
Highlights
-
Catalytic descriptors for oxygen evolution reaction under scaling relationship are comprehensively reviewed.
-
New oxygen evolution paradigms and design strategies aiming to circumvent the adsorption energy scaling relationship are summarized.
-
Challenges and perspectives for further improving oxygen evolution activity are discussed.
Abstract
The oxygen evolution reaction (OER) is the essential module in energy conversion and storage devices such as electrolyzer, rechargeable metal–air batteries and regenerative fuel cells. The adsorption energy scaling relations between the reaction intermediates, however, impose a large intrinsic overpotential and sluggish reaction kinetics on OER catalysts. Developing advanced electrocatalysts with high activity and stability based on non-noble metal materials is still a grand challenge. Central to the rational design of novel and high-efficiency catalysts is the development and understanding of quantitative structure–activity relationships, which correlate the catalytic activities with structural and electronic descriptors. This paper comprehensively reviews the benchmark descriptors for OER electrolysis, aiming to give an in-depth understanding on the origins of the electrocatalytic activity of the OER and further contribute to building the theory of electrocatalysis. Meanwhile, the cutting-edge research frontiers for proposing new OER paradigms and crucial strategies to circumvent the scaling relationship are also summarized. Challenges, opportunities and perspectives are discussed, intending to shed some light on the rational design concepts and advance the development of more efficient catalysts for enhancing OER performance.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The ever-increasing energy demands and climate change along with the exhausting fossil fuels require the modern society urgently to seek renewable and sustainable energy sources, predominantly the solar and wind. However, the distributed and intermittent nature of such renewable energy sources presents a formidable challenge toward their effective utilization [1,2,3]. Exploring high-performance energy conversion and storage (ECS) devices, such as small molecule (water, carbon dioxide and nitrogen) electrolyzers, rechargeable metal–air batteries, and regenerative fuel cells, that can harvest, convert and store the renewable energy in chemicals and then reconvert at the point of need, is therefore of essential importance but remains a great scientific challenge [4,5,6,7,8,9,10,11,12]. The oxygen evolution reaction (OER) is the essential module in these ECS devices since it supplies electrons required for electrochemical conversion cycles between renewable electricity and chemical fuels [13,14,15,16]. In an electrolyzer for chemical fuel generation by small molecules’ electrolysis, the OER occurs at the anode (Fig. 1a) [2]. Whereas, the OER proceeds on the cathode in metal–air battery (MAB in Fig. 1b); its activity and stability immediately determine the charging and discharging performances of MAB devices [17,18,19]. A regenerative fuel cell which operates in two modes of hydrogen production (electrolyzer cell mode) and power production (fuel cell mode) is able to provide an economical means for efficient long-term energy storage and on-demand conversion back to electrical energy only with the participation of powerful oxygen electrolysis (Fig. 1c) [20, 21]. Improving the OER efficiency is therefore crucial to realize a close-looped clean energy infrastructure based on the conversion and storage of renewable energy [22].

The OER in electrochemical energy conversion and storage devices. a Electrolyzers for small molecules’ electrolysis, b metal–air batteries, and c regenerative fuel cells
The OER that involves the transfer of four electrons and the formation of multiple intermediates is challenging. Electrocatalysts play a key role by facilitating the required electron transfer, as well as the formation and rupture of chemical bonds [14, 23]. Nowadays, the state-of-the-art OER catalysts are the precious metal Ir/Ru-based materials. Besides the high cost and low reserve, the benchmark OER catalyst IrO2 has a regular overpotential η10 of around 300–400 mV (at the current density of 10 mA cm−2), which, however, is still far from an ideal OER catalyst in terms of activity [14]. The first-row transition metal oxide (TMO) electrocatalysts (such as perovskite, spinels, rock salt, and rutile) sparked intensive interests for catalyzing the OER with impressive activity, thanks to their low cost and superior oxidation resistance in alkaline electrolyte and, particularly upon high oxidation potentials during the OER [19, 24,25,26,27,28]. Nonetheless, no one TMO can fully satisfy the requirements for commercial operations. Again, the grand challenge is to develop advanced electrocatalysts with high activity and stability based on non-noble metal materials to enable the widespread penetration of clean ECS technologies [14, 19, 29].
Central to the rational design of advanced and high-efficiency catalysts is the understanding of quantitative structure–activity relationships, which correlate the catalytic activities with structural and electronic parameters [30]. In the context of OER, the large intrinsic overpotential is actually at the very root of why widespread deployment of ECS devices is still largely hampered. The origin of high overpotential for the OER is the strong scaling relations among the three oxygen-containing intermediates (*OH, *O, and *OOH) that impose a minimum intrinsic overpotential of about 0.37 V on all OER catalysts. Under this scaling relationship, intensive efforts have been made to propose catalytic descriptors such as d-band center, bulk O-2p band center, eg occupancy, metal–oxygen covalency, electronegativity, coordination number, outer electron numbers, bulk thermochemistry and so on, aiming to bridge the structures and catalytic behaviors of catalysts and optimize the activity to reach the apex of the volcano plot. On the other hand, some recent advances have been achieved to circumvent the scaling relation limit for further improving the OER activity beyond the state-of-the-art. It is essential to have an in-depth understanding of the material-property relationships before designing advanced materials and structures with superior electrocatalytic properties [1, 31].
In view of this necessity, this paper comprehensively summarizes the benchmark descriptors for OER electrolysis to clarify the correlations between structures and catalytic behaviors of catalysts, intending to shed light on the origins of the electrocatalytic OER activity and further contribute to advancing rational catalyst design. This paper starts with the introduction of the conventional adsorbate evolution mechanism (AEM) and identify the intrinsic origin of large overpotential for the OER imposed by adsorption energy scaling relations. Then, catalytic descriptors under the scaling relationship are fully reviewed and standardized with the most recent research advances. Particular emphasis is placed on the newly proposed strategies to circumvent the scaling relationship toward designing novel catalysts with superior catalytic activities. The frontiers in this field such as lattice-oxygen participated OER and magnetic-dependent OER are highlighted. Based on the discussion above, a brief summary and outlook on the designing efficient OER catalysts are proposed and offered at the end.
2 OER Under Scaling Relations
In this section, we introduce the fundamental principle of the OER under scaling relationship and provide an insight into what gives rise to the high intrinsic overpotential and what descriptors have been proposed to rationalize the correlation between catalytic activity and electronic structures. Since TMOs are regularly employed given their low cost and stability in alkaline media, the alkaline OER mechanism is adopted in this paper.
2.1 Fundamental Principles of Adsorbate Evolution Mechanism
The well-acknowledged OER process is based on the adsorbate evolution mechanism (AEM), by which the oxygen-containing adsorbates undergo catalytic reactions on the surface-active TM cations of the catalyst. Therefore, the AEM could be regarded as the redox reactions of metal cations. On the whole, the oxygen generation in alkaline media follows Eq. 1, which is accompanied with the transfer of four electrons.
Theoretically, this process has a standard electrode potential (E0) of 0.401 V versus the standard hydrogen electrode (SHE) and a formal potential (\(E^{0\prime }\)) of 1.229 V versus the reversible hydrogen electrode (RHE) [22, 23]. In practical operations, however, it proceeds through multiple elementary steps and, thus, requires considerably high applied overpotential (\(\eta_{{{\text{OER}}}} = E - E_{{{\text{O}}^{2} {\text{/OH}}^{ - } }} > 0.3 \;{\text{V}}\)) [22, 23, 32,33,34]. Of note, the high applied potential to drive the OER usually leads to the oxidation of catalysts, thereby metal oxides are considered as the most potent catalysts for the OER regarding the long-term stability [35]. In the light of the computational prediction, the oxygen generation cycle involves four concerted electron-transfer steps as shown in Fig. 2 [20, 32]. The OER starts with adsorption of OH− at an active site (*) to generate *OH radical (Step I, Eq. 2). Then, *OH deprotonates to produce *O, which is accompanied with the release of an electron and a water molecule (Step II, Eq. 3). Thereafter, the nucleophilic attack of OH− on *O yields the intermediate *OOH (Step III, Eq. 4). Finally, a further proton-coupled-electron transfer process results in the generation of one oxygen molecule as well as a free active site (Step IV, Eq. 5) [36].

Any step in the process could restrain the overall OER performance. The adsorption, dissociation and desorption of oxygen-containing intermediates play a decisive role herein [35]. Though the multiple adsorbate-based steps have not been fully visualized experimentally, the common consensus is that the interactions between the catalyst surface and adsorbates unambiguously govern the OER catalytic activity. Accordingly, a systematic understanding of the interaction between the active sites and the oxygen-containing intermediates during the reaction is critical for developing more efficient OER electrocatalysts [34].
2.2 Scaling Relations
The electrocatalytic activity is to a large extent determined by the binding strength between the reaction intermediates and the catalyst surface, that is, the binding energy. Empirically, an ideal catalyst should be able to adsorb the intermediate with an optimal binding energy that is neither too strong nor too weak as stated by the Sabatier principle [1, 37]. Nowadays, advances in density functional theory (DFT) calculations make it possible to quantitatively determine the binding energy [37]. As schemed above in Fig. 2, the conventional AEM consists of four consecutive electron-transfer steps and three oxygen-containing intermediates, *OH, *O and *OOH, which are all adsorbed on the surface through O atoms. Under this circumstance, the binding energies of *OOH (EHOO*), *O (EO*), and *OH (EHO*) are linearly correlated [37,38,39]. EHOO*/EHO* shows a slope of approximately 1 and an intercept of 3.2 eV (Fig. 3a) [37]; analogously, both EHOO*/EO* and EHO*/EO* have slopes close to 0.5, since O* is double bonded to the surface, and HO* and HOO* are bound with a single bond [38,39,40]. This correlated binding energy relationship universally occurs on metal and metal oxide surfaces (including perovskite, spinel, rutile, rock salt, and bixbyite oxides and so on) independent of the binding strength to the surface, and regardless of binding sites. On the surfaces that bind oxygen too strongly, the potential is limited by the formation of *OOH species; whereas for the surfaces that bind oxygen too weakly, the potential is limited by the oxidation of *OH [37]. This gives rise to a volcano-shaped relationship between catalytic activity and the oxygen adsorption energy (Fig. 3b) [14, 16]. The catalysts that locate on the top of the volcano always have balanced binding energies and thus, demonstrate better OER performance. This paradigm has been acknowledged as a universal descriptor to successfully interpret and predict the OER activity of various catalysts. One can optimize the catalytic activity by approaching ΔGO*- ΔGHO* toward the apex of the volcano plot [41].

The adsorption energy scaling relations between oxygen-containing intermediates. a Plot of adsorption energies of HOO* and HO* on metal oxides. Hollow symbols represent the adsorption energy on the clean surfaces: perovskites (circle), rutiles (triangle), MnxOy (square), anatase (diamond), Co3O4 (+). The solid symbols represent the adsorption energies on high coverage surfaces [37]. Copyright © 2011 Wiley–VCH. b The volcano relationship between ΔGO*–ΔGHO* and OER activity for major oxides [14]. Copyright © 2017 American Association for the Advancement of Science
However, the problem raised by the scaling relationship is leaving all adsorption energies interdependent with each other. In other words, the adsorption energy of an intermediate cannot be freely tuned without affecting another [36, 37, 39, 41, 42]. Most importantly, the established scaling relationship (ΔGOOH = ΔGOH + 3.2 V) brings about an intrinsic overpotential as large as (3.2–2.46) eV/2e− = 0.37 V [2, 37]. This means that even the best OER catalysts still have a minimum theoretical overpotential of about 0.3–0.4 V, which sets up a limit for OER performance that cannot be further bypassed theoretically [41]. Novel paradigm to circumvent the scaling relationship is thus in urgent demand for OER electrocatalyst design.
2.3 Descriptors Under Scaling Relationship
A catalytic descriptor is established to identify the relations between the catalytic behavior and the catalyst, which should not only be able to explain and screen the activity trend but also allow for the exploration of more active catalysts. In the past decade, intensive efforts have been made to discover catalytic descriptors for electrocatalytic OER in terms of bulk and surface electronic structures [1, 30, 43].
2.3.1 d-Band Theory
The relationship between the electronic structure and the catalytic activity of TMs was first established with d-band theory in 1990s by Hammer and Nørskov [44]. The d-band theory describes the bond formation at a TM surface, as illustrated in Fig. 4a. To improve the description of the energy level of d band, the d-band center (i.e., the average energy of the band) was generally adopted. Upon adsorption on the TM surface, the valence states of the adsorbate couple with the TM s states and become broadened and shift downward, which then continue to interplay with the TM d states and form the filled bonding states and partially occupied antibonding states [45]. The antibonding states are above the d states, and its filling governs the bond strength in terms of the distance from the band center to the Fermi level (EF) [44]. In general, the higher the d states are in energy relative to the EF, the higher in energy the antibonding states are and the stronger the bond [31, 46, 47].

a Schematic illustration of bond formation at a transition-metal surface. The lower the d states are in energy relative to the EF, the more filled the antibonding states and the weaker the adsorption bond [45]. Copyright © 2019 Springer Nature. b The density of states (DOS) plots of RuO2, Cu-RuO2, and the corresponding schematic illustration of bond formation between the reaction surface and adsorbate [49]. Copyright © 2018 Wiley–VCH. c d-band centers of Ni2P and Fe-doped Ni2P; d Relationships of the calculated d-band centers and experimental overpotential of Fe-doped Ni2P [50]. Copyright © 2020 American Chemical Society
The d-band model has been widely adopted to understand bond formation and trends in reactivity among the TMs and their alloys. The facet, alloying, defects, strain and so forth greatly impact the d states and thus alter the reactivity [44, 45]. In context of the OER, the free-energy descriptor requires balanced adsorption and desorption of oxygen-containing intermediates, which thus necessitates an optimal Ed energy level for electrocatalyst to maximize the OER activity. Analogous to the alloying effect for TMs, doping TM-based compounds is the most effective pathway to impact the d states [46, 48]. The overly strong Ru–O bond in RuO2 regularly gives the high free energy of the rate determining step (RDS). With Cu being doped into RuO2 lattice, the d-band center shifts far away from the EF, leading to lowered antibonding states and weakened Ru–O bond strength (Fig. 4b) [49]. Conversely, the largely low Ed energy level of Ni2P (that is, the weak adsorption energy) makes oxygen-containing intermediates difficult to adsorb on the surface. Chen and coworkers found that Fe doping could raise the Ed energy levels closer to the EF (Fig. 4c) and thus, lifted the antibonding energy states and strengthened the interaction between adsorbates and catalyst surface, which enhanced the adsorption ability for intermediates during the OER process [50]. It is noted that a volcano-shaped OER performance against the d-band center was obtained by varying Fe concentrations (Fig. 4d). This means that a highly active OER catalyst should have an optimal Ed energy level to balance the adsorption and desorption of intermediates (*OH, *O, and *OOH) [50].
2.3.2 Bulk O-2p Band Center
Compared to TMs, electronic structure of TM cations in oxides undergoes significant changes upon being surrounded by an array of point charges generated by oxygen ions, where the five originally degenerated d orbitals lose the energy degeneracy (Fig. 5a). In an octahedrally coordinated field, the d orbitals split into two eg orbitals at the higher energy level and three t2g orbitals at lower energy level. The eg doublets have strong overlap with O 2p orbitals and generate σ-bonding and σ*-antibonding states. Whereas, the t2g triplets demonstrate weak overlap with O 2p orbital and form π-bonds and π*-antibonds. The molecular orbitals become bands in oxide crystals due to the translational symmetry of the unit cell, denoted as M d band and O 2p band [51]. The bonds develop through the hybridization of M d orbitals and O 2p orbitals, which could be flexibly adjusted given the abundant choices of TM ions and their oxidation states. Moreover, the electronic structure of oxides can also be influenced by the spin states, that is, the relative occupancy of eg and t2g states, which has been shown to influence electronic conductivity, thermal expansion, bulk modulus and catalytic activity [51]. The variable tunability on TM ions like number of d electron, oxidation state, electronegativity, coordination numbers, and arrangement and nature of ligands has sparked intensive explorations of useful descriptors on screening and predicting the efficient OER catalysts.

Bulk O 2p-band center relative to Fermi level (εO-2p) as a descriptor for OER. a Physical origin of shifts in constituent ion orbitals for oxides with octahedral oxygen coordination around transition metal ions. The dashed line represents the energy of free vacuum. b Schematic illustration of the density of states of perovskite oxides, showing the transition metal 3d and O 2p bands [51]. Copyright © 2015 Royal Chemical Society. c Correlation between the iR-corrected potential at 0.5 mA cm−2 and the εO-2p (eV) of (Ln0.5Ba0.5)CoO3−δ with Ln = Pr, Sm, Gd and Ho [4]. Copyright © 2013 Springer Nature
Morgan and coworkers demonstrated that the bulk O 2p-band center relative to the Fermi level (εO-2p) correlated strongly with the oxygen surface kinetics and vacancy formation energy in perovskites [52], which later indicated its effectiveness in explaining the associated energetics and kinetics of all oxygen point defects in Ruddlesden–Popper oxide and polycrystalline perovskite materials [53, 54]. This descriptor can be understood in a rigid band model (Fig. 5b). Typically, removing oxygen from the lattice corresponds to moving electrons from the O 2p states to the Fermi level, which leads to the uplift of the Fermi energy level. Conversely, moving electrons from Fermi level to O 2p band is equivalent to the addition of oxygen [4, 52]. The O 2p-band center reflects differences in the Fermi energy of the oxide, as the absolute energy of the O 2p-band largely depends on the Madelung potential and oxygen electron affinity [43, 51], which meanwhile can be regulated by varying the electronegativity and oxidation states of the transition metals [51, 55, 56]. Different to the d band model, the O 2p band demonstrates more electron delocalization and thus is able to more accurately capture the electronic structure characteristics in oxides [57, 58].
Grimaud and coworkers correlated the high activity and stability of some double perovskites ((Ln0.5Ba0.5)CoO3-δ, Ln = Pr, Sm, Gd, and Ho) with O 2p-band center being neither too close nor too far from the Fermi level (Fig. 5c) [4]. Moving the O 2p-band center closer to the Fermi level increased OER activities, while the overlifting (that is, too close to the Fermi level) led to the decreased stability because of the surface amorphization (BSCF, BSCF46 and SCF) [4]. Similarly, the amorphous surface after OER was also observed in BSCF82 with a high O 2p band center [59]. It was believed that the high O 2p band centers increase the oxygen-vacancy concentration due to much lower oxygen vacancy formation energy, which accelerates the surface oxygen exchange kinetic and facilitates lattice oxygen migration, thus leading to surface amorphization during the OER process [59, 60].
2.3.3 e g Occupancy
As stated above in the crystal field, the high-energy eg orbitals readily reveal strong overlap with O 2p orbital in octahedral coordination, thus leading to maximized p–d hybridization with oxygen-containing intermediates. In 1970s, Matsumoto and co-workers found that the catalytic activity of perovskite oxides for oxygen reduction reaction was influenced by the overlap between the eg orbital of the TM and the p orbital of oxygen adsorbates, and that the larger the overlap was, the higher the electrocatalytic activity [61, 62]. Suntivich and coworkers later theoretically and experimentally investigated the OER activities of more than 10 perovskite oxides against the eg occupancy and gave a volcano relationship between them, that is, the eg occupancy model (Fig. 6a) [63, 64]. The eg occupancy descriptor was proposed upon the fact that the σ-interaction of eg states dominates over the weaker π-interaction of the t2g states [51, 63]; meanwhile, the number of electrons in σ* states governs the binding strength of oxygen-containing intermediates with surface TM cations. The eg occupancy of near unity (slightly bigger than 1) leads to optimum OER activity. If eg < 1, the too strong M–O binding hinders the formation of M-OO2−; while if eg > 1, the weak interaction lowers the formation rate of O–O bond in M-OOH− [65]. The eg occupancy model that only takes account of electrons in eg orbital is different to those descriptors using all d electrons. [66, 67] With this model, the perovskite Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) was identified with the record intrinsic alkaline OER activity, though surface amorphization was later ascertained under OER conditions [4]. In case that oxides have similar eg occupancy, the OER activity has to be judged with a secondary descriptor, like the electronegativity of the TM cation [64, 68].

Correlation between eg occupancy and OER activity of TMOs, and strategies to regulate the eg occupancy. OER activity as a function of eg occupancy of the active center at octahedral site on a perovskite oxides [63], b spinel oxides [65], and c Heusler alloys [70], respectively. Copyright © 2011 American Association for the Advancement of Science. Copyright © 2017, 2021 Wiley–VCH. d The relationship between OER activity and spin configuration of differently oriented LCO films [73]. Copyright © 2017 Elsevier. e Double exchange interaction between Co and Ni at octahedral sites in spinels and the transitions of the eg occupancy [25]. Copyright © 2020 American Chemical Society. f Charge redistribution between Mn and Co in Co-Mn containing spinels via superexchange interaction [75]. Copyright © 2018 Wiley–VCH. g Oxygen vacancy and doping induced the increased conductivity and eg electrons filling status in perovskite CaMnO3 [77]. Copyright © 2015 Wiley–VCH
Besides perovskites (ABO3) [69], the eg occupancy model has been successfully applied to spinel oxides (AB2O4), mullites (AB2O5), and even Co2YZ-type Heusler compounds [70]. Xu and coworkers studied a series of MnCo2O4 spinels toward OER and oxygen reduction reaction (ORR) electrolysis, and identified the octahedrally coordinated Mn as the active sites. The correlation between OER/ORR activity (applied potential @ 25 μA cm−2ox) with eg filling of Mn at octahedral site gave a volcano shape (Fig. 6b), with the summit located at the Mn valency of ≈ + 3 (that is, t2g3eg1), which consolidated the decisive role of electron orbital filling in metal oxide catalysts for oxygen electrolysis [65]. Of significance, Tüysüz and coworkers recently demonstrated that the eg orbital filling of active Co sites could be precisely regulated by varying the Y and Z sites of the Co2YZ compounds, where the higher catalytic current was achieved for eg orbital filling approaching unity [70]. This further supports the effectiveness of the eg orbital filling model to modulate the M–O bonding strength towards more active OER catalysts (Fig. 6c).
Two parameters apparently regulate the eg occupancy: the number of d-electrons (namely, the oxidation state) and the spin state. For example, Mn3+ (3d4) typically affords the t2g3eg1 configuration, while Mn4+ (3d3) adopts t2g3eg0 configuration. Fe4+ (3d4, t2g3eg1) and Ni3+ (3d7, t2g6eg1) prefer to afford eg1 configuration, while eg orbitals in Fe3+ (3d5, t2g3eg2) and Ni2+ (3d8, t2g6eg2) are over-occupied. Meanwhile, the spin state is determined by the difference between the Hund exchange energy ΔEX and crystal field splitting energy ΔCF. If ΔEX > ΔCF, d electrons prefer to occupy the high-energy eg orbitals rather than pair at t2g orbitals; whereas, if ΔEX < ΔCF, d electrons will first pair at the low-energy t2g orbitals. One typical example is Co3+ (3d6) that has three different spin states: low-spin (LS, t2g6eg0), intermediate spin (IS, t2g5eg1) and high spin (HS, t2g4eg2). Under eg occupancy model, regulating Co3+ at octahedral site to possess an intermediate-spin electron configuration is therefore expected to improve the OER activity. Substitution/doping with cations having different valences and electronegativities, vacancies, strain and even nanostructuring can effectivity tailor the oxidation state and spin state of octahedral metal centers [71, 72]. Wu and co-workers demonstrated a spin-state regulation method to optimize the OER activity by controlling the lattice orientation of LaCoO3 film. The different lattice-oriented LaCoO3 films brought different degrees of distortion of the CoO6 octahedron (namely, the strain), which induced a spin-state transition of cobalt from a low spin state (LS, t2g6eg0) to an intermediate spin state (IS, t2g5eg1) and thus better OER performance (Fig. 6d) [73]. Oxygen vacancy also plays a significant role in altering the eg occupancy/spin states. In CoFe spinel oxide, electrons from the oxygen holes transferred to the neighboring cobalt sites, enabling the conversion of Co3+ from low spin to a stabilized higher spin state Co(3−δ)+ (t2g6egδ, δ is close to 1.2) [74]. Our recent study demonstrated a novel strategy to modulate the spin states in NiCo2O4 spinel via double-exchange interaction between octahedrally coordinated Ni and Co (Fig. 6e) [25]. Double exchange interaction was triggered by the co-actions of constructing a covalent nanoheterojunction and creating oxygen vacancies (VO) in NiCo2O4 and proceeded with the equation Co3+ + δNi2+ → Co(3−δ)+ + δNi3+ (0 < δ < 1). As a result, the high-spin Ni2+ (t2g6eg2) was oxidized to low-spin Ni3+ (t2g6eg1), whereas the low-spin Co3+ (t2g6eg0) switched to intermediate spin Co(3−δ)+ (t2g6−δ egδ, δ close to 1). Both octahedral sites thus had optimal eg occupancy close to 1 to readily improve OER activity. Doping with alien cations is another effective strategy to tune the electronic structure. Zhou and co-workers studied the Co–Mn containing spinel oxides ZnCoxMn2−xO4 (x = 0.0–2.0) and found that the eg occupancy of active Mn cations could be modulated through varying the Mn/Co ratio as a consequence of the superexchange effect between edge sharing CoO6 and MnO6 (Fig. 6f). The Mn/Co ratio of 0.43 gave the optimum eg occupancy and the best catalytic activity [75]. In practical operations, multiple actions always work cooperatively to modulate the eg occupancy. Guo and co-workers found that the introduction of oxygen vacancies in Yb-doped CaMnO3 through direct hydrogen treatment led to an increase in the eg filling of Mn and the improvement of conductivity (Fig. 6g). The optimized Yb0.1Ca0.9MnO3 catalyst demonstrated exceptional OER activity, 100 times higher than that of the pristine CaMnO3, which originated from the synergistic effects of doping and oxygen vacancies [76, 77].
2.3.4 Metal–Oxygen Covalency
Despite the great success of eg occupancy model as a useful activity descriptor, few weaknesses impede its universal effectiveness. First, it is technically difficult to ascertain the eg filling of active metal sites responsible for catalysis where the surface spin state is not well identified. The representative example is cobalt with three possible spin sates that have raised extensive debate [78]. Second, the eg occupancy model fails to screen the catalysts having the similar eg occupancy but different catalytic activities like LaBO3 (B = Mn, Co, Ni) [30]. Third, surface amorphization observed in some OER catalysts like BSCF is far beyond the explanation of eg occupancy. The major reason is ascribed to that the eg occupancy descriptor was established on the ionic model and was unable to effectively capture the metal–oxygen covalency or the sharing of electrons along the metal–oxygen bond [78]. Shao-Horn and coworkers therefore introduced the metal–oxygen covalency (charge transfer energy) as a more powerful descriptor because the metal–oxygen bonds in oxides have mixed ionic-covalent character, that is, the energetic similarity (covalency) and spatial overlap (hybridization) of metal 3d orbitals and O 2p states [78]. The metal–oxygen covalency has been shown to influence catalytic activities. It correlated linearly with experimental OER activity, surface exchange activity (such as the oxygen vacancy formation energy, oxygen binding energy, and electron transfer barrier associated with OER), as well as the stability in alkaline solution (Fig. 7a) [71, 78, 79]. This model well explained the activity of LaBO3 (B = Mn, Co, Ni) with an order of Ni > Co > Mn in terms of the TM electronegativity that is linearly correlated with M–O covalency [30, 64, 68]. Regarding the stability, there exists a positive correlation between covalency and oxygen vacancy formation: too high of a driving force for oxygen vacancy formation leads to structural loss of the perovskite phase (Fig. 7b) [78]. As a result, an optimum covalency, as measured by the O 2p band center model, gave rise to both high activity and stability in Pr0.5Ba0.5CoO3-δ (Fig. 5b). The covalency and the hybridization between M 3d and O 2p can be tuned by varying the electronegativity of the metal ions through the choice of TM and the oxidation state. In practice, metal substituents with higher electronegativity than the parent metal could reduce the energy of the d band, leading to enlarged metal–oxygen covalency [79, 80]. The incorporation of other low-valent metals or metal vacancy also increases the oxidation state of the metal, thereby lowering the metal 3d states into O 2p states and enlarging the metal–oxygen covalency [81, 82]. With this concept, Yagi and coworkers reported that the Fe4+-based quadruple perovskite CaCu3Fe4O12 with high OER activity [83]. Compared with Mn3+, the 3d-orbital energy levels of Fe4+ ion are lower than that of the O 2p orbitals, leading to enhanced metal–oxygen covalency (Fig. 7c). On the other hand, the large overlap between Cu (Fe) eg and O 2p orbitals in the square-planar (octahedral) coordination improves the structural stability of CaCu3Fe4O12. The covalent bonding network incorporating multiple Cu2+ and Fe4+ transition metal ions significantly enable the long-life structural stability and exceptional OER activity in CaCu3Fe4O12 [83].

Correlation between Metal–oxygen covalency and OER activities. a Correlations between charge-transfer energy and OER activity (red), surface exchange activity (blue), and the OER active site identity in the 3d transition metals. b Relationship of charge-transfer energy to relevant energetics and rate-determining steps: oxygen vacancy formation (blue), oxygen binding energy (red), and electron transfer energy (gray) [78]. Copyright © 2017 American Association for the Advancement of Science. c Schematic illustration of molecular orbitals for regular Mn3+O6 and Fe4+O6 octahedra. The Mn3+ and Fe4+ ion 3d-orbital energy levels are higher and lower than those of the O 2p orbitals, respectively [83]. Copyright © 2015 Springer Nature. d Molecular orbital diagrams of Co–OH (Co3+ and Co4+) bonding at the surface of spinel oxides. e OER activity as a function of the N–V parameter [84]. Copyright © 2018 Wiley–VCH. f Scheme for the charge-transfer energy: the relative energies of TM 3d and O 2p valence electronic states [79]. Copyright © 2017 Royal Society of Chemistry. g Current density (μA cmoxide−2) as a function of the ex situ electronic conductivity of the La1−xSrxCoO3 series; the red circle represents the SrCoO2.5 [88]. Copyright © 2015 American Chemical Society. (Color figure online)
The metal–oxygen covalency readily proved its effectiveness in other metal oxides like spinels [84], rocksalt oxide [85], and Ruddlesden–Popper oxide [86]. Xu and coworkers did intensive work on spinel oxides for OER [84, 87]. They presented a systematic study of spinel ZnFexCo2-xO4 oxides toward the OER, and found that Fe substitution (10–30 at% of Fe) promoted the formation of Zn vacancies and Co4+. Co4+ has a low energy 3d state and would produce higher hybridization due to the shortened energy distance between Co 3d and O 2p states, namely an enlarged Co–O covalency, which dominate the distinct OER activity (Fig. 7d–e) [84]. Later, with metal–oxygen covalency they further explained that the octahedral geometry in spinel oxides was more catalytically critical than the tetrahedral one [87].
Different to the O 2p-band center model that scales with the metal–oxygen covalency for semimetal oxides, but not for semiconducting oxides, the metal–oxygen covalency model tracks the energy difference between the M 3d and O 2p band centers (ε3d-2p), i.e., the charge transfer energy (Δ) [79]. The charge transfer energy plays a pronounced role in tuning the electronic properties of oxides that can impact the OER kinetics and mechanism. This has been widely confirmed in experiments with different TM substitution by varying the electronegativity and oxide states. Hong and coworkers found that reducing the charge transfer energy could greatly enhance the OER activity [79]. As shown in Fig. 7f, the reduced charge transfer energy Δ in PBCO relative to LaCrO3 leads to increased electrical conductivity and improved OER activity [79]. Sr doping into LaCrO3 (La1−xSrxCoO3) raised the realignment of Co–O–Co bonds and the oxidation of the Co cations, which enlarged the overlap between the occupied O 2p valence bands and the unoccupied Co 3d conduction bands, and improved the conductivity (Fig. 7g) [88]. It is significantly noted that the changes in metal–oxygen covalency are always accompanied with the variations in the O 2p band, d band and even the spin states of the involved TMs owing to their close relationships. Duan and coworkers found that Fe substitution in LaCoO3 resulted in the transition of Co3+ spin state from low-spin (LS: t2g6eg0) to a higher spin (IS: t2g5eg1). The strong hybridization between Co 3d states of eg symmetry and oxygen 2p states promoted the formation of broad σ* bands, which crossed over the EF and gave rise to the half-metal character [81]. Du and coworkers reached the same conclusion in Sr and Fe codop-LaCoO3 OER catalyst [82]. In this regard, switching from 3 to 4d and 5d metals, the orbitals will become spatially more extended, increasing the covalency of the M–O bond but also decreasing the on-site Coulomb interaction [89].
2.3.5 Other Descriptors
(1) Outer electron numbers. Otagawa and coworkers first employed the number of transition metal d electrons as a descriptor in their analysis of ABO3 electrocatalysts. They found the OER overpotential trended inversely with the enthalpy of hydroxide formation and d-electron number. Thus, it was concluded that the number of d electrons was the primary influence on the OH* bond strength via the occupancy of the metal-OH antibonding levels [66, 67]. Calle-Vallejo and coworkers later theoretically demonstrated that the binding strengths of possible OER intermediates scaled with the number of d electrons and oxidation state [51, 90,91,92]. As shown in Fig. 8a, the trends in adsorption energies of the intermediates of the OER on transition metals and their oxides are smoothly captured in terms of the number of outer electrons. Here, the outer electrons referred to the number of valence electrons remaining on the metal atom upon oxidation. This unique descriptor permits the construction of predictive adsorption-energy grids and explains the existence of scaling relationships among transition metals and their oxides [90].

Descriptors correlated with the oxygen electrolysis catalytic activity. a Adsorption-energy grid of *OH on metals (red), monoxides (blue), La/Y perovskites (green), and Sr/Ca/Ba perovskites (orange), as a function of the number of outer electrons [90]. Copyright © 2013 Royal Society of Chemistry. b Volcano-type activity plot for LaMO3 (violet circles), SrMO3 (orange squares), and MO (brown rhomboids) [93]. Copyright © 2015 American Chemical Society. c Correlation between 2p and reactivity of surface oxygen atoms adsorbed at various metal and metal-oxide surfaces [94]. Copyright © 2019 Elsevier d Overview of the overpotential dependence on surface MCUS for TMOs. The ηexp for Fe2O3 refers to current density of 1 mA cm−2, while that for the remaining TMOs corresponds to current density of 10 mA cm−2 [95]. Copyright © 2016 American Chemical Society. (Color figure online)
(2) Bulk Thermochemistry. Bulk thermochemistry has long been used to describe the trends in catalytic activity of oxide surface [91, 93]. Following the Outer electron numbers descriptor, Calle-Vallejo and coworkers later found that bulk thermochemistry scaled similarly as surface adsorption energetics with the number of outer electrons of the TMs in oxides [93]. This correspondence applied to a wide number of TMs and was responsible for the linear relationship between bulk and surface properties that enables the construction of volcano-type activity plots, and rationalizes the trends in catalytic activity through variations in bulk thermochemistry (Fig. 8b). The volcano curves demonstrate that the majority of the oxides lie on their left legs; thus, the most active compounds tend to be the least stable ones. Such volcano plots provide direct estimations of the chemical stability of the oxides, which is paramount in the search for catalysts with good trade-offs between activity and stability under reaction conditions [93].
(3) Average O-2p-State Energy (\({\overline{\varepsilon }}_{2p}\)). Unlike most descriptors that consider the surface of the catalyst, Dickens and coworkers examined the reactivity and electronic structure of adsorbed oxygen [37, 94]. Figure 8c shows the correlation between the average O 2p-state energy, \({\overline{\varepsilon }}_{2p}\), and ΔEO − ΔEOH (namely, the energy required to deprotonate adsorbed OH to create adsorbed O) for oxygen atoms at various metal and metal oxides. It is apparent that bound oxygen atoms with higher-lying 2p-states have higher ΔEO − ΔEOH, that is, stronger affinities towards binding hydrogen. In general, oxygen atoms with higher coordination to metal atoms tend to have weaker affinities toward binding hydrogen. The near-unity slope in Fig. 8c indicates that variations in \({\overline{\varepsilon }}_{2p}\) translate directly into variations in ΔEO − ΔEOH [94].
(4) Coordinatively Unsaturated Metal Cation. Tao and coworkers identified coordinatively unsaturated metal cations (MCUS) as a surface reactivity descriptor for the OER of TMOs [95]. In general, surface reactivity of a given TMO increases monotonically with the density of MCUS, and thus, the increase in MCUS improves the catalytic activity for weak-binding TMOs (right line in Fig. 8d) but impairs that for strong-binding ones (left line in Fig. 8d). The electronic origin for this descriptor can be explained as that the energy of the highest occupied d-states relative to the Fermi level (Ed–EF) serves as an electronic structure descriptor for the surface reactivity. This model is partially analogous to the d-band theory where the adsorption energy variations on TMs are mainly raised from the interaction of adsorbates with d-states of metal atoms; the difference is that the energy of highest occupied d-states relative to the Fermi level (Ed–EF) determines the bond strength of adsorbates on TMOs.
Overall, the OER activity is principally estimated through the adsorption energy of intermediates on the catalyst surface. This universal, but theoretically deduced, descriptor was then reified into variable electronic or geometric parameters of electrocatalysts that can be obtained experimentally to establish the correlation with the OER performance trend. However, it is worth noting that each sub-descriptor mentioned above normally has its own special application domain. d-band theory describes many reactions when using transition metals and their alloys as catalysts; nonetheless, it fails to embrace catalyst surface information and is not effective to non-metallic catalysts. [43, 46] Though eg occupancy model achieves great success by virtue of its tuning flexibility in describing the OER activity of various electrocatalysts, its ionic nature is unable to effectively capture the sharing of electrons along the metal–oxygen bond [78]. Metal–oxygen covalency model appears to be more powerful because it broadly not only involves the M 3d band and O 2p band, but also tackles with the energy difference between them (ε3d-2p). All of them are decisive parameters that give the electronic structure of a catalyst. It needs to take cautions in selecting an appropriate one for TMO catalysts under metal–oxygen covalency model. For example, O 2p band works for semimetal oxides, but is not effective for semiconducting oxides [79]. When considering charge transfer/electronic conductivity in designing OER catalyst, ε3d-2p, will be the first choice to correlate with OER activity. To the end, it is still challenging to deal with different catalysts using one experimental descriptor. The cooperative actions among descriptors should be taken into account to more precisely identify the catalytic activity and design advanced catalysts.
3 OER Beyond Scaling Relations
All aforementioned descriptors are useful in screening the catalytic activity and designing more efficient catalysts; however, they remain effective under the usual scaling relationship that gives rise to a large intrinsic overpotential. What remains as a relevant question is how much further adsorption energies can be tuned to correspond to the very top of the volcano plot [1, 96]. Another insight suggested by these descriptors for further improving OER catalysts beyond the state-of-the-art is to break the robust scaling relationship between the binding energies of OH* and OOH* [97]. For example, one basic idea is to engineer active sites that selectively stabilize the latter. Essentially, this boils down to strengthening surface oxygen’s affinity toward hydroxyl without affecting its affinity toward atomic hydrogen [42, 97]. Recent advances majorly include: (1) constructing multiple functional centers that will be selectivity active for different intermediates to independently optimize the binding energies; (2) applying external physical effects like strain, nanoscopic confinement and magnetic field to separately tune certain intermediate; and (3) lattice oxygen participated OER mechanism.
3.1 Constructing Multiple Functional Centers
The conventional concerted OER process was established on the redox reactions of a sole active cation. The interdependence between adsorption energies of intermediates reduces the degrees of freedom available for catalyst optimization [98]. Constructing multiple active centers would be a viable pathway that allows for the separate optimization of different intermediates, such as the recombination of oxygen adsorbates to form O2 or the dissociation of water [51]. Recent experimental and theoretical advances evidenced that the second active center could be either the TM cation or the non-metallic anions like C or the lattice oxygen.
3.1.1 Modified Active Centers Enabling O–O Direct Coupling
The O–O bond formation is considered as the rate-determining step (RDS) in the water oxidation reaction. As commonly observed in the natural photosystem II where the Mn4Ca clusters rationalize the four-electron oxidation of water, a multinuclear core typically is advantageous when reactions involve multi-electron transfer [99,100,101,102,103]. In these molecular cluster catalysts, the two neighboring metal centers with a suitable distance undergo *OH deprotonation to produce two metal-oxo species, which then will couple to release a O2 molecule (Fig. 9a) [41, 101]. Analogously, this concept has been successfully adopted in heterogeneous electrocatalysis for the OER. Yagi and coworkers proposed the direct O–O formation in perovskite CaCu3Fe4O12 (Fig. 9b). In the regular SrFeO3 perovskite (left in 9b), the OER proceeds with the conventional AEM. OH− is bound to B-site Fe ions, in which the RDS is the formation of the O–O bond (reaction 2) or the subsequent deprotonation (reaction 3) [83, 101]. Notably, after the incorporation of Fe4+ and Cu2+ in CaCu3Fe4O12, Fe–O-Fe bonds were heavily bent to give a shortened oxygen–oxygen (connected to the nearest neighboring Fe ions) distance of ~ 2.6 Å, which then enabled the occurrence of the direct formation of the O–O bond (right in Fig. 9b). In contrast, the oxygen–oxygen distance in sample SrFeO3 is ~ 3.9 Å, too large to interact with each other and form oxygen molecules. In this proposed pathway, the deprotonation of the oxyhydroxide group to form peroxide ions was skipped, resulting in the acceleration of the reaction and a lower overpotential due to the absence of a scaling relation. The catalyst CaCu3Fe4O12 demonstrated exceptional OER performance comparable to or exceeding those of state-of-the-art catalysts such as Ba0.5Sr0.5Co0.8Fe0.2O3−δ and the gold standard RuO2 [83]. Short oxygen–oxygen distance also exists in α-Mn2O3, where the direct O–O bond formation mechanism was thought to dominate [104]. Very recently, Lin and coworkers reported an electrocatalysts with Ru-atom-array patches supported on α-MnO2 for the OER, where Ru atoms substituting surface Mn atoms formed small and periodically arranged ensembles. The interatomic Ru–Ru distance in Ru/MnO2 (2.9 Å) is shorter than that in RuO2 (3.1 Å), facilitating direct O–O radical coupling for O2 evolution (Fig. 9c). The proposed electrocatalyst 12Ru/MnO2 shows high activity (161 mV at 10 mA cm−2) and outstanding stability [105]. This pathway has been denoted as the oxide path mechanism (OPM) for heterogeneous catalysis [99, 101, 106,107,108]. It is worth noting that this mechanism involves only O* and HO* species as intermediates without the generation of *OOH. Therefore, OER electrocatalysts proceeding with OPM pathway can break the scaling relationship without sacrificing stability. The stringent challenge to enable the OPM is the geometric configuration of metal active sites.

Oxygen generation through multiple functional centers. a Adjacent water-activation sites to promote intramolecular O–O bond formation [101]. Copyright © 2016 Springer Nature. b OH− adsorbates on Fe-terminated (100) planes of SFO (left) and CCFO (right) for Fe-mediated route [83]. Copyright © 2015 Springer Nature. c O–O radical coupling promoted by symmetric dual active sites [105]. Copyright © 2021 Springer Nature. d HRTEM images of cross-section of LDH nanosheets combined with NiO NPs. e Schematic illustration of the OER pathway at S1 site of the NiO/NiFe LDH intersection [109]. Copyright © 2019 Wiley. f Proposed reaction scheme with the intermediates having optimized geometry of the single-site and dual-site mechanisms towards OER. g Free energy diagram at 1.23 V for OER over Fe-NHGF, Co-NHGF and Ni-NHGF with a single-site mechanism, and Ni-NHGF with a dual-site mechanism [111]. Copyright © 2018 Springer Nature. h Differential charge densities of NiFe LDH with and without Au atom when one O atom is adsorbed on the Fe site. i Free energy diagram for the OER at different potentials on the surface of sAu/NiFe LDH model [112]. Copyright © 2018 American Chemical Society
3.1.2 Constructing Heterostructures and Heteroatoms
Separating *OH and *OOH on two different active sites would be expected to break the scaling relationship. An apparent way is to construct a heterostructure with fine interface engineering to ensure that the adsorption strength of each intermediate can be tuned independently. Gao and coworkers demonstrated the feasibility of this strategy in a NiO/NiFe layered double hydroxide heterostructure (Fig. 9d). In brief as shown in the proposed catalytic cycle (Fig. 9e), after OH− groups get adsorbed on S1, the O and H atoms in *OH form two hydrogen bonds with hydroxyl and lattice O in LDH, respectively. Then, *OH deprotonates to give rise to *O on both S1 and LDH. Subsequently, *OOH forms with *H captured by the lattice O in LDH and *OO adsorbed on S1, respectively. Meanwhile, one hydrogen bond is formed between *OO and the hydroxyl in LDH. Finally, when OH− captures *H and produces H2O, O2 is released. Based on the above, heterostructures exhibits two unique characteristics: (a) One or two additional chemical bonds always assist Ni cations (S1) in adsorbing intermediates; (b) The additional chemical bonds are different for each intermediate and vary dynamically during the whole OER process. As such, the binding energy of each intermediate can be adjusted independently, which provides an opportunity to break the scaling relationship [109].
Another approach is to incorporate heteroatoms in the crystal structure or on the surface by intervening in transition metal mediated catalysis with introduction of a proton acceptor to additionally stabilize *OOH. Halck and coworkers theoretically demonstrated that incorporation of Ni or Co into the surface on ruthenia could activate a proton donor–acceptor functionality on the conventionally inactive bridge sites (for *OOH) [110]. The calculation predicted that the optimized catalysts could achieve an overpotential of 0.1 and 0.25 V, much smaller than that for the conventional catalysts (0.37 V). However, the experimental results were much less significant than the theoretical predictions. It was believed that the clustering tendency violated the theoretical assumption. The recent advances in single atom catalysts (SACs) made the heteroatom structure possible to adjust the adjacent environments of the active centers. Fei and coworkers reported the synthesis of single-atom MN4C4 moieties (M = Fe, Co, Ni) for the OER, where both M and C atoms are possible adsorption sites for the different intermediates (Fig. 9f) [111]. Whether the C atoms participate in the OER process or not depends strongly on the number of d electrons of the metal in MN4C4 moieties. Specifically, for Fe and Co, the adsorption strength of all intermediates on the M site is stronger than that on the C site, and therefore, the adsorption-energy scaling between intermediates cannot be circumvented. On the other hand, for Ni, both intermediates of *O and *OH prefer to adsorb on the C site, whereas *OOH is favorably adsorbed on the M site, leading to circumvention of the scaling relationship (Fig. 9g). As a result, the NiN4C4 samples exhibited the best intrinsic activity, followed by FeN4C4 and CoN4C4 samples. Zhang and coworkers constructed single atom Au on NiFe layered double hydroxide (LDH) which revealed a sixfold enhancement for OER compared to pure LDH [112]. Upon the formation of Au = O bond (Fig. 9h), the spatially redistribution of d orbitals gives rise to the charge density around the ring region of Au parallel to the surface direction. The integrated charge density difference yields a net Au-to-LDH charge redistribution of 0.32 e, which transfers to surrounding O, Ni and Fe atoms, thus facilitates the adsorption of OH− and modifies the adsorption energies of O* and OOH* intermediates, resulting in low overpotential in the rate-limiting step from O* to OOH*. The free energy diagram for OER (Fig. 9i) shows the RDS is the formation of OOH* from O* with a small overpotential of 0.18 V. Of significance, the corresponding binding energies ΔEOH* and ΔEOOH* for Au decorated LDH sample are 0.93 and 3.46 eV, resulting in favorable Gibbs energies for four elementary steps, i.e., \(\Delta G_{1}^{0} = 1.33 \;{\text{eV}}\), \(\Delta G_{2}^{0} = 1.15 \;{\text{eV}}\), \(\Delta G_{3}^{0} = 1.41 \;{\text{eV}}\) and \(\Delta G_{4}^{0} = 1.03 \;{\text{eV}}\) at standard conditions, which is very close to the thermochemically ideal OER process (ΔEOH* = 0.86 eV and ΔEOOH* = 3.3 eV, with \(\Delta G_{1}^{0} = \Delta G_{2}^{0} = \Delta G_{3}^{0} = \Delta G_{4}^{0} = 1.23 \;{\text{eV}}\)) [37, 112].
3.2 Extrinsic Actions
3.2.1 Lattice Strain
Introducing lattice strain is one of the possible approaches with external forces to circumvent the scaling relations. Lattice strain, either comprehensive or tensile, can alter the surface electronic structure by modifying the distance between surface atoms [113]. Based on d-band theory, tensile strain leads to stronger oxygen binding energy; conversely, comprehensive strain leads to weaker binding [113,114,115]. Wang and coworkers reported to tune the catalytic activity of Pt catalyst by controlling its lattice strain [113]. Their calculation showed that change ranges of ΔGOH and ΔGOOH are the same, whereas ΔGO has a stronger dependence caused by its threefold coordination to the surface. Very similarly, Xie and coworkers, with N-doped graphene, theoretically demonstrated that tensile strain tends to stretch and break N–C* bond, which enhances the adsorption of *O while leaving that of *OH and *OOH unchanged [116]. Based on the results above, it appears that the O* adsorption can be controlled individually to potentially break the scaling relations among *O, *OH, and *OOH. Recently, Khorshidi and coworkers systematically described how strain can break this constraint by employing a mechanics-based eigenstress model to rationalize the effect of strain on adsorbate-catalyst bonding. In brief, when applying a uniaxial compression to a catalyst surface (Fig. 10a), if the energy level changes of the initial state (IS) and transition state (TS) resonate with the applied strain, the IS and TS are no longer correlated under applied strain, and the significantly decreased net reaction barrier upon strain can be expected to break scaling relation [117].

Copyright © 2018 Springer Nature. b Adsorption energies of OER intermediates (HO*, and HOO*) as a function of channel width for the OER relevant surface. c Volcano plot of the overpotential for OER; Arrows indicate trends in data as dmm decreases [118]. Copyright © 2015 Wiley. d The projected density of states (PDOS) of CoFe2O4 without and with spin alignment. e Schematic of spin-exchange mechanism for OER [122]. Copyright © 2021 Springer Nature. f Infrared images of NFO/NF under NIR irradiation. [125] Copyright © 2021 National Academy of Sciences. g Schematic electron transfer paths likely to occur in the Ni(OH)2-Au electrode under 532 nm laser irradiation responsible for the OER catalysis [129]. Copyright © 2016 American Chemical Society
Extinct actions on the OER to potentially break the scaling relations. a Applying uniaxial compression to the surface. When qualitative picture of the energy level changes of the IS and TS with the applied loading, a violation of transition-state scaling is expected [117].
3.2.2 Nanoscopic Confinement
This concept was introduced by Doyle and coworkers who hypothesized that a 3D nanoscopic channel would be able to provide a confined reaction environment that enables selective interaction between intermediates and catalyst (Fig. 10b) [118]. With rutile oxides for the OER as examples, they analyzed the bond formation by varying the chemical environment and found that the pronounced stabilization occurs for EOOH due to the hydrogen bond formation between HOO* and the opposite bridge site (Fig. 10b). Together with OER volcano based on the scaling-limited model (Fig. 10c), this model predicted that the overpotential for RuO2, on the left leg, could decrease to roughly 200 mV at dmm ≈ 7 Å, that is beyond the scaling limits. However, catalysts on the right leg, for example IrO2, were unaffected by confinement because the potential was limited by the deprotonation of HO* to O*.
3.2.3 Magnetic Field
Applying an external magnetic field is a viable approach to regulate the chemical reactions and thus the catalytic activity because (1) the electrochemical catalysis proceeds with charge transfer, and (2) most TMO catalysts display magnetic susceptibility that can be apparently impacted by magnetic field [119]. More insightfully in regard of the OER, the formation of O–O bond by breaking water molecules requires the spin conservation to yield the paramagnetic triplet state of molecular oxygen. Thus, spin polarization of the active catalyst surface, enabled by external magnetic field, may favor parallel spin alignment of oxygen atoms during the reaction to improve the efficiency of the process [120, 121]. Most recently, Garcés-Pineda and coworkers reported enhanced OER activity of magnetic oxides with an externally applied magnetic field [120]. Experimental data indicated that the magnetic field was affecting the reaction pathway, and the magneto-enhancement appeared to be proportional to the magnetic nature of the catalysts. Therefore, the highly magnetic TMO oxides like Fe–Ni oxides are the preferred spin-dependent OER catalysts. Later, Ren and coworkers further concluded that magnetic field effect worked on the ferromagnetic ordered catalysts (CoFe2O4 spinel) as the spin polarizer for spin selection, but did not apply to non-ferromagnetic catalysts [122]. Meanwhile, the authors pointed out that the key step of spin-polarized OER occurred at the first electron transfer step, where coherent spin exchange between the ferromagnetic catalyst and the adsorbed oxygen species enabled a fast kinetics under the magnetic field. As a result, the first electron transfer step was no longer the RDS. Analogously, Zhang and coworker also found that the magnetic field-induced spin-polarized kinetics was more distinct in the case that the first electron transfer step is the RDS during the OER [123]. With the projected density of states (PDOS) analysis, Ren and coworkers suggested that the spin alignment enabled a stronger 3d-2p hybridization in catalyst (Fig. 10d) and gave a higher spin density on the oxygen atoms. Then, the concomitant increment of the 3d–2p hybridization associate with FM ligand hole facilitated FM exchange between the ferromagnetic catalyst and the adsorbed oxygen species with smaller electron–electron repulsion (Fig. 10e), which gave rise to the improved spin conductivity and decreased bonding energies, making the first electron transfer step no longer the RDS. It is noteworthy that Zhou and coworkers reported a thermal-differentiated superlattice created by localized magnetic heating in CoMn metal–organic-frameworks (MOFs) [124]. In this particular case, the magnetic heating effect was mainly localized at magnetic layers instead of heating-insulating organic linkers, and the lattice expansion of the whole system was limited effectivity. With the increase in MS operation time, the temperature of magnetic layer can reach ~ 480 K, but the remaining regions keep unchanged at room temperature. This heating localization resulted in the rearrangement of the spin electron occupation and demonstrated a spin-dependent reaction pathway toward unprecedented OER performance, where the catalytic activity of OER is authentically related with spin configuration of reactive site. Overall, it is still in its very early stage; nonetheless, it opens a new strategy to manipulate the spin polarization in magnetic oxide catalysts for promoting the OER and is encouraging more detailed studies to understand how the magnetic field-induced spin polarization affects the OER process.
3.2.4 Photo-Induced Oxygen Evolution
Recently, it was found that photo-assisted OER improvement could be realized in photothermal electrocatalysts through photothermal effect [34, 125]. As well known, increasing the temperature improves the catalytic activity. Photothermal electrocatalysts allow the heating generation under the illumination of visible or near infrared light. Lin and coworkers studied the photothermal effect improved OER activity in NiFe2O4 nanoparticles [125]. Under a NIR-light irradiation (808 nm), infrared imaging clearly indicated the temperature increase on NFO catalyst while leaving the electrolyte temperature unchanged (Fig. 10f). The photothermal effect was demonstrated not only to reduce the energy barrier and improve the OER kinetics, but also to lower the potential for forming the catalytically active species. On the other hand, photo-generated charge holes are also helpful to improve the OER activity. Different to the regular semiconductor photocatalysis where the semiconductors directly absorb light illumination and generate charge carriers for chemical reactions [126], photo-assisted OER mainly refers to that the OER electrocatalysts accept photo-holes from a light-active donor. The representative is plasmonic particles which generate hot charger carriers (electrons and holes) under the resonant light illumination [127, 128]. Ye and coworkers reported Au-nanoparticle-decorated Ni(OH)2 nanosheets for catalyzing the OER. Under the irradiation of a 532 nm laser, the plasmon-generated hot holes inject into Ni(OH)2 and improve the oxidation of Ni2+ to Ni3+/4+, thus leading to the boosted OER activity (Fig. 10g) [129]. It is noted that photo-generated holes on the OER pathways were barely investigated yet. From this aspect, more in-depth understanding is needed in the future.
3.3 Lattice Oxygen-Mediated Mechanism (LOM)
3.3.1 Basics of the LOM
Recently, a new OER mechanism LOM has attracted intensive interests, which involves the participation of lattice oxygen for oxygen generation, and is able to rationalize the high intrinsic activity and surface reconstruction issues in some catalysts that were encountered and unexplained by conventional AEM. Compared to AEM (Fig. 2) that is based on cation redox chemistry, the LOM pathway cycle allows for the participation of lattice oxygen that must be active enough to escape and segregate from the lattice and interact with the adsorbed oxygen (*O) [130, 131]. In regular oxides, however, lattice oxygen is thermodynamically unfavorable given the deep location of the O 2p band caused by its large electronegativity (Fig. 11a) [130, 132, 133]. As shown as the rigid band diagram in Fig. 11a, the metal d-band of the metal oxides is generally located on the top of the oxygen p-band [134]. Anodic polarization causes an average increase in oxidation state of cations, then OER can be triggered when the O2/H2O redox potential aligns with the EF of the oxide. This is the commonly accepted catalytic cycle under the AEM in which the metal cation evidently plays a dominant role by changing oxidation states and concomitantly the bond strengths with the intermediates accordingly [1]. To initiate the oxygen redox reactions (that is, the lattice oxygen becomes thermodynamically favorable for oxygen generation), the EF has to move into the O 2p band and situates above the redox energy of the O2/H2O couple. Under this circumstance, EF gets pinned at the top of the O 2p band, and electronic states near the EF consist of substantial O 2p character. This electronic structure occurs in highly covalent oxides at which the overlap between M 3d and O 2p band is increased, i.e., the energy gap between O 2p center and d band center being minimized. Thus, it enables the electron transfer from lattice oxygen to the cation, resulting in the release of molecular oxygen and formation of oxygen vacancies [1, 132, 134,135,136]. In this regard, the later transition metal oxides with high metal–oxygen covalency are highly potential candidates for oxygen evolution with the LOM.

Copyright © 2017 Springer Nature. b Surface configurations of the intermediate after AEM Step 1 (I0) and the one after LOM Step 1 (I1). c The free energy change of I1 over I0 versus the O vacancy formation enthalpy for the cubic La1−xSrxCoO3−δ [134]. Copyright © 2016 Springer Nature. d Model of zinc-substituted MO2 and Schematic formation of ONB by extrapolating the molecular orbital energy diagram for octahedral MO6 [139]. Copyright © 2019 Springer Nature. e An illustration of how the oxygen 2p band center, and the relative band centers between oxygen 2p and active metal 3d, co-regulate the reaction mechanism of OER on spinel oxides. f The experimentally observed reaction activity as a function of the calculated Max(DT, DO). g The intrinsic activity of the as-synthesized Al0.5Mn2.5O4 and other reference OER catalysts in 0.1 M KOH at an overpotential of 300 mV [131]. Copyright © 2020 Springer Nature
Lattice oxygen mediated mechanism (LOM) for OER. a Scheme of the band positions to trigger the LOM. The position of the O2/H2O redox couple at pH 14 is 1.23 V versus RHE [132].
The release of lattice oxygen ligand from the catalyst surface is accompanied with the formation of oxygen vacancy (VO) that will be refilled and then act as a new active center. Mefford and coworkers presented a series of cobaltite perovskites and rationalized the roles of oxygen vacancies, M–O bond covalency, and redox activity of lattice oxygen through introducing Sr2+ substitution into La1-xSrxCoO3-δ [134]. With increasing Sr2+ substitution, higher Co–O covalency, more VO and faster bulk oxygen ion diffusion rate were observed. When x > 0.5 (that is, ΔG starts to be negative, Fig. 11c), the formation of superoxide-like –OO(VO) adsorbates (I1) is remarkably energetically favorable compared to –O (I0) in conventional AEM, which enables the switch of OER from AEM to LOM (Fig. 11b, c) [134, 137]. Analogously, this group later introduced Sr2+ into Ruddlesden–Popper oxide La0.5Sr1.5Ni1−xFexO4±δ, and observed the participation of lattice oxygen as well [138]. It was believed that the Sr2+ introduction improved the oxidation state of Ni and thus enhanced Ni–O covalency and electronic conductivity. Direct experimental observation for lattice oxygen participating the OER process was achieved by Grimaud and coworkers with in situ 18O isotope labelling mass DEMS [132]. The authors further confirmed that metal–oxygen covalency not only regulated the OER activity but also determined the reaction mechanism. The OER could be switched to LOM when the electronic states near the EF are filled with substantial O 2p character for highly covalent oxides.
Huang and coworker recently reported the possibility to trigger the LOM in oxyhydroxide through the inclusion of Zn2+ [139]. Different to perovskite, all three O 2p orbitals in the MOOH are engaged in M–O bond formation, while lacking the oxygen non-bonding (ONB) states that is essential for the formation of peroxo- or superoxo-like O–O dimers without the risk of structural destabilization [133, 140,141,142]. The authors proposed the incorporation of the low-valence and catalytically inactive Zn2+ into CoOOH to create accessible ONB states and increase Co–O covalency simultaneously, the two necessary conditions to permit LOM in oxide-based electrocatalysts (Fig. 11d) [133, 140]. The theoretical and experimental results showed that creating oxygen holes in ONB states along with the specific Zn–O2–Co–O2–Zn configuration is critical to regulating the OER mechanism; the OER proceeds via the LOM pathway only if the two neighboring oxidized oxygens can hybridize their oxygen holes without sacrificing metal–oxygen hybridization significantly.
Another representative case is spinel oxides AB2O4 with both A and B sites to be resided by transition metals, i.e., the co-existence of TMO6 and TMO4. Most recently, Sun and coworkers screened more than 300 spinel oxides and demonstrated two distinguishable energetical regions against the calculated oxygen 2p band center (ε2p) and the energy difference between the oxygen 2p and metal 3d band centers (ΔO2p-M3d): top-right suitable for LOM and bottom-left for AEM domains. ε2p and ΔO2p-M3d thus concurrently regulate the reaction mechanism of the OER (Fig. 11e) [131]. To achieve LOM, the oxygen 2p band center must be high enough (greater than − 1.75 eV) to guarantee its escape from the lattice, and meanwhile the oxygen p band level is higher than both cations for charge transfer from oxygen to cations. The authors further claimed that the OER activity on spinel oxides was intrinsically dominated by the covalency competition between tetrahedral and octahedral sites. The competition results in an asymmetric MT–O–MO backbone where the bond with weaker metal–oxygen covalency determines the exposure of cation sites and the activity. Of significance, the correlation of the OER activity with the calculated Max(DT, DO) (namely, the covalency between tetrahedral/octahedral cations and oxygen) displays a volcano-like shape (Fig. 11f) [131]. Spinel oxides on the very left side have low OER activity since the strong covalency for both M–O bonds makes them difficult to generate active sites. While on the very right part, the bond breakage results in the low-covalent M–O bond, which have no unpaired electrons to adsorb the hydroxyl groups for OER. As a result, the bonds at the middle part of the volcano are optimized to be neither too strong nor too polarized. Following this line, Al0.5Mn2.5O4 with spinel distribution [Mn]T[Al0.5Mn1.5]OO4 was predicted to locate at the summit of the established volcano plot with remarkable activity for OER. The experimental results demonstrated an overpotential of 240 mV for [Mn]T[Al0.5Mn1.5]OO4 and an intrinsic activity comparable to the state-of-the-art catalysts (Fig. 11g). Of note, no surface reconstructions to amorphous or hydroxide structures were observed after long-time OER cycling [131].
3.3.2 Reaction Pathways in the LOM
As discussed above, the LOM involves the lattice oxygen oxidization and the reversible formation of surface oxygen vacancies (Vo) in transition metal oxides. In a typical LOM catalytic cycle based on DFT calculations (Fig. 12), the dehydrogenation of *OH on the oxygen anion sites produces *OO species and VO (Step I, Eq. 6), then the *OO species evolves back to *OH, while releasing O2 and electrons (Step II, Eq. 7). During this step, VO is re-occupied by *OH and an adjacent surface lattice oxygen is protonated (Step III, Eq. 8). Finally, *OH is regenerated during the deprotonation process (Step IV, Eq. 9) [34, 134, 137, 138].

The LOM mechanism eliminates the concerted proton-electron transfer steps as occurred in conventional AEM. It generates the oxygen through the direct coupling of O–O bond (Eq. 7) and does not involve the formation of *OOH, suggesting a potent strategy to break the scaling relationship in AEM. Two approaches could potentially trigger the LOM in TMO-based electrocatalysts: (1) activating the lattice oxygen by strengthening the metal–oxygen covalency and lowering the formation energy of the defect oxygen, and (2) intentionally including oxygen defects that function as the active centers for the LOM.
3.3.3 Experimental Observations Evidencing the LOM
As a newly proposed OER paradigm, convincing experimental observations are required to evidence the participation of lattice oxygen during the oxygen evolution. Below, the commonly employed techniques to confirm the LOM mechanism are highlighted.
-
(1)
in situ 18O labelled electrochemical mass spectrometry (EMS). The most straightforward methodology is to monitor the products with labelled catalysts or electrolyte during the OER. On-line electrochemical mass spectroscopy (OLEMS) is regularly employed to quantitatively determine the volatile products during the electrochemical reactions [130]. Grimaud and coworkers carried out OLEMS measurements of 18O labelled perovskites and first provided direct experimental evidence of the oxidation of lattice oxygen in highly covalent oxides (La0.5Sr0.5CoO3−δ and LaCoO3−δ). Meanwhile, the results revealed that up to 37 monolayers of oxides (~ 14 nm) can be involved during the OER process [132]. Since then, 18O labelled EMS has been widely used as a powerful tool to monitor the participation of lattice oxygen [105, 130].
-
(2)
Operando spectroscopy. The operando spectroscopy techniques including infrared (IR) spectroscopy, Raman spectroscopy, X-ray absorption spectroscopy (XAS), and X-ray photoelectron spectroscopy (XPS) have been powerful tools for the investigation of electrocatalysts. The in situ use of these techniques for probing catalytic processes under reaction conditions gives accurate information regarding the dynamic changes occurring on electrode surface and deeper insight into basic mechanistic pathways [143]. As shown in the LMO catalytic pathways (Fig. 12), the lattice oxygen oxidation theoretically allows for the generation of ligand holes in O 2p orbitals, thus altering the electronic states. Also, the LMO process does not involve the formation of *OOH and generates the oxygen through the direct coupling of O–O bond. As such, in-situ monitoring of the catalyst surface during LOM-type OER with these operando techniques is viable to identify the reaction mechanism and the lattice oxygen redox chemistry in electrocatalysts [130]. Lin and coworkers carried out the operando synchrotron FT infrared (FTIR) spectroscopy because it is susceptible to the change of surface reaction intermediates. The formation of O–O bond at 1089 cm−1 could be clearly detected. Also, a unique peak at 1128 cm−1 is assigned to linearly bonded superoxol species, which are the intermediate before releasing O2 [105]. Several research groups have observed a shoulder peak of ~ 529 eV in O K-edge XAS spectra, that was assigned to positively charged oxygen species O(2−δ)−, and indicated the oxidation of lattice oxygen [144, 145]. Furthermore, coupling in situ Raman spectroscopy with 18O labeling, Hu and coworkers found that there existed a frequency variation at ~ 26 cm−1 with 18O isotope labeling, which was assigned to Co–O A1g vibration and consolidated the lattice oxygen exchange with electrolyte [146, 147]. Since either AEM- or LOM- medicated OER process takes place on the catalyst surface, we believe those operando spectroscopy techniques that are sensitive to the variations of surficial geometrical and electronic structures will play a critical role in elucidating the underlying mechanism.
-
(3)
Electrochemical pH-dependent OER activity. Grimaud and coworkers found that the oxides capable of LOM reaction like La0.5Sr0.5CoO3−δ, Pr0.5Ba0.5CoO3−δ and SrCoO3−δ showed pH-dependent OER activity on the scale of the RHE, while the intrinsic OER activity of LaCoO3 through the AEM is independent of pH (Fig. 13a) [132]. This was ascribed to the presence of non-concerted proton-electron transfer step during OER, originating from the mismatch between electron transfer kinetics and hydrogen affinity at the oxide/electrolyte interface [79, 132, 148, 149]. In the case that the OER proceeds with the LOM, the substantial decrease in charge transfer energy accelerates electron transfer and weakens hydroxide affinity, decoupling them in PDS with pH-dependent activity. It is noted that the pH dependent OER activity was also observed in spinel ZnFexCo2-xO4 oxides. However, that was believed to originate from the downshifted O p-band center relative to Fermi level induced by the spinel’s cation deficient nature and has nothing to do with the lattice oxygen oxidation [84, 150]. Therefore, the OER through the LOM can exhibit pH-dependent activity, nonetheless, the pH-dependent activity is not a decisive indicator for LOM reaction.
Fig. 13 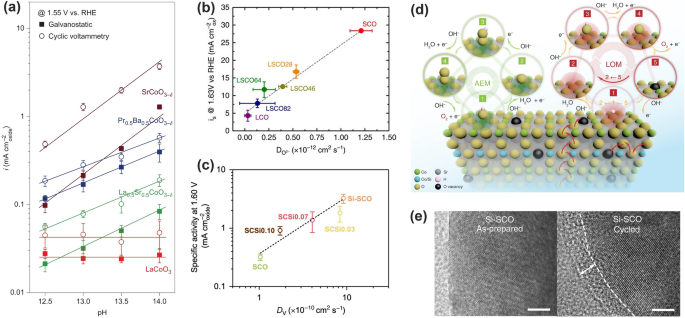
The pH-dependent activity, oxygen ion diffusion rate and surface stability for the OER in LOM pathways. a Specific OER activity of some perovskites at 1.55 V versus RHE after iR correction as a function of pH [132]. Copyright © 2017 Springer Nature. b Correlation of oxygen evolution activity with the oxygen ion diffusion rate in La1−xSrxCoO3−δ perovskites [134]. Copyright © 2016 Springer Nature. c Correlation of intrinsic OER activity in 0.1 M KOH with the oxygen anion diffusion rate. d A schematic illustration of the AEM and LOM reaction pathways on Si-incorporated strontium cobaltites. e HRTEM images for as-prepared and OER-Cycled Si-SCO [2]. Copyright © 2020 Springer Nature
-
(4)
Surface Amorphization. Given the participation of lattice oxygen for oxidation, migration and release under the LOM mediated OER, a high oxygen ions diffusion rate (DO) is vital to improve OER performance. Mefford and coworkers found that the catalytic activity towards the OER was strongly correlated with the oxygen diffusion rate in La1−xSrxCoO3−δ perovskite with a linear relationship (Fig. 13b) [134]. The SCO with the best OER activity through LOM pathway displays a DO of ~ 1.2 × 10–12 cm2 s−1 is nearly two orders of magnitude higher than that of LaCoO3 (~ 3 × 10–12 cm2 s−1) with AEM for oxygen evolution. The DO was further improved to 12.04 × 10–11 cm2 s−1 by introducing Si into SrCo1-xSixO3-δ, which is ~ 12.8 times faster than SCO and correlated well with the tenfold improvement in intrinsic activity (Fig. 13c) [2]. Based on this correlation, Pan and coworker proposed that the surface oxygen vacancy left after oxygen evolution could be quickly replenished by oxygen ions diffusing from the bulk of the electrocatalyst, instead by the refilling of OH−/H2O from the electrolyte (Fig. 13d) [2]. In this regard, increasing the oxygen ion diffusion rate is able to facilitate the refilling of the oxygen vacancies left by surface lattice oxygen, consequently promoting the OER catalysis.

The involvement of lattice oxygen in the OER suggests that surface exchange kinetics could be a key catalyst design factor. Including bulk oxygen deficiency could switch the OER mechanism from AEM to LOM, which is beneficial for OER efficiency [137]. However, a large oxygen deficiency decreases bulk stability, especially in cases of high activity and could possibly result in cation leaching and surface amorphization, as in the case of BSCF and Si-SCO though with different refilling species (Fig. 13e). Under current LOM, therefore, balancing the surface oxygen vacancy refilling rate and the surface vacancy formation rate will be helpful. Given the goal to achieve higher activity, constructing surfaces which allow fast enough kinetics for oxygen vacancy refilling appears to be one direct means to address this issue [2, 89]. On the other hand, as demonstrated in some spinel oxides (Al0.5Mn2.5O4 in Fig. 11) and perovskites (CaCu3Fe4O12 in Fig. 9b), and oxyhydroxide (Zn incorporated MOOH in Fig. 11), the high OER activity could be achieved while keeping the stability without surface amorphization. Further understanding the underlying mechanism will be another direction for high-active OER catalyst design.
4 Summary and Outlooks
As discussed above, the OER plays a vital role in determining the performance of ECS devices in utilization of renewable and sustainable energies. Therefore, developing high-efficiency and low-cost OER electrocatalysts to replace the noble metal-based Ru/Ir compounds is in urgent demand but remains a great technical challenge. In the last decade, we have witnessed lots of innovations in the catalyst fabrication and structure design, which was built on an in-depth understanding of the correlation between the catalyst and its catalytic behavior. Such fundamental understanding still needs to move forward before ideal catalyst could be reached. The OER activity is principally determined by the adsorption energy of intermediates on the catalyst surface. According to the conventional AEM pathway, the OER involves three oxygen-containing intermediates (*OH, *O, and *OOH). Tremendous research efforts have been made to build up the relationship between catalytic performance and catalysts’ electronic structures. For example, d-band theory, eg occupancy, metal–oxygen covalency models have successfully manifested their effectiveness in interpreting the catalytic activity trend and guiding the design of more efficient catalysts. However, the intrinsic adsorption energy scaling relationship between intermediates imposes an overpotential of as large as 300–40 mV. Therefore, research emphasis should be placed on experimentally and theoretically explore innovative strategies to circumvent the energy scaling limit based on the previous understanding of AME-type OER. Significant advances have been achieved recently in (1) developing new OER mechanisms like the LOM and the OPM, (2) constructing multifunctional active centers to separate the adsorption of different intermediates, and (3) applying extrinsic physical actions that potentially impacts certain intermediates while not affecting others. Regardless of those significant accomplishments, challenges still remain in this area. To be specific, we bring up several perspectives for further advancing the development.
-
(1)
Universal descriptor. One powerful descriptor that bridges the structure and catalytic activity should be able to not only explain the current catalytic behavior but also precisely predict the catalytic trend, which is essential for the rational design of novel catalysts with superior activity. Unfortunately, each individual descriptor shows its limitation to accurately describing the structure–activity relationship. For example, eg occupancy model was established on the ionic model and was unable to effectively capture the metal–oxygen covalency [78]. Moreover, the difficulty and sensitivity in determining the surface, sub-surface and bulk electronic is also an obstacle. Of very importance, to improve the descriptor-based design strategies, Hong and coworkers with statistical learning aggregated 101 observations in perovskites and identified five basic descriptor families, where covalency and electron occupancy hold the strongest influence on the OER activity [30]. The analysis suggested that combination of descriptors is necessary for concluding the best prediction. Besides, there remains a strong need to explore more physically meaningful approaches to fully understand the effects of chemistry on the OER activity. On the other hand, the most popular catalytic descriptors were developed in terms of bulk properties; whereas, catalytic reactions occur on the catalyst surface, the surface electronic structure information and the coordination chemistry should be fully taken into account in the future research. Overall, it is still a great challenge to discover a universal and measurable descriptor to accurately rationalize the structure–activity relationship [43].
-
(2)
New OER paradigms. The conventional OER paradigm (AEM) is hampered by the adsorption energy scaling relationship. The LOM skips the OOH* step and allows for the direct O–O formation with lattice oxygen and, thus, is a promising strategy to break the scaling relationship limit toward more efficient OER catalysts. Strengthened metal–oxygen covalency and oxygen vacancies are two prerequisites to permit the LOM in oxide-based electrocatalysts. Given the multimetallic positions in most oxides like perovskites, spinels, and LDHs, cation substitution has been a routine approach to modulate the OER toward the LOM pathway. In this regard, later TMs with large electronegativity will be potent dopants. Meanwhile, partially replacing oxygen with anions having larger electronegativity like F could also be able to improve the covalency and enable lattice oxygen oxidation during the OER [151, 152]. Additionally, introducing oxygen vacancy intentionally into the lattice matrix is highly expected to accelerate the lattice oxygen redox chemistry because the presence of Vo can tune the relative band position of TM and O 2p bands of the catalyst host and promote the metal–oxygen covalency [130]. Challenge remaining for the LOM is the long-term stability due to the cation leaching and surface amorphization. Constructing surfaces which allows fast enough kinetics for oxygen vacancy refilling appears to be one direction to address this issue [2, 89]. Of significance, the spinel Al0.5Mn2.5O4 [131], CaCu3Fe4O12 perovskites [83], and oxyhydroxide Zn incorporated MOOH [139] did not show surface amorphization while achieving high OER activity. Further understanding the underlying mechanism will be another direction for high-active OER catalyst design under the LOM to couple the stability and high activity. Analogous to the homogeneous oxo–oxo coupling mechanism, oxide path mechanism (OPM) has been proposed for heterogeneous catalysts [99, 101, 105]. Different to the LOM, the OPM pathway allows direct O–O radical coupling without the generation of oxygen vacancies and the participation of lattice oxygen. Therefore, the OPM is another highly promising candidate for further improving OER performance. However, the OPM has very stringent requirements for the geometric configuration of active sites. Engineering electrocatalysts to have symmetric dual-metal sites with appropriate atomic distances are technically challenging, but expected to promote O–O radical coupling with a low energy barrier [105].
-
(3)
Operando characterizations. Probing active sites with operando techniques will be a direct tool to identify the active center and more accurately understand the underlying reaction mechanisms. Given the surface reconstruction commonly observed during the OER because of the high applied potential on which the active sites are sensitive to the local environmental conditions [2, 58], this will be more useful to clarify what parameter governs the participation of lattice oxygen and what pathway is more thermodynamically favorable for oxygen refilling and oxygen ions diffusion and so on when combining in situ techniques, such as scanning probe microscopy, Raman spectroscopy, infrared spectroscopy, and X-ray absorption fine structure (XAFS) technique under electrochemical operation conditions [41]. For example, Grimaud and coworkers used OLEMS measurements and demonstrated the participation of lattice oxygen in perovskites [132]. Lin and coworkers employed operando synchrotron FT infrared (FTIR) spectroscopy and identified the distinctive absorption peak at 1128 cm−1 for linearly bonded superoxo species (metal-O–O) in 12Ru/MnO2 catalyst, which is the evidence for the direct O–O radical coupling pathway during the OER [105]. With operando X-ray absorption spectroscopy, Bai and coworkers revealed that the single-atom Co can be in situ transformed into Co-Fe dimeric moiety as the active center for the boosted OER activity [153]. As discussed above, the OER takes places on catalyst surface, which is very susceptible to the surrounding environments and cannot be tracked by ex situ technologies. Monitoring the catalyst surface with operando spectroscopic techniques during the reactions will be essential to shed light on the fundamental origin that triggers the OER and governs the reaction kinetics.
-
(4)
Novel catalysts and advanced synthetic methodologies. Despite the tremendous accomplishments in the last decades, there still exists a wide gap between the state-of-the-art OER catalysts and the ideal ones. Novel materials and structures along with the advanced.
Overall, due to the sluggish reaction kinetics, the OER has long been the bottleneck for the electrochemical energy conversion and storage devices. This paper comprehensively reviewed the benchmark catalytic descriptors for OER electrocatalysis under the adsorption-energy scaling relationship among oxygen-containing intermediates. Given current achievements and limitations on the AEM, a summary of the physiochemical fundamentals and recent progress on the LOM, the OPM and other extrinsic actions to break the scaling relationship has been provided, targeting to help the researchers gain an in-depth understanding. Clarifying the new OER paradigms like the LOM and the OPM is in urgent demand to advance the further development by accelerating the OER kinetics and lowering the overpotentials. Integrating advanced operando techniques, novel synthetic methodologies and cutting-edge computational simulations will be essential to advance the OER fundamental research toward commercial ECS devices.
References
A.R. Zeradjanin, J. Masa, I. Spanos, R. Schlögl, Activity and stability of oxides during oxygen evolution reaction—from mechanistic controversies toward relevant electrocatalytic descriptors. Front. Energy Res. 8, 613092 (2021). https://doi.org/10.3389/fenrg.2020.613092
Y. Pan, X. Xu, Y. Zhong, L. Ge, Y. Chen et al., Direct evidence of boosted oxygen evolution over perovskite by enhanced lattice oxygen participation. Nat. Commun. 11, 2002 (2020). https://doi.org/10.1038/s41467-020-15873-x
Y. Yang, Y. Yu, J. Li, Q. Chen, Y. Du et al., Engineering ruthenium-based electrocatalysts for effective hydrogen evolution reaction. Nano-Micro Lett. 13, 160 (2021). https://doi.org/10.1007/s40820-021-00679-3
A. Grimaud, K.J. May, C.E. Carlton, Y. Lee, M. Risch et al., Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution. Nat. Commun. 4, 2439 (2013). https://doi.org/10.1038/ncomms3439
M. Cui, X. Ding, X. Huang, Z. Shen, T. Lee et al., Ni3+-induced hole states enhance the oxygen evolution reaction activity of nixco3−xo4 electrocatalysts. Chem. Mater. 31(18), 7618–7625 (2019). https://doi.org/10.1021/acs.chemmater.9b02453
W.H. Lee, M.H. Han, Y. Ko, B.K. Min, K.H. Chae et al., Electrode reconstruction strategy for oxygen evolution reaction: maintaining Fe–CoOOH phase with intermediate-spin state during electrolysis. Nat. Commun. 13, 605 (2022). https://doi.org/10.1038/s41467-022-28260-5
D.A. Cullen, K.C. Neyerlin, R.K. Ahluwalia, R. Mukundan, K.L. More et al., New roads and challenges for fuel cells in heavy-duty transportation. Nat. Energy 6, 462–474 (2021). https://doi.org/10.1038/s41560-021-00775-z
X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng et al., Fe–N–C electrocatalyst with dense active sites and efficient mass transport for high-performance proton exchange membrane fuel cells. Nat. Catal. 2, 259–268 (2019). https://doi.org/10.1038/s41929-019-0237-3
W.H. Lee, Y. Ko, J.H. Kim, C.H. Choi, K.H. Chae et al., High crystallinity design of Ir-based catalysts drives catalytic reversibility for water electrolysis and fuel cells. Nat. Commun. 12, 4271 (2021). https://doi.org/10.1038/s41467-021-24578-8
J. Li, D. Chu, D.R. Baker, R. Jiang, Seamless separation of OHad and Had on a Ni–O catalyst toward exceptional alkaline hydrogen evolution. J. Mater. Chem. A 10(3), 1278–1283 (2022). https://doi.org/10.1039/D1TA07303F
H. Nan, Y. Su, C. Tang, R. Cao, D. Li et al., Engineering the electronic and strained interface for high activity of PdMcore@Ptmonolayer electrocatalysts for oxygen reduction reaction. Sci. Bull. 65(16), 1396–1404 (2020). https://doi.org/10.1016/j.scib.2020.04.015
P. Rao, Y. Liu, Y. Su, M. Zhong, K. Zhang et al., S, N co-doped carbon nanotube encased Co NPs as efficient bifunctional oxygen electrocatalysts for zinc–air batteries. Chem. Eng. J. 422, 130135 (2021). https://doi.org/10.1016/j.cej.2021.130135
T.M. Gür, Review of electrical energy storage technologies, materials and systems: challenges and prospects for large-scale grid storage. Energy Environ. Sci. 11(10), 2696–2767 (2018). https://doi.org/10.1039/C8EE01419A
Z. Seh, J. Kibsgaard, C.F. Dickens, I. Chorkendorff, J.K. Norskov et al., Combining theory and experiment in electrocatalysis: insights into materials design. Science 355(6321), eaad4998 (2017). https://doi.org/10.1126/science.aad4998
L.C. Seitz, C.F. Dickens, K. Nishio, Y. Hikita, J. Montoya et al., A highly active and stable IrOx/SrIrO3 catalyst for the oxygen evolution reaction. Science 353(6303), 1011–1014 (2016). https://doi.org/10.1126/science.aaf5050
H.N. Nong, T. Reier, H. Oh, M. Gliech, P. Paciok et al., A unique oxygen ligand environment facilitates water oxidation in hole-doped IrNiOx core-shell electrocatalysts. Nat. Catal. 1, 841–851 (2018). https://doi.org/10.1038/s41929-018-0153-y
J. Zhang, Q. Zhou, Y. Tang, L. Zhang, Y. Li, Zinc–air batteries: are they ready for prime time? Chem. Sci. 10(39), 8924–8929 (2019). https://doi.org/10.1039/C9SC04221K
W. Sun, F. Wang, B. Zhang, M. Zhang, V. Küpers et al., A rechargeable zinc–air battery based on zinc peroxide chemistry. Science 371(6524), 46–51 (2021). https://doi.org/10.1126/science.abb9554
Y. Zhu, K. Yue, C. Xia, S. Zaman, H. Yang et al., Recent advances on MOF derivatives for non-noble metal oxygen electrocatalysts in zinc–air batteries. Nano-Micro Lett. 13, 137 (2021). https://doi.org/10.1007/s40820-021-00669-5
W. Xia, A. Mahmood, Z. Liang, R. Zou, S. Guo, Earth-abundant nanomaterials for oxygen reduction. Angew. Chem. Int. Ed. 55(8), 2650–2676 (2016). https://doi.org/10.1002/anie.201504830
Y.N. Regmi, X. Peng, J.C. Fornaciari, M. Wei, D.J. Myers et al., A low temperature unitized regenerative fuel cell realizing 60% round trip efficiency and 10,000 cycles of durability for energy storage applications. Energy Environ. Sci. 13(7), 2096–2105 (2020). https://doi.org/10.1039/C9EE03626A
J.T. Mefford, A.R. Akbashev, M. Kang, C.L. Bentley, W.E. Gent et al., Correlative operando microscopy of oxygen evolution electrocatalysts. Nature 593, 67–73 (2021). https://doi.org/10.1038/s41586-021-03454-x
H.N. Nong, L.J. Falling, A. Bergmann, M. Klingenhof, H.P. Tran et al., Key role of chemistry versus bias in electrocatalytic oxygen evolution. Nature 587, 408–413 (2020). https://doi.org/10.1038/s41586-020-2908-2
J. Li, D. Chu, D.R. Baker, A. Leff, P. Zheng et al., Earth-abundant Fe and Ni dually doped Co2P for superior oxygen evolution reactivity and as a bifunctional electrocatalyst toward renewable energy-powered overall alkaline water splitting. ACS Appl. Energy Mater. 4(9), 9969–9981 (2021). https://doi.org/10.1021/acsaem.1c01926
J. Li, D. Chu, H. Dong, D.R. Baker, R. Jiang, Boosted oxygen evolution reactivity by igniting double exchange interaction in spinel oxides. J. Am. Chem. Soc. 142(1), 50–54 (2020). https://doi.org/10.1021/jacs.9b10882
R. Xie, Z. Nie, X. Hu, Y. Yu, C. Aruta et al., Pr-doped LaCoO3 toward stable and efficient oxygen evolution reaction. ACS Appl. Energy Mater. 4(9), 9057–9065 (2021). https://doi.org/10.1021/acsaem.1c01348
J.S. Kim, B. Kim, H. Kim, K. Kang, Recent progress on multimetal oxide catalysts for the oxygen evolution reaction. Adv. Energy Mater. 8(11), 1702774 (2018). https://doi.org/10.1002/aenm.201702774
H. Tian, L. Zeng, Y. Huang, Z. Ma, G. Meng et al., In situ electrochemical Mn(III)/Mn(IV) generation of Mn(II)O electrocatalysts for high-performance oxygen reduction. Nano-Micro Lett. 12, 161 (2020). https://doi.org/10.1007/s40820-020-00500-7
G. Wu, A. Santandreu, W. Kellogg, S. Gupta, O. Ogoke et al., Carbon nanocomposite catalysts for oxygen reduction and evolution reactions: from nitrogen doping to transition-metal addition. Nano Energy 29, 83 (2016). https://doi.org/10.1016/j.nanoen.2015.12.032
W.T. Hong, R.E. Welsch, Y. Shao-Horn, Descriptors of oxygen-evolution activity for oxides: a statistical evaluation. J. Phys. Chem. C 120(1), 78–86 (2016). https://doi.org/10.1021/acs.jpcc.5b10071
B. Hammer, J.K. Nørskov, Theoretical surface science and catalysis-calculations and concepts. Adv. Catal. 45, 71–129 (2000). https://doi.org/10.1016/S0360-0564(02)45013-4
H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan et al., The mechanism of water oxidation: from electrolysis via homogeneous to biological catalysis. ChemCatChem 2(7), 724–771 (2010). https://doi.org/10.1002/cctc.201000126
H. Lv, D. Li, D. Strmcnik, A.P. Paulikas, N.M. Markovic et al., Recent advances in the design of tailored nanomaterials for efficient oxygen reduction reaction. Nano Energy 29, 149–165 (2016). https://doi.org/10.1016/j.nanoen.2016.04.008
L. Gao, X. Cui, C. Sewell, J. Li, Z. Lin, Recent advances in activating surface reconstruction for the high-efficiency oxygen evolution reaction. Chem. Soc. Rev. 50(15), 8428–8569 (2021). https://doi.org/10.1039/D0CS00962H
S. Yuan, X. Duan, J. Liu, Y. Ye, F. Lv et al., Recent progress on transition metal oxides as advanced materials for energy conversion and storage. Energy Storage Mater. 42, 317–369 (2021). https://doi.org/10.1016/j.ensm.2021.07.007
H. Sun, X. Xu, Y. Song, W. Zhou, Z. Shao, Designing high-valence metal sites for electrochemical water splitting. Adv. Funct. Mater. 31(16), 2009779 (2021). https://doi.org/10.1002/adfm.202009779
I.C. Man, H. Su, F. Calle-Vallejo, H.A. Hansen, J.I. Martínez et al., Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 3(7), 1159–1165 (2011). https://doi.org/10.1002/cctc.201000397
J. Rossmeisl, A. Logadottir, J.K. Nørskov, Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 319(1–3), 178–184 (2005). https://doi.org/10.1016/j.chemphys.2005.05.038
J. Rossmeisl, Z.W. Qu, H. Zhu, G.J. Kroes, J.K. Nørskov, Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 607(1–2), 83–89 (2007). https://doi.org/10.1016/j.jelechem.2006.11.008
J.K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J.R. Kitchin et al., Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108(46), 17886–17892 (2004). https://doi.org/10.1021/jp047349j
Z. Huang, J. Song, S. Dou, X. Li, J. Wang et al., Strategies to break the scaling relationtoward enhanced oxygen electrocatalysis. Matter 1(6), 1494–1518 (2019). https://doi.org/10.1016/j.matt.2019.09.011
X. Li, S. Duan, E. Sharman, Y. Zhao, L. Yang et al., Exceeding the volcano relationship in oxygen reduction/evolution reactions using single-atom-based catalysts with dual-active-sites. J. Mater. Chem. A 8(20), 10193–10198 (2020). https://doi.org/10.1039/D0TA01399D
J. Liu, H. Liu, H. Chen, X. Du, B. Zhang et al., Progress and challenges toward the rational design of oxygen electrocatalysts based on a descriptor approach. Adv. Sci. 7(1), 1901614 (2020). https://doi.org/10.1002/advs.201901614
J.K. Nørskov, F. Abild-Pedersen, F. Studt, T. Bligaard, Density functional theory in surface chemistry and catalysis. PNAS 108(3), 937–943 (2011). https://doi.org/10.1073/pnas.1006652108
Z. Zhao, S. Liu, S. Zha, D. Cheng, F. Studt et al., Theory-guided design of catalytic materials using scaling relationships and reactivity descriptors. Nat. Rev. Mater. 4, 792–804 (2019). https://doi.org/10.1038/s41578-019-0152-x
K. Zhang, R. Zou, Advanced transition metal-based OER electrocatalysts: current status, opportunities, and challenges. Small 17(37), 2100129 (2021). https://doi.org/10.1002/smll.202100129
J.K. Nørskov, T. Bligaard, J. Rossmeisl, C.H. Christensen, Towards the computational design of solid catalysts. Nat. Chem. 1, 37–46 (2009). https://doi.org/10.1038/nchem.121
Z. Wang, J. Huang, L. Wang, Y. Liu, W. Liu et al., Cation-tuning induced d-band center modulation on Co-based spinel oxide for oxygen reduction/evolution reaction. Angew. Chem. Int. Ed. (2022). https://doi.org/10.1002/anie.202114696
K. Yang, P. Xu, Z. Lin, Y. Yang, P. Jiang et al., Ultrasmall Ru/Cu-doped RuO2 complex embedded in amorphous carbon skeleton as highly active bifunctional electrocatalysts for overall water splitting. Small 14(41), 1803009 (2018). https://doi.org/10.1002/smll.201803009
S. Sun, X. Zhou, B. Cong, W. Hong, G. Chen, Tailoring the d-band centers endows (NixFe1−x)2P nanosheets with efficient oxygen evolution catalysis. ACS Catal. 10(16), 9086–9097 (2020). https://doi.org/10.1021/acscatal.0c01273
W.T. Hong, M. Risch, K.A. Stoerzinger, A. Grimaud, J. Suntivich et al., Toward the rational design of non-precious transition metal oxides for oxygen electrocatalysis. Energy Environ. Sci. 8(5), 1404–1427 (2015). https://doi.org/10.1039/C4EE03869J
Y. Lee, J. Kleis, J. Rossmeisl, Y. Shao-Horn, D. Morgan, Prediction of solid oxidefuel cell cathode activity with first-principles descriptors. Energy Environ. Sci. 4(10), 3966–3970 (2011). https://doi.org/10.1039/C1EE02032C
Y. Lee, D. Lee, X.R. Wang, H.N. Lee, D. Morgan et al., Kinetics of oxygen surface exchange on epitaxial Ruddlesden–Popper phases and correlations to first-principles descriptors. J. Phys. Chem. Lett. 7(2), 244–249 (2016). https://doi.org/10.1021/acs.jpclett.5b02423
W. Xie, Y. Lee, Y. Shao-Horn, D. Morgan, Oxygen point defect chemistry in Ruddlesden–Popper oxides (La1−xSrx)2MO4±δ (M=Co, Ni, Cu). J. Phys. Chem. Lett. 7(10), 1939–1944 (2016). https://doi.org/10.1021/acs.jpclett.6b00739
A. Fujimori, Electronic structure of metallic oxides: band-gap closure and valence control. J. Phys. Chem. Solids 53(12), 15951602 (1992). https://doi.org/10.1016/0022-3697(92)90149-8
E. Antolini, J. Salgado, E. Gonzalez, The stability of Pt–M (M = first row transition metal) alloy catalysts and its effect on the activity in low temperature fuel cells: a literature review and tests on a Pt–Co catalyst. J. Power Sources 160(2), 957–968 (2006). https://doi.org/10.1016/j.jpowsour.2006.03.006
T. Wang, Y. Sun, Y. Zhou, S. Sun, X. Hu et al., Identifying influential parameters of octahedrally coordinated cations in spinel ZnMnxCo2−xO4 oxides for the oxidation reaction. ACS Catal. 8(9), 8568–8577 (2018). https://doi.org/10.1021/acscatal.8b02376
H. Ding, H. Liu, W. Chu, C. Wu, Y. Xie, Structural transformation of heterogeneous materials for electrocatalytic oxygen evolution reaction. Chem. Rev. 121(21), 13174–13212 (2021). https://doi.org/10.1021/acs.chemrev.1c00234
K.J. May, C.E. Carlton, K.A. Stoerzinger, M. Risch, J. Suntivich et al., Influence of oxygen evolution during water oxidation on the surface of perovskite oxide catalysts. J. Phys. Chem. Lett. 3(22), 3264–3270 (2012). https://doi.org/10.1021/jz301414z
B. Han, K.A. Stoerzinger, V. Tileli, A.D. Gamalski, E.A. Stach et al., Nanoscale structural oscillations in perovskite oxides induced by oxygen evolution. Nat. Mater. 16, 121–126 (2017). https://doi.org/10.1038/nmat4764
Y. Matsumoto, H. Yoneyama, H. Tamura, Catalytic activity for electrochemical reduction of oxygen of lanthanum nickel oxide and related oxides. J. Electroanal. Chem. Interfacial Electrochem. 79(2), 319–326 (1977). https://doi.org/10.1016/S0022-0728(77)80453-1
Y. Matsumoto, H. Yoneyama, H. Tamura, Influence of the nature of the conduction band of transition metal oxides on catalytic activity for oxygen reduction. J. Electroanal. Chem. Interfacial Electrochem. 83(2), 237–243 (1977). https://doi.org/10.1016/S0022-0728(77)80169-1
J. Suntivich, K.J. May, H.A. Gasteiger, J.B. Goodenough, Y. Shao-Horn, A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science 334(6061), 1383–1385 (2011). https://doi.org/10.1126/science.1212858
J. Suntivich, H.A. Gasteiger, N. Yabuuchi, H. Nakanishi, J.B. Goodenough et al., Design principles for oxygen-reduction activity on perovskite oxide catalysts for fuel cells and metal–air batteries. Nat. Chem. 3, 546–550 (2011). https://doi.org/10.1038/nchem.1069
C. Wei, Z. Feng, G.G. Scherer, J. Barber, Y. Shao-Horn et al., Cations in octahedral sites: a descriptor for oxygen electrocatalysis on transition-metal spinels. Adv. Mater. 29(23), 1606800 (2017). https://doi.org/10.1002/adma.201606800
J. Bockris, T. Otagawa, Mechanism of oxygen evolution on perovskites. J. Phys. Chem. 87(15), 2960–2971 (1983). https://doi.org/10.1021/j100238a048
J. Bockris, T. Otagawa, The electrocatalysis of oxygen evolution on perovskites. J. Electrochem. Soc. 131, 290 (1984). https://doi.org/10.1149/1.2115565
J. Suntivich, W.T. Hong, Y. Lee, J.M. Rondinell, W. Yang et al., Estimating hybridization of transition metal and oxygen states in perovskites from O K-edge X-ray absorption spectroscopy. J. Phys. Chem. C 118(4), 1856–1863 (2013). https://doi.org/10.1021/jp410644j
X. Wang, X.J. Gao, L. Qin, C. Wang, L. Song et al., eg occupancy as an effective descriptor for the catalytic activity of perovskite oxide-based peroxidase mimics. Nat. Commun. 10, 704 (2019). https://doi.org/10.1038/s41467-019-08657-5
M. Yu, G. Li, C. Fu, E. Liu, K. Manna et al., Tunable eg Orbital occupancy in heusler compounds for oxygen evolution reaction. Angew. Chem. Int. Ed. 60(11), 5800–5805 (2021). https://doi.org/10.1002/anie.202013610
J. Song, C. Wei, Z. Huang, C. Liu, L. Zeng et al., A review on fundamentals for designing oxygen evolution electrocatalysts. Chem. Soc. Rev. 49(7), 2196–2214 (2020). https://doi.org/10.1039/C9CS00607A
Y. Li, H. Jiang, Z. Cui, S. Zhu, Z. Li et al., Spin state tuning of the octahedral sites in Ni–Co-based spinel toward highly efficient urea oxidation reaction. J. Phys. Chem. C 125(17), 9190–9199 (2021). https://doi.org/10.1021/acs.jpcc.1c02116
Y. Tong, Y. Guo, P. Chen, H. Liu, M. Zhang et al., Spin-state regulation of perovskite cobaltite to realize enhanced oxygen evolution activity. Chem 3(5), 812–821 (2017). https://doi.org/10.1016/j.chempr.2017.09.003
H. Liu, S. Liu, Y. Zou, X. Zhang, P. Guo et al., Synergistic doping and tailoring: realizing in depth modulation on valence state of CoFe spinel oxide for high-efficiency water oxidation. Appl. Surf. Sci. 572, 151388 (2022). https://doi.org/10.1016/j.apsusc.2021.151388
Y. Zhou, S. Sun, S. Xi, Y. Duan, T. Sritharan et al., Superexchange effects on oxygen reduction activity of edge-sharing [CoxMn1−xO6] octahedra in spinel oxide. Adv. Mater. 30(11), 1705407 (2018). https://doi.org/10.1002/adma.201705407
A. Badreldin, A.E. Abusrafa, A. Abdel-Wahab, Oxygen-deficient cobalt-based oxides for electrocatalytic water splitting. Chemsuschem 14(1), 10–32 (2021). https://doi.org/10.1002/cssc.202002002
Y. Guo, Y. Tong, P. Chen, K. Xu, J. Zhao et al., Engineering the electronic state of a perovskite electrocatalyst for synergistically enhanced oxygen evolution reaction. Adv. Mater. 27(39), 5989–5994 (2015). https://doi.org/10.1002/adma.201502024
J. Hwang, R.R. Rao, L. Giordano, Y. Katayama, Y. Yu et al., Perovskites in catalysis and electrocatalysis. Science 358(6364), 751–756 (2017). https://doi.org/10.1126/science.aam7092
W.T. Hong, K.A. Stoerzinger, Y. Lee, L. Giordano, A. Grimaud et al., Charge-transfer-energy-dependent oxygen evolution reaction mechanisms for perovskite oxides. Energy Environ. Sci. 10(10), 2190–2200 (2017). https://doi.org/10.1039/C7EE02052J
D.A. Kuznetsov, B. Han, Y. Yu, R.R. Rao, J. Hwang et al., Tuning redox transitions via inductive effect in metal oxides and complexes, and implicationsin oxygen electrocatalysis. Joule 2(2), 225–244 (2018). https://doi.org/10.1016/j.joule.2017.11.014
Y. Duan, S. Sun, S. Xi, X. Ren, Y. Zhou et al., Tailoring the Co 3d-O 2p covalency in LaCoO3 by Fe substitution to promote oxygen evolution reaction. Chem. Mater. 29(24), 10534–10541 (2017). https://doi.org/10.1021/acs.chemmater.7b04534
D. Du, R. Zheng, X. Chen, W. Xiang, C. Zhao et al., Adjusting the covalency of metal–oxygen bonds in LaCoO3 by Sr and Fe cation codoping to achieve highly efficient electrocatalysts for aprotic lithium–oxygen batteries. ACS Appl. Mater. Interfaces 13(28), 33133–33146 (2021). https://doi.org/10.1021/acsami.1c08586
S. Yagi, I. Yamada, H. Tsukasaki, A. Seno, M. Murakami et al., Covalency-reinforced oxygen evolution reaction catalyst. Nat. Commun. 6, 8249 (2015). https://doi.org/10.1038/ncomms9249
Y. Zhou, S. Sun, J. Song, S. Xi, B. Chen et al., Enlarged Co–O covalency in octahedral sites leading to highly efficient spinel oxides for oxygen evolution reaction. Adv. Mater. 30(32), 1802912 (2018). https://doi.org/10.1002/adma.201802912
F. Liu, M. Yu, X. Chen, J. Li, H. Liu et al., Defective high-entropy rocksalt oxide with enhanced metal-oxygen covalency for electrocatalytic oxygen evolution. Chin. J. Catal. 43(1), 122–129 (2022). https://doi.org/10.1016/S1872-2067(21)63794-4
Y. Zhu, H.A. Tahini, Z. Hu, Y. Yin, Q. Lin et al., Boosting oxygen evolution reaction by activation of lattice-oxygen sites in layered Ruddlesden–Popper oxide. EcoMat 2(2), e12021 (2020). https://doi.org/10.1002/eom2.12021
S. Sun, Y. Sun, Y. Zhou, S. Xi, X. Ren et al., Shifting oxygen charge towards octahedral metal: a way to promote water oxidation on cobalt spinel oxides. Angew. Chem. Int. Ed. 58(18), 6042–6047 (2019). https://doi.org/10.1002/anie.201902114
X. Cheng, E. Fabbri, M. Nachtegaal, I.E. Castelli, M.E. Kazzi et al., Oxygen evolution reaction on L1−xSrxCoO3 perovskites: a combined experimental and theoretical study of their structural, electronic, and electrochemical properties. Chem. Mater. 27(22), 7662–7672 (2015). https://doi.org/10.1021/acs.chemmater.5b03138
C. Yang, A. Grimaud, Factors controlling the redox activity of oxygen in perovskites: from theory to application for catalytic reactions. Catalysts 7(5), 149 (2017). https://doi.org/10.3390/catal7050149
F. Calle-Vallejo, N.G. Inoglu, H. Su, J.I. Martínez, I.C. Man et al., Number of outer electrons as descriptor for adsorption processes on transition metals and their oxides. Chem. Sci. 4(3), 1245–1249 (2013). https://doi.org/10.1039/C2SC21601A
F. Calle-Vallejo, J.I. Martínez, J.M. García-Lastra, J. Rossmeisl, M.T.M. Koper, Physical and chemical nature of the scaling relations between adsorption energies of atoms on metal surfaces. Phys. Rev. Lett. 108, 116103 (2012). https://doi.org/10.1103/PhysRevLett.108.116103
F. Calle-Vallejo, J.I. Martínez, J.M.G. Lastra, E. Abad, M.T.M. Koper, Oxygen reduction and evolution at single-metal active sites: comparison between functionalized graphitic materials and protoporphyrins. Surf. Sci. 607, 47–53 (2013). https://doi.org/10.1016/j.susc.2012.08.005
F. Calle-Vallejo, O. Diaz-Morales, M.J. Kolb, M.T.M. Koper, Why is bulk thermochemistry a good descriptor for the electrocatalytic activity of transition metal oxides? ACS Catal. 5(2), 869–873 (2015). https://doi.org/10.1021/cs5016657
C.F. Dickens, J.H. Montoya, A.R. Kulkarni, M. Bajdich, J.K. Nørskov, An electronic structure descriptor for oxygen reactivity at metal and metaloxide surfaces. Surf. Sci. 681, 122–129 (2019). https://doi.org/10.1016/j.susc.2018.11.019
H.B. Tao, L. Fang, J. Chen, H.B. Yang, J. Gao et al., Identification of surface reactivity descriptor for transition metal oxides in oxygen evolution reaction. J. Am. Chem. Soc. 138(31), 9978–9985 (2016). https://doi.org/10.1021/jacs.6b05398
N. Govindarajan, M.T.M. Koper, E.J. Meijer, F. Calle-Vallejo, Outlining the scaling-based and scaling-free optimization of electrocatalysts. ACS Catal. 9(5), 4218–4225 (2019). https://doi.org/10.1021/acscatal.9b00532
C.F. Dickens, C. Kirk, J.K. Nørskov, Insights into the electrochemical oxygen evolution reaction with ab initio calculations and microkinetic modeling: beyond the limiting potential volcano. J. Phys. Chem. C 123(31), 18960–18977 (2019). https://doi.org/10.1021/acs.jpcc.9b03830
N.B. Halck, V. Petrykin, P. Krtil, J. Rossmeisl, Beyond the volcano limitations in electrocatalysis–oxygen evolution reaction. Phys. Chem. Chem. Phys. 16(27), 13688–13688 (2014). https://doi.org/10.1039/C4CP00571F
M. Kodera, Y. Kawahara, Y. Hitomi, T. Nomura, T. Ogura et al., Reversible O–O bond scission of peroxodiiron(III) to high-spin oxodiiron(IV) in dioxygen activation of a diiron center with a bis-tpa dinucleating ligand as a soluble methane monooxygenase model. J. Am. Chem. Soc. 134(32), 13236–13239 (2012). https://doi.org/10.1021/ja306089q
S. Kundu, E. Matito, S. Walleck, F.F. Pfaff, F. Heims et al., O–O bond formation mediated by a hexanuclear iron complex supported on a stannoxane. Chem. Eur. J. 18(10), 2787–2791 (2012). https://doi.org/10.1002/chem.201102326
M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai et al., A pentanuclear iron catalyst designed for water oxidation. Nature 530, 465–468 (2016). https://doi.org/10.1038/nature16529
Y. Umena, K. Kawakami, J. Shen, N. Kamiya, Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 473, 55–60 (2011). https://doi.org/10.1038/nature09913
G.C. Dismukes, R. Brimblecombe, G.A.N. Felton, R.S. Pryadun, J.E. Sheats et al., Development of bioinspired Mn4O4-cubane water oxidation catalysts: lessons from photosynthesis. Acc. Chem. Res. 42(12), 1935–1943 (2009). https://doi.org/10.1021/ar900249x
A. Ramírez, P. Hillebrand, D. Stellmach, M.M. May, P. Bogdanoff et al., Evaluation of MnOx, Mn2O3, and Mn3O4 electrodeposited films for the oxygen evolution reaction of water. J. Phys. Chem. C 118(26), 14073–14081 (2014). https://doi.org/10.1021/jp500939d
C. Lin, J. Li, X. Li, S. Yang, W. Luo et al., In-situ reconstructed Ru atom array on α-MnO2 with enhanced performance for acidic water oxidation. Nat. Catal. 4, 1012–1023 (2021). https://doi.org/10.1038/s41929-021-00703-0
J.O. Bockris, Kinetics of activation controlled consecutive electrochemical reactions: anodic evolution of oxygen. J. Chem. Phys. 24, 817 (1956). https://doi.org/10.1063/1.1742616
F. Song, M.M. Busch, B. Lassalle-Kaiser, C. Hsu, E. Petkucheva et al., An unconventional iron nickel catalyst for the oxygen evolution reaction. ACS Cent. Sci. 5(3), 558–568 (2019). https://doi.org/10.1021/acscentsci.9b00053
P. Garrido-Barros, C. Gimbert-Suriñach, R. Matheu, X. Sala, A. Llobet, How to make an efficient and robust molecular catalyst for water oxidation. Chem. Soc. Rev. 46(20), 6088–6098 (2017). https://doi.org/10.1039/C7CS00248C
Z. Gao, J. Liu, X. Chen, X. Zheng, J. Mao et al., Engineering NiO/NiFe LDH intersection to bypass scaling relationship for oxygen evolution reaction via dynamic tridimensional adsorption of intermediates. Adv. Mater. 31(11), 1804769 (2019). https://doi.org/10.1002/adma.201804769
N.B. Halck, V. Petrykin, P. Krtil, J. Rossmeisl, Beyond the volcano limitations in electrocatalysis—oxygen evolution reaction. Phys. Chem. Chem. Phys. 16(27), 13682–13688 (2014). https://doi.org/10.1039/C4CP00571F
H. Fei, J. Dong, Y. Feng, C.S. Allen, C. Wan et al., General synthesis and definitive structural identification of MN4C4 single-atom catalysts with tunable electrocatalytic activities. Nat. Catal. 1, 63–72 (2018). https://doi.org/10.1038/s41929-017-0008-y
J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen et al., Single-atom Au/NiFe layered double hydroxide electrocatalyst: probing the origin of activity for oxygen evolution reaction. J. Am. Chem. Soc. 140(11), 3876–3879 (2018). https://doi.org/10.1021/jacs.8b00752
H. Wang, S. Xu, C. Tsai, Y. Li, C. Liu et al., Direct and continuous strain control of catalysts with tunable battery electrode materials. Science 354(6315), 1031–1036 (2016). https://doi.org/10.1126/science.aaf7680
L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li et al., Biaxially strained PtPb/Pt core/shell nanoplate boosts oxygen reduction catalysis. Science 354(6318), 1410–1414 (2016). https://doi.org/10.1126/science.aah6133
M. Luo, S. Guo, Strain-controlled electrocatalysis on multimetallic nanomaterials. Nat. Rev. Mater. 2, 17059 (2017). https://doi.org/10.1038/natrevmats.2017.59
Y. Xie, Z. Wang, T. Zhua, D. Shu, Z. Hou et al., Breaking the scaling relations for oxygen reduction reaction on nitrogen-doped graphene by tensile strain. Carbon 139, 129–136 (2018). https://doi.org/10.1016/j.carbon.2018.06.026
A. Khorshidi, J. Violet, J. Hashemi, A.A. Peterson, How strain can break the scaling relations of catalysis. Nat. Catal. 1, 263–268 (2018). https://doi.org/10.1038/s41929-018-0054-0
A.D. Doyle, J.H. Montoya, A. Vojvodic, Improving oxygen electrochemistry through nanoscopic confinement. ChemCatChem 7(5), 738–742 (2015). https://doi.org/10.1002/cctc.201402864
Y. Li, L. Zhang, J. Peng, W. Zhang, K. Peng, Magnetic field enhancing electrocatalysis of Co3O4/NF for oxygen evolution reaction. J. Power Sources 433, 226704 (2019). https://doi.org/10.1016/j.jpowsour.2019.226704
F.A. Garcés-Pineda, M. Blasco-Ahicart, D. Nieto-Castro, N. López, J.R. Galán-Mascarós, Direct magnetic enhancement of electrocatalytic water oxidation in alkaline media. Nat. Energy 4, 519–525 (2019). https://doi.org/10.1038/s41560-019-0404-4
W. Mtangi, F. Tassinari, K. Vankayala, A.V. Jentzsch, B. Adelizzi et al., Control of electrons’ spin eliminates hydrogen peroxide formation during water splitting. J. Am. Chem. Soc. 139(7), 2794–2798 (2017). https://doi.org/10.1021/jacs.6b12971
X. Ren, T. Wu, Y. Sun, Y. Li, G. Xian et al., Spin-polarized oxygen evolution reaction under magnetic field. Nat. Commun. 12, 2608 (2021). https://doi.org/10.1038/s41467-021-22865-y
Y. Zhang, P. Guo, S. Li, J. Sun, W. Wang et al., Magnetic field assisted electrocatalytic oxygen evolution reaction of nickel-based materials. J. Mater. Chem. A 10(4), 1760–1767 (2022). https://doi.org/10.1039/D1TA09444K
G. Zhou, P. Wang, H. Li, B. Hu, Y. Sun et al., Spin-state reconfiguration induced by alternating magnetic field for efficient oxygen evolution reaction. Nat. Commun. 12, 4827 (2021). https://doi.org/10.1038/s41467-021-25095-4
L. Gao, X. Cui, Z. Wang, C.D. Sewell, Z. Li et al., Operando unraveling photothermal-promoted dynamic active-sites generation in NiFe2O4 for markedly enhanced oxygen evolution. PNAS 118(7), e2023421118 (2021). https://doi.org/10.1073/pnas.2023421118
J. Li, N. Wu, Semiconductor-based photocatalysts and photoelectrochemical cells for solar fuel generation: a review. Catal. Sci. Technol. 5(3), 1360–1384 (2015). https://doi.org/10.1039/C4CY00974F
J. Li, S. Cushing, F. Meng, T. Senty, A.D. Bristow et al., Plasmon-induced resonance energy transfer for solar energy conversion. Nat. Photon. 9, 601–607 (2015). https://doi.org/10.1038/nphoton.2015.142
J. Li, S.K. Cushing, P. Zheng, F. Meng, D. Chu et al., Plasmon-induced photonic and energy-transfer enhancement of solar water splitting by a hematite nanorod array. Nat. Commun. 4, 2651 (2013). https://doi.org/10.1038/ncomms3651
G. Liu, P. Li, G. Zhao, X. Wang, J. Kong et al., Promoting active species generation by plasmon-induced hot-electron excitation for efficient electrocatalytic oxygen evolution. J. Am. Chem. Soc. 138(29), 9128–9136 (2016). https://doi.org/10.1021/jacs.6b05190
N. Zhang, Y. Chai, Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Enviorn. Sci. 14(9), 4647–4671 (2021). https://doi.org/10.1039/D1EE01277K
Y. Sun, H. Liao, J. Wang, B. Chen, S. Sun et al., Covalency competition dominates the water oxidation structure–activity relationship on spinel oxides. Nat. Catal. 3, 554–563 (2020). https://doi.org/10.1038/s41929-020-0465-6
A. Grimaud, O. Diaz-Morales, B. Han, W.T. Hong, Y. Lee et al., Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9, 457–465 (2017). https://doi.org/10.1038/nchem.2695
G. Assat, J. Tarascon, Fundamental understanding and practical challenges of anionic redox activity in Li–ion batteries. Nat. Energy 3, 373–386 (2018). https://doi.org/10.1038/s41560-018-0097-0
J.T. Mefford, X. Rong, A.M. Abakumov, W.G. Hardin, S. Dai et al., Water electrolysis on La1−xSrxCoO3−δ perovskite electrocatalysts. Nat. Commun. 7, 11053 (2016). https://doi.org/10.1038/ncomms11053
X. Li, H. Wang, Z. Cui, Y. Li, S. Xin et al., Exceptional oxygen evolution reactivities on CaCoO3 and SrCoO3. Sci. Adv. 5(8), eaav6262 (2019). https://doi.org/10.1126/sciadv.aav6262
T. Maiyalagan, K.A. Jarvis, S. Therese, P.J. Ferreira, A. Manthiram, Spinel-type lithium cobalt oxide as a bifunctional electrocatalyst for the oxygen evolution and oxygen reduction reactions. Nat. Commun. 5, 3949 (2014). https://doi.org/10.1038/ncomms4949
X. Rong, J. Parolin, A.M. Kolpak, A fundamental relationship between reaction mechanism and stability in metal oxide catalysts for oxygen evolution. ACS Catal. 6(2), 1153–1158 (2016). https://doi.org/10.1021/acscatal.5b02432
R.P. Forslund, W.G. Hardin, X. Rong, A.M. Abakumov, D. Filimonov et al., Exceptional electrocatalytic oxygen evolution via tunable charge transfer interactions in La0.5Sr1.5Ni1−xFexO4±δ Ruddlesden–Popper oxides. Nat. Commun. 9, 3150 (2018). https://doi.org/10.1038/s41467-018-05600-y
Z. Huang, J. Song, Y. Du, S. Xi, S. Dou et al., Chemical and structural origin of lattice oxygen oxidation in Co–Zn oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy 4, 329–338 (2019). https://doi.org/10.1038/s41560-019-0355-9
D. Seo, J. Lee, A. Urban, R. Malik, S. Kang et al., The structural and chemical origin of the oxygen redox activity in layered and cation-disordered Li-excess cathode materials. Nat. Chem. 8, 692–697 (2016). https://doi.org/10.1038/nchem.2524
J. Lee, D.A. Kitchaev, D. Kwon, C. Lee, J.K. Papp et al., Reversible Mn2+/Mn4+ double redox in lithium-excess cathode materials. Nature 556, 185–190 (2018). https://doi.org/10.1038/s41586-018-0015-4
J. Hong, W.E. Gent, P. Xiao, K. Lim, D. Seo et al., Metal–oxygen decoordination stabilizes anion redox in Li-rich oxides. Nat. Mater. 18, 256–265 (2019). https://doi.org/10.1038/s41563-018-0276-1
V.M.V.G. Nageswaran, Operando X-ray spectroscopic techniques: a focus on hydrogen and oxygen evolution reactions. Front. Chem. 8, 23 (2020). https://doi.org/10.3389/fchem.2020.00023
N. Zhang, X. Feng, D. Rao, X. Deng, L. Cai et al., Lattice oxygen activation enabled by high-valence metal sites for enhanced water oxidation. Nat. Commun. 11, 4066 (2020). https://doi.org/10.1038/s41467-020-17934-7
J. Shan, T. Ling, K. Davey, Y. Zheng, S. Qiao, Transition-metal-doped RuIr bifunctional nanocrystals for overall water splitting in acidic environments. Adv. Mater. 31(17), 1900510 (2019). https://doi.org/10.1002/adma.201900510
A. Moysiadou, S. Lee, C. Hsu, H.M. Chen, X. Hu, Mechanism of oxygen evolution catalyzed by cobalt oxyhydroxide: cobalt superoxide species as a key intermediate and dioxygen release as a rate-determining step. J. Am. Chem. Soc. 142(27), 11901–11914 (2020). https://doi.org/10.1021/jacs.0c04867
S. Lee, K. Banjac, M. Lingenfelder, X. Hu, Oxygen isotope labeling experiments reveal different reaction sites for the oxygen evolution reaction on nickel and nickel iron oxides. Angew. Chem. Int. Ed. 58(30), 10295–10299 (2019). https://doi.org/10.1002/anie.201903200
M.T.M. Koper, Theory of multiple proton-electron transfer reactions and its implications for electrocatalysis. Chem. Sci. 4(7), 2710–2723 (2013). https://doi.org/10.1039/C3SC50205H
L. Giordano, B. Han, M. Risch, W.T. Hong, R.R. Rao et al., pH dependence of OER activity of oxides: current and future perspectives. Catal. Today 262, 2–10 (2016). https://doi.org/10.1016/j.cattod.2015.10.006
C. Roy, B. Sebok, S.B. Scott, E.M. Fiordaliso, J.E. Sørensen et al., Impact of nanoparticle size and lattice oxygen on water oxidation on NiFeOxHy. Nat. Catal. 1, 820–829 (2018). https://doi.org/10.1038/s41929-018-0162-x
B. Hua, M. Li, W. Pang, W. Tang, S. Zhao et al., Activating p-blocking centers in perovskite for efficient water splitting. Chem 4(12), 2902–2916 (2018). https://doi.org/10.1016/j.chempr.2018.09.012
P. Wang, Q. Cheng, C. Mao, W. Su, L. Yang et al., Regulation of oxygen vacancy within oxide pyrochlores by F-doping to boost oxygen-evolution activity. J. Power Sources 502, 229903 (2021). https://doi.org/10.1016/j.jpowsour.2021.229903
L. Bai, C. Hsu, D.T.L. Alexander, H.M. Chen, X. Hu, A cobalt–iron double-atom catalyst for the oxygen evolution reaction. J. Am. Chem. Soc. 141(36), 14190–14199 (2019). https://doi.org/10.1021/jacs.9b05268
Acknowledgements
The author gratefully acknowledges support from the U. S. Department of the Army and U.S. Army Future Command. J. Li also acknowledges support from the U.S. Army Research Laboratory Senior Research Fellowship Program.
Funding
Open access funding provided by Shanghai Jiao Tong University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visithttp://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, J. Oxygen Evolution Reaction in Energy Conversion and Storage: Design Strategies Under and Beyond the Energy Scaling Relationship. Nano-Micro Lett. 14, 112 (2022). https://doi.org/10.1007/s40820-022-00857-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40820-022-00857-x