.1002/adma.201502005
Z. Sun, T. Liao, Y. Dou, S.M. Hwang, M.S. Park et al., Generalized self-assembly of scalable two-dimensional transition metal oxide nanosheets. Nat. Commun. 5, 3813 (2014). https://doi.org/10.1038/ncomms4813
X. Zhang, L. Hou, A. Ciesielski, P. Samorì, 2D materials beyond graphene for high-performance energy storage applications. Adv. Energy Mater. 6(23), 1600671 (2016). https://doi.org/10.1002/aenm.201600671
A.A. AbdelHamid, Y. Yu, J. Yang, J.Y. Ying, Generalized synthesis of metal oxide nanosheets and their application as Li-ion battery anodes. Adv. Mater. 29(32), 1701427 (2017). https://doi.org/10.1002/adma.201701427
S.L. Wang, X. Luo, X. Zhou, Y. Zhu, X. Chi et al., Fabrication and properties of a free-standing two-dimensional titania. J. Am. Chem. Soc. 139(43), 15414–15419 (2017). https://doi.org/10.1021/jacs.7b08229
M. Chen, X.Z. Xiao, M. Zhang, J.F. Mao, J.G. Zheng et al., Insights into 2D graphene-like TiO2 (B) nanosheets as highly efficient catalyst for improved low-temperature hydrogen storage properties of MgH2. Mater. Today Energy 16, 100411 (2020). https://doi.org/10.1016/j.mtener.2020.100411
Z. Hao, Q. Chen, W. Dai, Y. Ren, Y. Zhou et al., Oxygen-deficient blue TiO2 for ultrastable and fast lithium storage. Adv. Energy Mater. 10(10), 1903107 (2020). https://doi.org/10.1002/aenm.201903107
G. Ou, Y. Xu, B. Wen, R. Lin, B. Ge et al., Tuning defects in oxides at room temperature by lithium reduction. Nat. Commun. 9, 1302 (2018). https://doi.org/10.1038/s41467-018-03765-0
G. Yin, X. Huang, T. Chen, W. Zhao, Q. Bi et al., Hydrogenated blue titania for efficient solar to chemical conversions: preparation, characterization, and reaction mechanism of CO2 reduction. ACS Catal. 8(2), 1009–1017 (2018). https://doi.org/10.1021/acscatal.7b03473
L. Ren, W. Zhu, Q. Zhang, C. Lu, F. Sun et al., MgH2 confinement in MOF-derived N-doped porous carbon nanofibers for enhanced hydrogen storage. Chem. Eng. J. 434, 134701 (2022). https://doi.org/10.1016/j.cej.2022.134701
C. Lu, Y. Ma, F. Li, H. Zhu, X. Zeng et al., Visualization of fast “hydrogen pump” in core–shell nanostructured Mg@Pt through hydrogen-stabilized Mg3Pt. J. Mater. Chem. A 7(24), 14629–14637 (2019). https://doi.org/10.1039/c9ta03038g
J. Yang, Y.L. Jiang, L.J. Li, E. Muhire, M.Z. Gao, High-performance photodetectors and enhanced photocatalysts of two-dimensional TiO2 nanosheets under UV light excitation. Nanoscale 8(15), 8170–8177 (2016). https://doi.org/10.1039/c5nr09248e
D. Pukazhselvan, N. Narendar, C. Pedro, C.A. Enrique, O.I. Gonzalo et al., Evolution of reduced Ti containing phase(s) in MgH2/TiO2 system and its effect on the hydrogen storage behavior of MgH2. J. Power Sourc 362, 174–183 (2017). https://doi.org/10.1016/j.jpowsour.2017.07.032
D. Pukazhselvan, N. Narendar, K.S. Sandhya, S. Budhendra, B. Igor et al., Role of chemical interaction between MgH2 and TiO2 additive on the hydrogen storage behavior of MgH2. Appl. Surf. Sci. 420, 740–745 (2017). https://doi.org/10.1016/j.apsusc.2017.05.182
D. Pukazhselvan, N. Narendar, Y. Tao, R. Devaraj, S. Aliaksandr et al., Chemically transformed additive phases in Mg2TiO4 and MgTiO3 loaded hydrogen storage system MgH2. Appl. Surf. Sci. 472, 99–104 (2019). https://doi.org/10.1016/j.apsusc.2018.04.052
O. Friedrichs, J.C. Sánchez-López, C. López-Cartes, T. Klassen, R. Bormann et al., Nb2O5 “pathway effect” on hydrogen sorption in Mg. J. Phys. Chem. B 110(15), 7845–7850 (2006). https://doi.org/10.1021/jp0574495
X. Chen, L. Liu, P.Y. Yu, S.S. Mao, Increasing solar absorption for photocatalysis with black hydrogenated titanium dioxide nanocrystals. Science 331(6018), 746–750 (2011). https://doi.org/10.1126/science.1200448
H. Zhang, Q. Kong, S. Hu, D. Zhang, H. Chen et al., Engineering the oxygen vacancies in Na2Ti3O7 for boosting its catalytic performance in MgH2 hydrogen storage. ACS Sustain. Chem. Eng. 10(1), 363–371 (2021). https://doi.org/10.1021/acssuschemeng.1c06444
D. Pukazhselvan, G. Otero-Irurueta, J. Pérez, B. Singh, I. Bdikin et al., Crystal structure, phase stoichiometry and chemical environment of MgxNbyOx+y nanoparticles and their impact on hydrogen storage in MgH2. Int. J. Hydrog. Energy 41(27), 11709–11715 (2016). https://doi.org/10.1016/j.ijhydene.2016.04.029
G. Liang, J. Huot, S. Boily, A.V. Neste, R. Schulz, Catalytic effect of transition metals on hydrogen sorption in nanocrystalline ball milled MgH2-Tm (Tm=Ti, V, Mn, Fe and Ni) systems. J. Alloys Compd. 292(1), 247–252 (1999). https://doi.org/10.1016/S0925-8388(99)00442-9
J. Zhang, S. Yan, L.P. Yu, X.J. Zhou, T. Zhou et al., Enhanced hydrogen storage properties and mechanisms of magnesium hydride modified by transition metal dissolved magnesium oxides. Int. J. Hydrog. Energy 43(48), 21864–21873 (2018). https://doi.org/10.1016/j.ijhydene.2018.10.017
H. Yike, A. Cuihua, Z. Qiuyu, Z. Lei, S. Huaxu et al., Cost-effective mechanochemical synthesis of highly dispersed supported transition metal catalysts for hydrogen storage. Nano Energy 80, 105535 (2020). https://doi.org/10.1016/j.nanoen.2020.105535
Acknowledgements
We would like to thank the support from the National Natural Science Foundation (No. 52171186), the Science and Technology Commission of Shanghai Municipality under No. 19511108100, Shanghai Education Commission “Shuguang” scholar Project (16SG08). The authors also appreciate the financial support from the Center of Hydrogen Science, Shanghai Jiao Tong University.
Funding
Open access funding provided by Shanghai Jiao Tong University.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ren, L., Zhu, W., Li, Y. et al. Oxygen Vacancy-Rich 2D TiO2 Nanosheets: A Bridge Toward High Stability and Rapid Hydrogen Storage Kinetics of Nano-Confined MgH2. Nano-Micro Lett. 14, 144 (2022). https://doi.org/10.1007/s40820-022-00891-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40820-022-00891-9