
Abstract
Background
Many patients with allergic rhinitis (AR) have moderate-to-severe persistent AR. Meda Pharma’s AzeFlu (MP-AzeFlu®) is an intranasal AR treatment comprising a novel formulation of azelastine hydrochloride and fluticasone propionate in a single device.
Methods
This prospective observational study of 214 adults and adolescents in Austria with moderate-to-severe persistent AR assessed the effectiveness of MP-AzeFlu (one spray/nostril twice daily; daily doses: azelastine hydrochloride 548 μg; and fluticasone propionate 200 μg) for AR control in clinical practice using the visual analog scale. Symptom severity was reported on days 0, 1, 3, 7, 14, 21, 28, 35, and 42. Patient demographics, AR phenotype, allergen sensitization, symptomatology, AR treatments in the previous year, and the reason for the MP-AzeFlu prescription were recorded.
Results
MP-AzeFlu treatment was associated with a rapid and statistically significant reduction in the visual analog scale score from baseline to each timepoint measured, including day 1 (all p < 0.0001). Mean (standard deviation) visual analog scale score was 53.5 mm (26.3) at baseline, 25.3 mm (21.0) on day 28, and 19.6 mm (17.4) on day 42, a mean overall reduction from baseline of 41.4 (23.9) mm for completers. Results were consistent irrespective of patient age, gender, severity, or traditional AR phenotype. Prior to MP-AzeFlu prescription, congestion was considered the most bothersome symptom. The majority of patients reported using at least two AR therapies in the past year, including oral antihistamines, intranasal corticosteroids, and intranasal antihistamines.
Conclusions
Many patients in Austria live with uncontrolled persistent AR despite treatment. MP-AzeFlu provides effective and rapid control of persistent AR in a real-world Austrian setting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Allergic rhinitis (AR) is a highly prevalent allergic respiratory disease affecting the European population. |
The primary objective of this real-world study was to assess the effectiveness of Meda Pharma’s AzeFlu (MP-AzeFlu®) nasal spray in routine clinical practice among Austrian patients with persistent AR. |
Assessment of patient profiles showed that many Austrian patients with persistent AR live with uncontrolled symptoms despite treatment with monotherapies and multiple therapies. |
A more effective treatment option, such as MP-AzeFlu, may improve AR control for patients with persistent AR. |
This region-specific study would help the physicians to take a more robust treatment decision. |
1 Introduction
Allergic rhinitis (AR) is a highly prevalent allergic respiratory disease affecting approximately 24% of the European population [1], and many patients with AR have moderate-to-severe persistent disease [2]. Allergic rhinitis symptoms include sneezing, nasal obstruction, and mucous discharge [3]. The increasing prevalence of AR [4, 5] is troubling because of associated morbidities that reduce patients’ quality of life [6, 7] and impair school and work performance [8,9,10]. Furthermore, AR interferes with sleep in 50–80% of patients, leading to fatigue, depression, irritability, and difficulty concentrating [11]. Alone and in combination with frequent comorbidities (e.g., asthma, sinusitis, upper respiratory tract infection, otitis media with effusion, and nasal polyposis) [12, 13], AR is accompanied by substantial direct and indirect medical costs [14,15,16,17].
Meda Pharma’s (now Viatris) AzeFlu (MP-AzeFlu®) is a novel intranasal AR treatment [18] comprising intranasal antihistamine (azelastine hydrochloride [AZE]) and intranasal corticosteroid (fluticasone propionate [FP]). MP-AzeFlu was approved in Europe in 2013 and is indicated for the relief of symptoms of moderate-to-severe seasonal AR (SAR) and perennial AR (PAR) if monotherapy with either an intranasal antihistamine or a glucocorticoid is not considered sufficient [18]. The efficacy and safety of MP-AzeFlu have been repeatedly assessed in several thousand adult and adolescent patients with AR [19,20,21,22,23]. Results suggest MP-AzeFlu is effective and well tolerated, and provides superior overall nasal and ocular symptom relief and more complete and rapid symptom control compared with intranasal corticosteroids or intranasal H1-antihistamines [22].
Noninterventional studies (NIS) are considered expedient tools to analyze the use of a treatment under real-world conditions. The primary objective of this national, multi-center, prospective NIS was to assess the effectiveness of MP-AzeFlu nasal spray in routine clinical practice among Austrian patients with persistent AR (PER) using a simple visual analog scale (VAS) and by analyzing sleep quality. Recent guidelines from MACVIA-ARIA (Contre les Maladies Chroniques pour un Vieillissement Actif-Allergic Rhinitis and Its Impact on Asthma) endorsed using a VAS to assess AR control and guide treatment decisions [24]. The secondary objective was to gather information on the real-world use of MP-AzeFlu nasal spray among these patients, including the reason for prescribing MP-AzeFlu; type, duration, and severity of current symptoms; and previous treatments for AR. From these data, conclusions can be drawn about patients with PER who are treated with MP-AzeFlu nasal spray in routine clinical practice.
2 Methods
2.1 Ethics and Good Clinical Practice
The current NIS are compliant with European regulations [7, 8, 10] such that rules imposed for this study did not interfere with the physician's common therapy. Study conduct was in accordance with Austrian drug laws and regulations for conducting NIS [9, 11]. All patients or designated caretakers provided written informed consent.
2.2 Study Design, Patients, and Treatments
The multi-center prospective NIS was conducted in 29 sites in Austria (EU PAS Register Number, EUPAS22774) during September 2015 to June 2016. The participating physicians were general practitioners, allergists, ear, nose, and throat specialists, and pediatricians. Eligible patients were adults or adolescents (≥ 12 years of age) with moderate-to-severe PER (i.e., symptoms > 4 days/week, for > 4 consecutive weeks) who were prescribed MP-AzeFlu (one spray/nostril twice daily [morning and evening]; daily doses: AZE 548 μg; FP 200 μg) according to the summary of product characteristics [18]. A valid diagnosis verified by standard local practice, such as a skin prick test or serum-specific immunoglobulin E measures, was also required. Exclusion criteria included known allergic reactions to MP-AzeFlu or any of its ingredients; pregnancy (or planned pregnancy during the NIS) or breastfeeding; and missing consent for collection, archiving or transfer of personal data in the context of this study and in accordance with the observational plan. There were no restrictions regarding concomitant treatments other than those contraindicated for use of MP-AzeFlu.
Physicians usually involved in the management of AR were invited to participate. Each physician (center) was to document treatment with MP-AzeFlu in three patients. The planned duration of observation to assess treatment effectiveness was approximately 6 weeks (42 days) per patient, allowing flexibility depending on usual clinical practice. The study consisted of an inclusion visit (day 0) and an optional follow-up visit after approximately 42 days. The decision to include patients in the study was made by physicians independently from and after the decision to prescribe MP-AzeFlu. After a patient provided informed consent, the physician documented the patient’s demographic data, first diagnosis of PER, and type of perennial allergens (i.e., dust mites, pet dander [cat, dog, and other], mold, and other) to which the patient was sensitized. Allergic rhinitis symptoms, such as nasal itching, congestion, sneezing, runny nose, and allergic rhinoconjunctivitis, were also recorded. Additionally, data on the patient’s predominant symptom, bothersome symptoms, type of symptoms, number and duration of symptom flares, and the presence of ARIA-qualifying criteria were collected [25]. Levels of sleep disturbance and impairment of daily activities, leisure and/or sport activities, and school or work abilities were assessed. Previous treatments for AR, including current immunotherapy, in the last calendar year prior to an MP-AzeFlu prescription were also noted. Data for each patient were entered into an electronic case report form.
Data on symptom severity, level of disease control, and assessment of sleep were recorded by patients on patient diary cards, to be handed back to the physician at the day 42 visit. Alternatively, the card could be sent back by mail to the physician after 6 weeks. After receipt of the card, the physician transcribed the information into the electronic case report form. Because of the nature of the study, good clinical practice rules regarding monitoring did not apply.
2.3 Effectiveness and Safety Assessments
Patients assessed symptom severity using a VAS from 0 mm (not at all bothersome) to 100 mm (very bothersome) on the patient diary card in the morning prior to MP-AzeFlu use on days 0, 1, 3, 7, 14, 21, 28, 35, and 42 after the start of the treatment. Three parts of the patient diary card were dispensed to the patient: card 1 up to day 14, card 2 up to day 28, and card 3 up to day 42. During the inclusion visit (day 0), an additional VAS assessment reflecting the severity of the symptoms experienced in the preceding > 4-week period was made.
In the morning of day 1 after the start of the treatment, patients assessed their level of disease control within the previous 24 h according to the assessment categories given on the patient diary card (symptoms well controlled, symptoms partly controlled, symptoms uncontrolled, unknown). On days 7, 14, 21, 28, 35, and 42 after the start of treatment, patients assessed their sleep quality during the previous 7 nights using a 5-point rating scale (very good, good, fair, bad, very bad).
Adverse events, adverse drug reactions (ADRs), and serious adverse events were recorded throughout the study. Adverse drug reactions were coded using Medical Dictionary for Regulatory Activities version 19.0.
2.4 Statistical Methods
This NIS is part of a set of international observational studies with a similar design, which are intended to form a pooled database. This study was designed to provide insight into the background of patients with PER receiving an MP-AzeFlu nasal spray in routine clinical practice.
All analyses were based on the safety population (SAF), which included all patients who were treated with MP-AzeFlu and for whom the physician had confirmed data validity. Separate analyses of effectiveness variables were performed for the following subpopulations: patients with PAR but without SAR; patients with SAR and PAR; and subgroups regarding age, sex, and symptom severity. As recommended by ARIA, disease control was defined by a VAS score cut-off of 50 mm.
Descriptive statistics included continuous variables presented by number (N), mean, median, standard deviation (SD), and standard error of the mean. Categorical variables are presented by N and frequency. Missing data were not replaced. Analysis of covariance for repeated measures (SAS [Cary, NC, USA], proc mixed) was used to analyze the change of AR symptom severity from baseline to day 1, 3, 7, 14, 21, 28, 35, and 42 using baseline (day 0) as a covariate and a VAS change from baseline as a dependent variable repeated in time. Analysis of covariance was analyzed for those in the SAF with at least one valid post-baseline assessment in the patient diary card only.
3 Results
3.1 Baseline Patient Characteristics
Of 231 enrolled patients, 17 were excluded from the data analysis because of unconfirmed data documentation (electronic signature of the treating physician was missing). The remaining 214 adult and adolescent patients with moderate-to-severe PER who were prescribed MP-AzeFlu and enrolled at 29 sites were included in the SAF. Mean treatment duration was 35.1 days. Demographic data (e.g., sex and age) were available for all 214 patients (Table 1). The SAF was evenly divided between men and women (each n = 107, 50%). Most patients were adults aged 18–65 years (n = 178; 83.2%); mean (± SD) age was 39.5 ± 16.8 years (median, 40 years; range, 12–82 years). The mean (± SD) duration of history of AR at the time of inclusion in the study was 9.8 ± 10.1 years (n = 197; median, 7 years; range, 0–60 years). Based on the types of allergens sensitized, 52.8% of the patients were sensitized to seasonal and perennial allergens, and the remainder were sensitized to perennial allergens only (Table 1). According to ARIA classification, all patients in the SAF (N = 214) had documented moderate-to-severe AR.
3.2 Patient Profiles
House dust mites was the most frequently documented category of perennial allergen (76.2%), followed by “other,” cat dander, mold, dog dander, and other pet dander (Fig. 1A). Bothersome symptoms were reported for 132 (61.7%) patients (Fig. 1B). More than half of the patients suffered from sleep disturbances, just less than half reported impairments in daily activities, leisure and/or sport, and roughly one-third noted school or work impairments. The most frequent predominant AR symptom (53.7%) was nasal congestion (Fig. 1C).

Patient and symptom characteristics reviewed included perennial allergen types (A), distribution of Allergic Rhinitis and Its Impact on Asthma (ARIA) criteria (B), predominant symptoms reported (C), and the number of allergic rhinitis (AR) treatments prior to Meda Pharma’s AzeFlu (MP-AzeFlu®) prescription (D); percentages refer to total population (N = 214). *Multiple answers were permitted. AH antihistamine, Decon decongestant, IN intranasal, INAH intranasal antihistamine, INS intranasal corticosteroid, LTRA leukotriene receptor antagonist, MCS mast cell stabilizer, OAH oral antihistamine, Sys Cort systemic corticosteroid
In the preceding year, 55.1%, 45.3%, and 33.2% of patients were treated with at least an oral antihistamine, intranasal corticosteroid, or intranasal antihistamine, respectively (Fig. 1D). The most frequent reason for prescribing MP-AzeFlu was that alternative therapies were not sufficient in the past to treat symptoms (59.3%; 127/214), followed by alternative therapies were not considered sufficient to treat symptoms (23.8%; 51/214). At the start of the study, 15.0% (32/214) of patients were undergoing immunotherapy and 10.7% (23/214) had undergone immunotherapy in the past. Two or more allergy medications were used previously by 55.1% (118/214) of patients, and 15.4% (33/214) received no treatment (Fig. 2).

Number of previous allergic rhinitis (AR) treatments used by patients with persistent AR attending routine clinical care prior to Meda Pharma’s AzeFlu (MP-AzeFlu®) prescription (N = 214)
3.3 Effectiveness
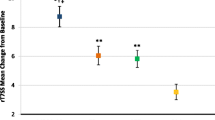
MP-AzeFlu treatment was associated with a significant reduction in VAS scores (Fig. 3). At baseline (day 0, prior to start of treatment), mean (± SD) VAS score in the total SAF population was 53.5 ± 26.3 mm (n = 211; median, 59 mm; Table 1). The mean and median VAS scores decreased during the treatment period, with the most rapid decrease occurring during the first week of treatment, indicating a rapid improvement of symptoms. Mean (± SD) VAS scores were 47.7 ± 25.6 mm on day 1, 41.2 ± 25.6 mm on day 3, 34.7 ± 24.5 mm on day 7, 29.0 ± 23.6 mm on day 14, 26.5 ± 21.0 mm on day 21, 25.3 ± 21.0 mm on day 28, 22.5 ± 18.3 mm on day 35, and 19.6 ± 17.4 mm on day 42. The symptoms (demonstrated in VAS scores) declined continually with the duration of treatment. A first symptom improvement was observed already on day 1, with a mean (± SD) change from baseline in VAS of − 6.6 ± 14.6 mm. The mean (± SD) change from baseline in VAS scores for patients with available data on the corresponding day on day 3 was − 13.1 ± 18.5 mm, − 19.9 ± 19.5 mm on day 7, − 25.8 ± 21.8 mm on day 14, − 30.8 ± 22.7 mm on day 21, − 32.3 ± 23.7 mm on day 28, − 37.8 ± 24.5 mm on day 35, and − 41.4 ± 23.9 mm on day 42. The analysis of covariance for repeated measures demonstrated that all post-baseline changes were statistically significant (all p < 0.0001, day 1 to day 42).

Mean visual analog scale score (VAS) reduction after the use of Meda Pharma’s AzeFlu (MP-AzeFlu®) for a period of 42 days by Austrian patients with persistent allergic rhinitis (*p < 0.0001 vs day 0). ARIA Allergic Rhinitis and Its Impact on Asthma, Last Day data are aggregated results from all patients’ last day of assessment; patients who dropped out of the study early had a last day assessment prior to day 42, SEM standard error of the mean
Results were relatively consistent irrespective of age group (12–17 years, 18–65 years, > 65 years; Fig. 4A), gender (Fig. 4B), baseline disease severity (less severe, baseline VAS score 50–74 mm; more severe, baseline VAS score 75–100 mm; Fig. 4C), or traditional AR phenotype classification (PAR only or SAR and PAR; Fig. 4D). Changes from baseline to day 42 indicated similar therapeutic effects between subgroups. A smaller change was observed for adolescents, with little VAS data available from day 21 onward (fewer than ten patients; mean change [± SD] from baseline to day 42, − 32.4 ± 23.3 mm). For the subgroup with a high baseline severity score (baseline VAS 75–100 mm; baseline mean [± SD], 85.3 ± 7.7 mm), the high baseline provided more room for improvement, with a mean change (± SD) from baseline to day 42 of − 60.6 ± 20.9 mm.

Mean visual analog scale (VAS) score reduction after the use of Meda Pharma’s AzeFlu (MP-AzeFlu®) for a period of 42 days by Austrian patients with persistent allergic rhinitis according to patient age (A), gender (B), baseline disease severity (C), and phenotype classification (D). ARIA Allergic Rhinitis and Its Impact on Asthma, PAR perennial allergic rhinitis, SAR seasonal allergic rhinitis (allergy to at least one pollen allergen), SAR and PAR allergy to at least one pollen allergen and at least one nonpollen allergen, SEM standard error of the mean, y years
On day 1, only 16.9% (31/183) patients rated their AR symptoms during the last 24 h as well controlled, 64.5% (118/183) indicated that their symptoms were partly controlled, and 18.6% (34/183) indicated that their symptoms were uncontrolled. Percentages are related to the total number of patients with available symptom control data. The optimal VAS cut-off score was 33 mm for differentiating between well-controlled versus partly controlled or uncontrolled symptoms (Youden index, 0.556) and 67 mm for differentiating between well-controlled or partly controlled versus uncontrolled symptoms (Youden index, 0.420).
Patient-assessed sleep quality improved continuously up to day 42 (Fig. 5). The percentage of patients with very good/good sleep quality increased from 27.5% (58/211) at baseline (day 0) to 61.3% (111/181) on day 7, 70.8% (126/178) on day 14, 81.4% (118/145) on day 21, 79.9% (111/139) on day 28, 83.8% (93/111) on day 35, and 87.2% (95/109) on day 42. Conversely, bad/very bad sleep quality decreased from 36.0% (76/211) at baseline to 3.7% (4/109) on day 42. Percentages are related to the total number of patients with available sleep quality assessment at each timepoint. The Wilcoxon rank test revealed statistically significant differences between baseline (day 0) and all post-baseline assessment days (p < 0.0001 from day 7 to day 42). Comparable results were obtained for the subpopulations (e.g., PAR only and SAR and PAR).

Patient assessment of quality of sleep (*p < 0.0001 vs day 0 per Wilcoxon rank test)
3.4 Safety
MP-AzeFlu nasal spray was well tolerated. A total of five ADRs were reported in two patients (0.9%). None of the ADRs was serious. Fatigue and dizziness were reported by one patient and increased intraocular pressure, pruritus, and headache occurred in another patient. The safety results of this study are generally consistent with the summary of product characteristics for MP-AzeFlu, as no other ADRs occurred in this study.
4 Discussion
Persistent AR is a distinct category of AR associated with substantial comorbidities, including asthma [3, 26] emphasizing the important of AR treatment. Because of potential adverse effects with systemic treatment, topical (intranasal) treatments are preferred for PER [3]. The combination intranasal therapy, MP-AzeFlu, was previously found to be safe and effective over 1 year of treatment in a large clinical trial [19, 23]. Our study assessed the effectiveness of MP-AzeFlu for patients with PER in real-world settings in Austria.
Our study population corresponded well with the MP-AzeFlu nasal spray summary of product characteristics specifications regarding the indication and target population [18]. The majority of patients were prescribed MP-AzeFlu because alternative therapies had not been sufficient in the past and/or were considered insufficient to treat acute symptoms. All patients had documented moderate-to-severe AR according to the ARIA classification.
Compared with patients with moderate-to-severe SAR in the clinical studies of MP-AzeFlu, patients in the current study had a shorter average history of AR (9.8 vs 20–22 years) and were slightly older (39.5 vs 35.6–38.8 years of age) [20]. Approximately 25% had undergone and/or were undergoing immunotherapy at the study start and the majority of patients reported using at least two AR therapies in the past year, including oral antihistamines, intranasal corticosteroids, and intranasal antihistamines. Thus, the proportion of patients with prior or ongoing immunotherapy was consistent with the Forsa survey, in which 28% of patients with allergies had been or were undergoing medical treatment [13]. The average last day of treatment or documentation in the patient diary card was 35.1 days after the start of treatment (day 0); the median of 42 days shows most patients adhered to the 42-day treatment period. The lower average value was likely a result of patients not completing and/or returning the diary after day 0 (11.2% without any post-baseline value). The post-baseline visit was optional, which may have contributed to this missing proportion.
The severity of AR symptoms decreased from the start of MP-AzeFlu treatment to day 42 by a mean of − 41 mm on the VAS, which has a 0–100 mm scale (not at all to very bothersome). Demoly and colleagues suggested that a threshold of 23 mm can be considered a clinically important change in the VAS score [27]. In another study, Bousquet and colleagues considered a difference in the VAS score of more than 10 mm to be significant [28]. In the present study, a significant improvement in symptom severity was observed after 3 days of treatment in half of the patients, with a median decrease in the VAS score of − 10 mm. Additionally, on day 3, a clinically important improvement in symptom severity of 20 mm was observed in approximately 25% of patients. On day 42, 75% of 109 patients with valid data had a clinically important decrease in the VAS score of at least 25 mm. Our findings are consistent with previous real-world studies of European patients with moderate-to-severe SAR who experienced substantial reductions in VAS scores over 14 days of treatment [21, 29,30,31]. In a similar study in the Swedish population with PER, 89% of patients had their AR symptoms “well controlled” or “partly controlled” after day 1 of treatment [32]. The VAS improvement was also similar to previous studies conducted in Irish and Swedish populations [32, 33].
Results were similar between patients with documentation up to day 42 and patients with treatment discontinuation and/or incomplete documentation. On day 1, 81.4% (149/183) of patients with symptom control data assessed their symptoms as well controlled or partly controlled and 18.6% (34/183) assessed them as uncontrolled. These results indicate most patients experienced symptom control after 1 day of MP-AzeFlu treatment. In a recent study utilizing a ragweed pollen challenge in an environmental exposure chamber, the onset of action of MP-AzeFlu was as short as 5 min among patients with AR [34]. Our study thus adds to a body of evidence demonstrating the rapid onset of action of this agent.
The estimated optimal VAS cut-off scores were 33 mm for differentiating between well-controlled and partly/uncontrolled symptoms and 67 mm for differentiating between well/partly controlled and uncontrolled symptoms. The corresponding Youden indices of 0.556 and 0.420, respectively, indicated moderate accuracy of cut-off determinations. Using the 33-mm cut-off value, half of the patients had well-controlled symptoms (median VAS, 33 mm) on day 7.
Sleep quality improved continuously throughout this NIS, which was reflected by increasing rates of very good/good and decreasing rates of fair/bad/very bad sleep quality. Comparable results were obtained for the PAR-only and SAR-and-PAR subgroups. Abundant research has demonstrated the effects of poor sleep quality on the quality of life of patients with AR, highlighting the importance of sleep quality improvements experienced in this study [11, 35,36,37,38,39].
Missing data may have biased the data analysis. Data were complete for most variables at baseline, except for the duration of AR (7.9% of patients missing data) and the number and duration of symptom flares (76.2% missing data), but this did not impact effectiveness results. Data were missing for 14.0% of patients on day 1 and 49.1% on day 42. Missing post-baseline data may be due to treatment discontinuation, incomplete diary entry and/or the patient card not returned to the physician. However, the values obtained for day 42 and the day of last documentation were very similar. This suggests the time course was likely not biased by the selective loss of patients because of a lack of treatment effectiveness.
The actual sample size of 214 patients was considered sufficient to draw general conclusions about the patient background and effectiveness and safety of MP-AzeFlu treatment in routine clinical practice. Further limitations are those associated with noninterventional observational studies, including a lack of a control group, a lack of random assignment, and confounding.
5 Conclusions
Collectively, these results as assessed by the VAS indicate that MP-AzeFlu provides effective and rapid control of PER in real-world settings in Austria. Symptom improvement was noted from day 1 and sustained for 42 days. Assessment of patient profiles showed that many Austrian patients with PER live with uncontrolled symptoms despite treatment with monotherapies and multiple therapies. A more effective treatment option, such as MP-AzeFlu, may improve AR control for patients with PER. Because of the limitations of the study, it is difficult to conclude that the benefits seen are due to the medication or a clinical improvement that would have happened spontaneously. The results will include regression to the mean due to missing data.
References
Valovirta E, Myrseth SE, Palkonen S. The voice of the patients: allergic rhinitis is not a trivial disease. Curr Opin Allergy Clin Immunol. 2008;8(1):1–9.
Small P, Kim H. Allergic rhinitis. Allergy asthma. Clin Immunol. 2011;7(1):S3.
Bousquet J, Khaltaev N, Cruz AA, Denburg J, Fokkens WJ, Togias A, et al. Allergic Rhinitis and its Impact on Asthma (ARIA) 2008 update (in collaboration with the World Health Organization, GA(2)LEN and AllerGen). Allergy. 2008;63(Suppl 86):8–160.
Aberg N, Hesselmar B, Aberg B, Eriksson B. Increase of asthma, allergic rhinitis and eczema in Swedish schoolchildren between 1979 and 1991. Clin Exp Allergy. 1995;25(9):815–9.
Maziak W, Behrens T, Brasky TM, Duhme H, Rzehak P, Weiland SK, et al. Are asthma and allergies in children and adolescents increasing? Results from ISAAC phase I and phase III surveys in Munster, Germany. Allergy. 2003;58(7):572–9.
Bousquet PJ, Demoly P, Devillier P, Mesbah K, Bousquet J. Impact of allergic rhinitis symptoms on quality of life in primary care. Int Arch Allergy Immunol. 2013;160(4):393–400.
Small M, Piercy J, Demoly P, Marsden H. Burden of illness and quality of life in patients being treated for seasonal allergic rhinitis: a cohort survey. Clin Transl Allergy. 2013;3(1):33.
Blaiss MS, Allergic Rhinitis in Schoolchildren Consensus Group. Allergic rhinitis and impairment issues in schoolchildren: a consensus report. Curr Med Res Opin. 2004;20(12):1937–52.
Szeinbach SL, Seoane-Vazquez EC, Beyer A, Williams PB. The impact of allergic rhinitis on work productivity. Prim Care Respir J. 2007;16(2):98–105.
Walker S, Khan-Wasti S, Fletcher M, Cullinan P, Harris J, Sheikh A. Seasonal allergic rhinitis is associated with a detrimental effect on examination performance in United Kingdom teenagers: case-control study. J Allergy Clin Immunol. 2007;120(2):381–7.
Storms W. Allergic rhinitis-induced nasal congestion: its impact on sleep quality. Prim Care Respir J. 2008;17(1):7–18.
Nathan RA. The burden of allergic rhinitis. Allergy Asthma Proc. 2007;28(1):3–9.
Schatz M, Zeiger RS, Chen W, Yang SJ, Corrao MA, Quinn VP. The burden of rhinitis in a managed care organization. Ann Allergy Asthma Immunol. 2008;101(3):240–7.
Blaiss MS. Cognitive, social, and economic costs of allergic rhinitis. Allergy Asthma Proc. 2000;21(1):7–13.
de la Hoz CB, Rodriguez M, Fraj J, Cerecedo I, Antolin-Amerigo D, Colas C. Allergic rhinitis and its impact on work productivity in primary care practice and a comparison with other common diseases: the Cross-sectional study to evAluate work Productivity in allergic Rhinitis compared with other common dIseases (CAPRI) study. Am J Rhinol Allergy. 2012;26(5):390–4.
Hellgren J, Cervin A, Nordling S, Bergman A, Cardell LO. Allergic rhinitis and the common cold: high cost to society. Allergy. 2010;65(6):776–83.
Lamb CE, Ratner PH, Johnson CE, Ambegaonkar AJ, Joshi AV, Day D, et al. Economic impact of workplace productivity losses due to allergic rhinitis compared with select medical conditions in the United States from an employer perspective. Curr Med Res Opin. 2006;22(6):1203–10.
emc. SPC Dymista nasal spray; 2015. https://www.medicines.org.uk/emc/medicine/27579.
Berger WE, Shah S, Lieberman P, Hadley J, Price D, Munzel U, et al. Long-term, randomized safety study of MP29-02 (a novel intranasal formulation of azelastine hydrochloride and fluticasone propionate in an advanced delivery system) in subjects with chronic rhinitis. J Allergy Clin Immunol Pract. 2014;2(2):179–85.
Carr W, Bernstein J, Lieberman P, Meltzer E, Bachert C, Price D, et al. A novel intranasal therapy of azelastine with fluticasone for the treatment of allergic rhinitis. J Allergy Clin Immunol. 2012;129:1282–9.
Klimek L, Bachert C, Mosges R, Munzel U, Price D, Virchow JC, et al. Effectiveness of MP29-02 for the treatment of allergic rhinitis in real-life: results from a noninterventional study. Allergy Asthma Proc. 2015;36(1):40–7.
Meltzer E, Ratner P, Bachert C, Carr W, Berger W, Canonica GW, et al. Clinically relevant effect of a new intranasal therapy (MP29-02) in allergic rhinitis assessed by responder analysis. Int Arch Allergy Immunol. 2013;161:369–77.
Price D, Shah S, Bhatia S, Bachert C, Berger W, Bousquet B, et al. A new therapy (MP29-02) is effective for the long-term treatment of chronic rhinitis. J Investig Allergol Clin Immunol. 2013;23(7):495–503.
Bousquet J, Schünemann HJ, Hellings PW, Arnavielhe S, Bachert C, Bedbrook A, et al. MACVIA clinical decision algorithm in adolescents and adults with allergic rhinitis. J Allergy Clin Immunol. 2016;138(2):367–74.
Bousquet J, Khaltaev N, Cruz AA, Denburg J, Fokkens WJ, Togias A, et al. Allergic rhinitis and its impact on asthma (ARIA) 2008*. Allergy. 2008;63:8–160.
Bousquet J, Annesi-Maesano I, Carat F, Leger D, Rugina M, Pribil C, et al. Characteristics of intermittent and persistent allergic rhinitis: DREAMS study group. Clin Exp Allergy. 2005;35(6):728–32.
Demoly P, Bousquet PJ, Mesbah K, Bousquet J, Devillier P. Visual analogue scale in patients treated for allergic rhinitis: an observational prospective study in primary care: asthma and rhinitis. Clin Exp Allergy. 2013;43(8):881–8.
Bousquet PJ, Combescure C, Klossek JM, Daures JP, Bousquet J. Change in visual analog scale score in a pragmatic randomized cluster trial of allergic rhinitis. J Allergy Clin Immunol. 2009;123(6):1349–54.
Agache I, Doros IC, Leru PM, Bucur I, Poenaru M, Sarafoleanu C. MP-AzeFlu provides rapid and effective allergic rhinitis control: results of a non-interventional study in Romania. Rhinology. 2018;56(1):33–41.
Dollner R, Lorentz Larsen P, Dheyauldeen S, Steinsvag S. A multicenter, prospective, noninterventional study in a Norwegian cohort of patients with moderate-to-severe allergic rhinitis treated with MP-AzeFlu. Allergy Rhinol (Providence). 2017;8(3):148–56.
Klimek L, Bachert C, Stjarne P, Dollner R, Larsen P, Haahr P, et al. MP-AzeFlu provides rapid and effective allergic rhinitis control in real-life: a pan-European study. Allergy Asthma Proc. 2016;37:376.
Stjarne P, Nguyen DT, Kuhl HC. Real-life effectiveness of MP-AzeFlu (Dymista®) in Swedish patients with persistent allergic rhinitis, assessed by the visual analogue scale. Pragmat Obs Res. 2023;14:1–11.
Kaulsay R, Nguyen DT, Kuhl HC. Real-life effectiveness of MP-AzeFlu in Irish patients with persistent allergic rhinitis, assessed by visual analogue scale and endoscopy. Immun Inflamm Dis. 2018;6(4):456–64.
Bousquet J, Meltzer E, Couroux P, Koltun A, Kopietz F, Munzel U, et al. Onset of action of the fixed combination intranasal azelastine-fluticasone propionate in an allergen exposure chamber. J Allergy Clin Immunol Pract. 2018;6(5):1726-32.e6.
Benninger MS, Benninger RM. The impact of allergic rhinitis on sexual activity, sleep, and fatigue. Allergy Asthma Proc. 2009;30(4):358–65.
Craig TJ, Sherkat A, Safaee S. Congestion and sleep impairment in allergic rhinitis. Curr Allergy Asthma Rep. 2010;10(2):113–21.
Ferguson BJ. Influences of allergic rhinitis on sleep. Otolaryngology. 2004;130(5):617–29.
Stull DE, Roberts L, Frank L, Heithoff K. Relationship of nasal congestion with sleep, mood, and productivity. Curr Med Res Opin. 2007;23(4):811–9.
Thompson A, Sardana N, Craig TJ. Sleep impairment and daytime sleepiness in patients with allergic rhinitis: the role of congestion and inflammation. Ann Allergy Asthma Immunol. 2013;111(6):446–51.
Acknowledgements
An abstract of the data has been previously presented at the European Academy of Allergy and Clinical Immunology Congress (P654 and 1173) held on 17–21 June, 2017, in Helsinki, Finland. Tina Rideout, MS, Roger J. Hill, PhD, and Lisa Baker, PhD (Ashfield Healthcare Communications, Middletown, CT, USA) provided writing support, and Paula Stuckart and Shannon Davis (Ashfield Healthcare Communications) copy edited and styled the manuscript per journal requirements. The authors also acknowledge medical writing support from Arghya Bhattacharya, PhD (Viatris). All persons named in the acknowledgments have given permission to be named in the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study and medical writing support in the development of this article was funded by Mylan Inc. (Canonsburg, PA, USA), now Viatris.
Conflict of Interest
Katharina Marth has conducted received speaker/consultancy fees and financial support for conference attendance from GSK, Novartis, Chiesi, ThermoFisher, Medmedia, Teva, and Meda. Andreas Renner has received speaker/consultancy fees from Meda Pharma GmbH & Co. KG. Georg Langmayr (now Viatris) has no conflicts of interest that are directly relevant to the content of this article. Duc Tung Nguyen is an employee of Meda Pharma GmbH & Co. KG (a Mylan Company, now Viatris). Wolfgang Pohl.
Ethics Approval
The study was performed in accordance with the standards of ethics outlined in the Declaration of Helsinki. This investigation represented a NIS as defined by European regulations (EU 2001; ICH E2E 2004; EMA 2012), i.e., the rules imposed for this observational plan did not interfere with the physician’s common therapy. The study was approved on 30 July, 2015 by the Ethics Committee “Ethikkommision der Stadt Wien”. The study was carried out in accordance with the national laws and guidelines current at that time: the current Austrian drug law and the Austrian regulations for conducting noninterventional studies (Available from: https://www.ris.bka.gv.at/Dokument.wxe?Abfrage=BgblAuth&Dokumentnummer=BGBLA_2010_II_180. [Accessed 5 Dec 2023].
Consent to Participate
Written informed consent by the patient and (if applicable) in addition by the caregiver for patients below 18 years of age were taken before enrollment in the study.
Consent for Publication
Not applicable.
Availability of Data and Material
The datasets generated during the current study are not publicly available because the data reside in a proprietary database maintained by Meda Pharma GmbH & Co. KG (a Mylan Company, now Viatris); however, data are available from the corresponding author on reasonable request and with permission of Mylan Inc. (now Viatris).
Code Availability
Not applicable.
Author Contributions
HCK and DTN contributed to the design and implementation of the research, analysis of the results, interpretation of the data, and writing of the manuscript. KM, AR, GL, and WP performed the experiments. All authors extensively reviewed and contributed to the manuscript and approved the final version for publication.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Marth, K., Renner, A., Langmayr, G. et al. An Observational Study to Determine the Real-Life Effectiveness of MP-AzeFlu® in Austrian Patients with Persistent Allergic Rhinitis. Drugs - Real World Outcomes 11, 231–240 (2024). https://doi.org/10.1007/s40801-023-00412-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40801-023-00412-z