
Abstract
Background
Plerixafor was approved in Japan in 2016 for peripheral blood stem cell (PBSC) mobilization in autologous stem cell transplantation (A-SCT).
Objective
Our objective was to evaluate the safety and effectiveness of plerixafor in Japanese patients undergoing A-SCT for various indications in real-world practice.
Patients and Methods
This post-marketing surveillance study included Japanese patients initiating PBSC mobilization with plerixafor for A-SCT. Safety assessments included the incidence of adverse events (AEs) including serious AEs, adverse drug reactions (ADRs), and laboratory variables. Effectiveness assessments were the proportion of patients with the target CD34+ cell yield (≥2 × 106 cells/kg) ≤4 days after plerixafor administration and the number of days required to reach the target CD34+ cell yield.
Results
In total, 785 patients were registered, and the safety and effectiveness analysis sets comprised 764 and 717 patients, respectively. ADRs occurred in 12.2% of patients, with gastrointestinal disorders (5.5%), laboratory investigations (4.5%), and blood and lymphatic system disorders (3.0%) being the most common. A total of 71.1% of patients had the target CD34+ cell yield within ≤4 days of treatment, with a mean (standard deviation) of 1.3 (0.7) days to reach the target CD34+ cell yield. Over 80% of patients with a baseline CD34+ cell count >2 cells/μL had a target CD34+ cell yield within ≤4 days of treatment.
Conclusions
This large post-marketing surveillance study provided real-world evidence detailing the safety and effectiveness of plerixafor for PBSC mobilization in Japanese patients undergoing A-SCT. Importantly, no new safety concerns were identified, and the safety profile of plerixafor was consistent with the established profile of this drug.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
This post-marketing study demonstrated the safety and effectiveness of plerixafor for peripheral blood stem cell mobilization in Japanese patients undergoing autologous stem cell transplantation for various indications. |
Adverse drug reactions (ADRs) occurred in a relatively low percentage of patients, with the most common being gastrointestinal disorders, laboratory investigations, and blood and lymphatic system disorders. |
A high proportion of patients reached the target CD34+ cell yield within ≤4 days of treatment, with a mean of 1.3 (standard deviation 0.7) days to reach the target CD34+ cell yield. |
1 Introduction
High-dose chemotherapy with autologous stem cell transplantation (A-SCT) is an established treatment for hematopoietic malignancies, such as multiple myeloma (MM) and malignant lymphoma (ML), including Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL) [1,2,3,4,5]. As outcomes and safety with the procedure have improved, its use has evolved to include other serious conditions that may benefit, including autoimmune diseases [6, 7]. The vast majority of A-SCT procedures are now performed using peripheral blood stem cells (PBSCs) [8], which necessitates mobilization of hematopoietic stem cells (HSCs) and progenitor cells from bone marrow to the peripheral blood for collection via apheresis prior to transplantation.
At present, most patients undergo stem cell mobilization with hematopoietic growth factors, such as granulocyte colony-stimulating factor (G-CSF), alone or in combination with chemotherapy [9,10,11]. However, despite recent advancements in PBSC mobilization and collection techniques, approximately 20% of patients fail to have an adequate number of cells collected for SCT [13,14,14]. These patients are termed “poor mobilizers” [15] and may require repeated mobilization attempts, which is associated with diminishing stem cell yields [16] and may ultimately mean collection of sufficient stem cells for transplantation is impossible. Further, the delay in or inability to attain the target stem cell yield that results from a failure to mobilize increases the likelihood of disease progression and mortality [17, 18].
Although no consensus exists on the definition of stem cell mobilization failure, the generally accepted minimum CD34+ cell yield is 2 × 106 cells/kg [17, 19, 20]. However, higher yields of 4–5 × 106 CD34+ cells/kg may be targeted as they are associated with faster neutrophil and platelet engraftment [26,27,23], improved survival rates [24, 25], and reduced resource utilization [26]. A pheresis is initiated when peak peripheral blood CD34+ levels are attained, typically day 4–5 of mobilization with G-CSF only or day 10–20 with G-CSF/chemotherapy [27, 28].
The chemokine receptor CXCR4 and its ligand, CXCL12 [29], play a crucial role in the homing and retention of stem and progenitor cells in the bone marrow niche [35,36,37,33]. Plerixafor, a small-molecule antagonist of CXCR4, selectively and reversibly inhibits the direct binding of CXCR4 on stem cells to CXCL12 secreted from bone marrow stromal cells, thereby promoting their mobilization into the peripheral blood [32, 34]. Approval for its use was granted in 2008 by the US FDA [34] and in 2009 by the European Medicines Agency (EMA) [35] in combination with G-CSF for the mobilization of HSCs to peripheral blood for A-SCT in patients with NHL and MM. The EMA recently expanded this indication to include pediatric patients with lymphoma or solid malignant tumors [36].
In 2016, plerixafor was approved for use in Japan in combination with G-CSF for peripheral stem cell mobilization in patients undergoing A-SCT based on the results of the global phase III studies [37, 38] and domestic phase II studies [39, 40]. These studies showed that patients receiving plerixafor in combination with G-CSF increased mobilization of the target number of CD34+ cells required for SCT compared with G-CSF alone, with fewer days of apheresis required. Although two domestic phase II clinical trials evaluated plerixafor for PBSC mobilization in Japanese patients with MM and NHL [39, 40], the number of patients enrolled in these studies was low, and comprehensive data evaluating the real-world safety and effectiveness of plerixafor in this population are lacking. We therefore conducted a post-marketing surveillance study to evaluate the safety and effectiveness of plerixafor for PBSC mobilization in patients undergoing A-SCT in Japan.
2 Methods
2.1 Study Design, Patients, and Data Collection
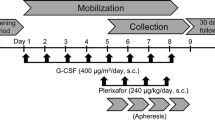
This was an observational, multicenter post-marketing all-case surveillance study conducted between February 2017 and December 2017 in Japan. Patients who initiated plerixafor for PBSC mobilization prior to A-SCT were included. Patients were managed under normal clinical conditions, and study visits were not prespecified. The observation period was defined as the period between the start of plerixafor treatment to 30 days after the first dose of plerixafor or the day prior to radiotherapy or chemotherapy, whichever occurred first. Treatment with plerixafor was initiated after the patient had received G-CSF. The dosage of plerixafor was based on 0.24 mg/kg actual body weight, with administration by subcutaneous injection, as per the approved product information [34]. Patient data were collected via case report form or electronic data capture system at baseline and at routine clinic visits during the observation period.
The study was conducted in accordance with the Japanese regulatory requirements of good post-marketing study practice. As this was a mandatory post-marketing study, informed consent from individual patients was not required.
2.2 Assessments
Upon entry to the study, baseline data were collected on demographics (age, sex, body weight), disease-related characteristics (disease type, clinical stage, Eastern Cooperative Oncology Group Performance Status [ECOG PS], presence of complications), prior treatments (chemotherapy, HSC collection, and G-CSF use), and concomitant medications. Complications were defined as any disease that the investigator deemed insufficient to infer causal correlation with the primary disease. The creatinine clearance rate (CCr), white blood cell (WBC) count, and presence/absence of renal function impairment and liver dysfunction were assessed at baseline and during the observation period. The CD34+ cell count was measured from peripheral blood via apheresis prior to study drug administration at baseline and approximately 11 h after plerixafor administration. Details regarding plerixafor treatment, including its reason for use, and average dose and total number of doses, were also collected at baseline and during the observation period. The number of days of G-CSF use was also collected.
Safety assessments included the incidence of adverse events (AEs), including serious AEs, and adverse drug reactions (ADRs), which were AEs for which a causal relationship to plerixafor could not be denied. AEs and ADRs were evaluated and categorized by system organ class (SOC) and preferred term (PT) using Medical Dictionary for Regulatory Activities (MedDRA)/Japanese version 22.1. Details regarding AEs were collected on the patient survey form by the treating physician at the study site. The incidence of ADRs was also compared amongst specific subgroups (elderly patients, diagnosis, and those with renal function impairment or liver dysfunction). ADRs of special interest included allergic and hypersensitivity reactions, leukocytosis, thrombocytopenia, interstitial lung disease, myocardial infarction, tumor cell mobilization, and splenomegaly/splenic rupture. Safety in patients with renal function impairment was also evaluated.
Vital signs and laboratory variables were monitored as part of the safety assessment and included blood pressure, liver function tests (alanine aminotransferase, aspartate aminotransferase, lactate dehydrogenase, gamma-glutamyltransferase, serum alkaline phosphatase), uric acid, C-reactive protein, lymphocyte count, neutrophil count, platelet count, and WBC count.
The effectiveness analysis comprised the proportion of patients with a CD34+ cell yield of ≥2 × 106 cells/kg within ≤4 days after plerixafor administration and the number of days of apheresis required to reach a CD34+ cell yield of ≥2 × 106 cells/kg. Subgroup comparisons were performed for each patient background factor (age, diagnosis, sex, previous HSC collection, presence/absence of a medical history, presence/absence of complications, renal function impairment and liver dysfunction, number of days of study drug and G-CSF administration, baseline CD34+ cell count, and CCr).
2.3 Statistical Analysis
The safety analysis set comprised all patients who received plerixafor treatment and had no registration violations. The effectiveness analysis set included all patients in the safety analysis set who had CD34+ data available and were administered plerixafor according to the approved dosage and administration schedule [34]. Based on the results of the global phase III clinical trials [37, 38], a sample size of 150 patients was estimated to detect an AE incidence rate of ≥2.7% with a confidence level of 95%, when accounting for withdrawals, missing data, and other study deviations. The sample size was determined with a precision-based approach, and no formal hypothesis testing was conducted in the study. Rather, P values were used as a reference only for the purposes of signal detection, with the primary interpretation of results made by medical consideration.
Baseline demographics were summarized using descriptive statistics. The mean, standard deviation (SD), and median (min; max) were calculated for continuous variables, and the frequency number and proportion were calculated for categorical variables. A chi-squared (χ2) test was used to compare the incidence of ADRs and the effectiveness of plerixafor in specific subgroups, with a two-sided significance level of 0.05 as supplemental information. The frequency of AEs was summarized descriptively overall and for each individual event (by SOC and PT). The effectiveness of plerixafor was also assessed in patients with prior HSC collection versus no prior HSC collection by baseline CD34+ cell count. As part of this analysis, the mean (SD) and median (range) CD34+ cell count prior to study drug administration was calculated for the overall population and according to prior HSC collection status and baseline CD34+ count cutoffs. The frequency and proportion of patients achieving the target CD34+ cell count at days 1, 2, 3, and 4, and within 2 and 4 days, was tabulated for the total population and categorized according to prior HSC collection status and baseline CD34+ count. Kaplan–Meier analysis was performed to determine the median time to reach the target CD34+ cell yield by baseline CD34+ cell count, with results compared using a proportional hazards model.
The possibility of a relationship between risk factors, such as patient background and treatment factors, and reaching the target CD34+ cell yield within 4 days was initially analyzed by univariate analysis stratified by patient background and treatment factors using the χ2 test, Fisher’s exact test, or the Cochran–Armitage test according to the nature (nominal/ordinal) of the categorical variable analyzed. Factors in the univariate analysis that showed P < 0.05 were included in the multivariable logistic regression to estimate the odds ratio (OR) for reaching the target CD34+ cell yield with 95% confidence intervals (CIs) and P values by the Wald χ2 test. The final models were built using a stepwise procedure with P values of <0.15 set for entry and staying in the model.
All statistical analyses were conducted using SAS software version 9.4 (SAS Institute Inc.; Cary, NC, USA).
3 Results
3.1 Patient Disposition
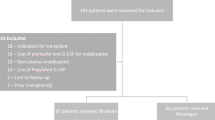
Details regarding patient disposition are presented in Fig. 1. A total of 785 patients were registered from 224 institutions across Japan. Survey forms were collected for 765 patients, one of whom was registered by an uncontracted physician and so was excluded. The safety analysis set therefore comprised 764 patients. For the effectiveness analysis set, 47 patients were excluded because of off-label study drug dosage and administration (n =26) and incomplete effectiveness evaluation (n = 23). The effectiveness analysis set therefore comprised 717 patients.

Patient disposition. *Some patients were excluded for more than one reason
Among the patients whose survey forms were collected (n = 765), a total of 745 patients (97.4%) completed treatment, and 20 patients (2.6%) discontinued treatment with plerixafor. Reasons for discontinuation were death (n = 4 [0.5%]) due to primary disease (n = 3 [0.4%]) or another unspecified cause (n = 1 [0.1%]), hospital non-attendance (n = 10 [1.3%]) and other reasons not specified (n = 6 [0.8%]).
3.2 Patient Characteristics
Baseline demographic and clinical characteristics of patients included in the safety and effectiveness analysis sets are presented in Table 1. In the safety analysis set, the median (min; max) age was 61.0 (1; 76) years, and 54.6% of patients were male. By age category, 2.4% (n = 18) of patients were aged <15 years, 68.3% (n = 522) were aged between 15 and <65 years, and 29.3% (n = 224) were aged ≥65 years. The majority of patients were undergoing A-SCT for MM (43.3% [n = 331]) or ML (47.1% [n = 360]: NHL 45.0% [n = 344]; HL 2.1% [n = 16]). A smaller proportion of patients (9.6% [n = 73]) were undergoing A-SCT for other diseases, including AL amyloidosis (2.6% [n = 20]); <1% of patients were undergoing A-SCT for other diseases.
Regarding treatment, 83.6% (n = 639) of patients received chemotherapy immediately prior to administration of plerixafor, including 78.9% (n = 261) of patients with MM and 88.3% (n = 318) of patients with ML. A total of 7.7% (n = 59) had renal function impairment, and the CCr was ≤50 mL/min in 28.8% (n = 17) of patients at baseline. A total of 3.8% (n = 29) of patients had liver dysfunction during the study.
Prior to study drug administration, among 237 patients whose CD34+ cell count was evaluated, the CD34+ cell count was <1 cell/μL in 6.3% (n = 15) of patients, 1 to <2 cells/μL in 6.3% (n = 15), 2 to <5 cells/μL in 24.5% (n = 58), 5 to <10 cells/μL in 22.8% (n = 54), 10 to <20 cells/μL in 18.1% (n = 43), and ≥20 cells/μL in 21.9% (n = 52) of patients. The CD34+ cell count prior to study drug administration was unknown or not evaluated in 14 and 512 patients, respectively.
3.3 Treatment
The mean (SD) daily dose of plerixafor was 0.24 mg/kg (0.03), and the mean (SD) number of administrations was 1.5 (0.7). No difference in mean (SD) daily dose was observed in patients with renal function impairment (0.23 mg/kg [0.05]). Most patients received one (64.1%) or two (27.9%) doses of study drug, with a smaller proportion receiving three (6.2%) or four (1.8%) doses. All patients (100.0%) received plerixafor in combination with G-CSF, which was most commonly administered for 5 days (38.1% of patients) or 6 days (27.0% of patients).
3.4 Safety
3.4.1 Incidence of Adverse Events and Adverse Drug Reactions
The overall incidence of AEs and the incidence of ADRs classified by SOC and PT in the 764 patients in the safety analysis set are presented in Table 2. AEs occurred in 138 patients (18.1%), and serious AEs occurred in 34 patients (4.5%) of the safety analysis set. All patients recovered or were in remission from serious adverse reactions, and no new concerns were noted. Overall, ADRs occurred in 93 patients (12.2%), with major ADRs by SOC (occurring in ≥1% of patients) consisting of gastrointestinal disorders (n = 42 [5.5%]), laboratory investigations (n = 34 [4.5%], including blood lactate dehydrogenase increased, n = 18 [2.4%]; WBC count increased, n = 10 [1.3%]; and serum alkaline phosphatase increased, n = 14 [1.8%]), and blood and lymphatic system disorders (n = 23 [3.0%]). Other ADRs occurred in <1% of patients.
By PT, major ADRs included diarrhea (n = 28 [3.7%]), leukocytosis (n = 18 [2.4%]), blood lactate dehydrogenase increased (n = 18 [2.4%]), serum alkaline phosphatase increased (n = 14 [1.8%]), vomiting (n = 12 [1.6%]), and nausea (n = 11 [1.4%]). Of the patients who developed diarrhea, ten (34.5%) were treated with loperamide hydrochloride capsules orally. As the classification of ‘leukocytosis’ was at the discretion of the attending physician, WBC counts were examined separately. Three patients had a WBC count >100,000/mm3 during the study.
Musculoskeletal and connective tissue disorders occurred infrequently during the study (n = 4 [0.5%]) and included back pain (n = 2 [0.3%]), bone pain (n = 1 [0.1%]), and flank pain (n = 1 [0.1%]), which were reported in <1% of patients.
3.4.2 Patient Background Factors Associated With the Incidence of Adverse Drug Reactions
Patient background factors potentially associated with the incidence of ADRs are presented in Table 3. Patient risk factors associated with an increased incidence of ADRs were age (P = 0.0011), diagnosis (P = 0.0027), and the presence of complications (P = 0.0064).
The overall incidence of ADRs by age group was 38.9% (7/18) in patients aged <15 years compared with 12.5% (65/522) in patients aged 15 to <65 years and 9.4% (21/224) in patients aged ≥65 years.
When evaluated by diagnosis, patients with a diagnosis other than MM and ML had a numerically higher incidence of ADRs. The incidence of ADRs by diagnosis was 10.6% (35/331) for those with MM, 11.1% (40/360) for those with ML, and 24.7% (18/73) for those with other diagnoses.
The incidence of adverse reactions was 15.9% (52/327) in patients with complications compared with 9.4% (41/437) in those without complications.
The incidence of ADRs was also numerically higher in patients with renal impairment (17.0% [10/59]) than in those without renal impairment (11.8% [83/705]), higher in those with liver dysfunction (17.2% [5/29]) than in those without liver dysfunction (12.0% [88/735]), and higher in non-elderly patients (aged <65 years; 13.3% [72/540]) than in elderly patients (aged ≥65 years; 9.4% [21/224]). However, no dosage adjustments were required in these patients.
3.4.3 Adverse Drug Reactions of Special Interest
Among ADRs of special interest, incidences of allergic and hypersensitivity reactions, leukocytosis, and thrombocytopenia were 0.1% (n = 1), 3.7% (n = 28), and 1.8% (n = 14), respectively. Serious ADRs, including allergic and hypersensitivity reactions, occurred in one patient (blood pressure decreased by PT), and thrombocytopenia occurred in eight patients (all were thrombocytopenia by PT). Although ‘blood pressure decreased’ was attributed to an allergic reaction/hypersensitivity to plerixafor, primary diseases and episodic diseases were also reported as other possible factors. The outcomes of leukocytosis (nine events) and thrombocytopenia (two events) events were remission, and all other events were resolved. No cases of interstitial lung disease, myocardial infarction, tumor cell mobilization, or splenomegaly/splenic rupture occurred. In patients with renal impairment (n = 59), the incidence of ADRs was 17.0% (n = 10), and two patients had serious ADRs.
When evaluating the time to onset of ADRs of special interest with an incidence of ≥1%, almost all occurred either during or in the week following administration. Leukocytosis developed in 1.8% (n = 14) of patients during and in the week following administration, and thrombocytopenia developed in 0.4% (n = 3) of patients during administration and in 1.4% (n = 11) of patients in the week following administration. In patients developing leukocytosis, by PT, 1.3% (n = 10) of patients developed leukocytosis and 0.5% (n = 4) developed increased WBC count during administration, and 1.1% (n = 8) of patients developed leukocytosis and 0.8% (n = 6) of patients developed increased WBC count in the week following administration. Of those who developed thrombocytopenia, by PT, thrombocytopenia developed during administration in 0.4% (n = 3) of patients, and thrombocytopenia or decreased platelet count developed in the week following administration in 0.8% (n = 6) and 0.7% (n = 5) of patients, respectively. A similar pattern was observed in patients with renal function impairment, with most patients experiencing ADRs of special interest either during administration (n = 4 [6.8%]) or in the week following administration (n = 6 [10.2%]).
3.5 Effectiveness
3.5.1 Mean CD34+ Cell Count Prior to Study Drug Administration and Proportion of Patients With a CD34+ Cell Yield of ≥2 × 106 cells/kg Following Study Drug Administration
The CD34+ cell count prior to study drug administration and the proportion of patients with a CD34+ cell yield ≥2 × 106 cells/kg after study drug administration are presented in Table 4. Of 227 patients with CD34+ cell count results available at baseline, the mean (SD) CD34+ cell count prior to study drug administration was 25.3 (69.6) cells/μL and the median (min; max) CD34+ cell count was 7.0 (0.0; 668.0) cells/μL. Following study drug administration, 71.1% (n = 504) had a CD34+ cell yield ≥2 × 106 cells/kg within ≤4 days of treatment.
3.5.2 Proportion of Patients With a CD34+ Cell Yield ≥2 × 106 cells/kg Following Study Drug Administration by Specific Subgroups
When examined by disease type, the mean (SD) CD34+ cell count prior to study drug administration was 18.7 (47.6) cells/μL in patients with MM (n = 113) and 29.6 (86.7) in patients with ML (n = 93). Following study drug administration, the proportion of patients with MM (75.8% [n = 235]), ML (65.7% [n = 215]), and other disease types (75.0% [n = 54]) achieving a CD34+ cell yield of ≥2 × 106 cells/kg within ≤4 days was comparable with the overall population, although a numerical difference was observed among the patients with a different disease type (P = 0.0148) (Table 5). The proportion of patients with a CD34+ cell yield of ≥2 × 106 cells/kg within ≤4 days was slightly lower in patients with ML than in those with MM (OR 0.81 [95% CI 0.68–0.98]) and was comparable between patients with MM and other disease types (OR 0.99 [95% CI 0.74–1.33]) (Table S1 in the electronic supplementary material [ESM]). When examined by rare disease type (Table 4), the mean (SD) CD34+ cell count prior to study drug administration was 10.6 (6.5) cells/μL in patients with AL amyloidosis (n = 8) and 4.0 (5.7) cells/μL in patients with POEMS syndrome (n = 2). Following study drug administration, 18 (90.0%) of 20 patients with AL amyloidosis and four of seven patients with POEMS syndrome had a CD34+ cell count ≥2 × 106 cells/kg within ≤4 days of treatment.
The CD34+ cell count prior to study drug administration and the proportion of patients with a CD34+ cell yield ≥2 × 106 cells/kg after study drug administration were also analyzed by CD34+ cell count prior to study drug administration overall (Table S2 in the ESM) and in patients with prior HSC collection versus no prior HSC collection (Table S3 in the ESM). When examined by baseline CD34+ cell count, patients with a baseline CD34+ count ≥2 cells/μL were more likely to reach the target CD34+ cell yield, and in a shorter time frame, than patients with a baseline CD34+ count <2 cells/μL (Table S2 in the ESM; Fig. 2). Predictably, patients with a prior history of HSC collection were less likely to have a target CD34+ cell yield within ≤4 days of study drug administration than patients without a prior history of HSC collection (OR 0.46 [95% CI 0.35–0.59]). Nevertheless, the fact that 43.8% of patients with a prior history of HSC collection had a target CD34+ cell yield was still clinically significant when considering this difficult-to-mobilize population.

Time to reach target CD34+ cell yield by baseline CD34+ cell count (Kaplan–Meier analysis)
3.5.3 Number of Days of Apheresis Required to Reach the Target CD34+ Cell Yield ≥2 × 106 cells/kg After Study Drug Administration
Of those who had a target CD34+ cell yield ≥2 × 106 cells/kg (n = 506) in the overall population, the mean (SD) time to reach the target CD34+ cell yield was 1.3 (0.7) days. By disease subtype, the mean (SD) number of days to reach the target CD34+ cell yield was 1.3 (0.7) days in patients with MM and 1.3 (0.6) days in patients with ML. The majority of patients (76.5% [n = 387]) reached a target CD34+ cell yield within the first day of study drug administration, and almost all (99.4% [n = 503]) reached a target CD34+ cell yield within 3 days of study drug administration.
3.5.4 Time From Study Drug Administration to First Apheresis Procedure
Of 681 patients who received first apheresis within 24 h after study drug administration with available data, the mean (SD) time to first apheresis following study drug administration was 11.5 (1.8) h, and the median (min; max) was 11.0 (7.0; 24.0) h.
3.5.5 Patient Background Factors Associated With a CD34+ Cell Count ≥2 × 106cells/kg After Study Drug Administration
Patient background factors potentially associated with a CD34+ cell yield ≥2 × 106 cells/kg within ≤4 days of study drug administration are presented in Table 5. Patient background factors associated with increased effectiveness of plerixafor were a diagnosis of MM (75.8%; P = 0.0148), no prior collection of HSCs (78.6%; P < 0.0001), fewer study drug administrations (1 [78.6%]; P < 0.0001), fewer days of G-CSF administration (3 [75.0%]; P < 0.0001), and a CD34+ cell count ≥2 prior to study drug administration (Table 5; P = 0.0031). When examining the effect of G-CSF administration on subsequent target CD34+ cell yield with plerixafor, pretreatment with G-CSF for 3−5 days appeared most efficacious (Table 5). Notably, successful mobilization was achieved within 4 days in elderly patients aged ≥65 years (67.8% [139/205] vs. 72.4% [365/504] aged < 65 years; P = 0.4043), those with renal function impairment (70.2% [40/57] vs. 71.2% [464/652] without renal function impairment; P = 0.8744), and in those with liver dysfunction (60.7% [17/28] vs. 71.5% [487/681] without liver dysfunction; P = 0.2168).
By multivariable regression analysis, patient background factors associated with a CD34+ cell yield ≥2 × 106 cells/kg within ≤4 days of study drug administration were no prior history of HSC collection (78.8% [438/556] vs. 44.4% [68/153] with a prior history of HSC collection; OR 0.53 [95% CI 0.32–0.89]; P = 0.0154) and a baseline CD34+ cell count of 2 to <5 (80.4% [45/56]; OR 3.71 [95% CI 1.43–9.66]; P = 0.0071), 5 to <10 (83.0% [44/53]; OR 3.76 [95% CI 1.44–9.79]; P = 0.0068), 10 to <20 (85.7% [36/42]; OR 4.35 [95% CI 1.62–11.64]; P = 0.0035), or ≥20 (84.0% [42/50]; OR 4.74 [95% CI 1.79–12.57]; P = 0.0018) cells/μL.
4 Discussion
This large post-marketing surveillance study demonstrated the safety and effectiveness of plerixafor in combination with G-CSF for stem cell mobilization in Japanese patients undergoing A-SCT. ADRs occurred in a relatively low percentage of patients (12.2%), and 71.1% of patients had a target CD34+ cell yield of ≥2 × 106 cells/kg within ≤4 days of treatment. Importantly, no new safety concerns were identified in the current study, and the safety profile of plerixafor was consistent with the established profile of this drug [34, 35].
Although a direct comparison between studies is difficult because of differences in study designs and patient characteristics, the incidence rate of ADRs reported in our study was substantially lower than those reported in the phase II studies conducted in Japanese patients (75.0–85.7%) [39, 40]. The exact reasons for this discrepancy are unclear; however, the relatively small sample size in both phase II Japanese studies (N = 14 and N = 32) compared with our study (N = 764) and the potential for underreporting of ADRs in the real-world setting may have contributed to the differences between these studies.
The most common ADRs reported in the current study were gastrointestinal disorders (5.5%), laboratory investigations (4.5%), and blood and lymphatic system disorders (3.0%). By PT, major ADRs included diarrhea (n = 28 [3.7%]), leukocytosis (n = 18 [2.4%]), blood lactate dehydrogenase increased (n = 18 [2.4%]), serum alkaline phosphatase increased (n = 14 [1.8%]), vomiting (n = 12 [1.6%]), and nausea (n = 11 [1.4%]). ADRs were broadly consistent with those reported in the pivotal phase III clinical studies [37, 38] and the phase II Japanese clinical studies [39, 40], although the incidence was relatively lower. When the incidence of ADRs was assessed in terms of background factors that may affect the safety of plerixafor, younger age (<15 years [38.9%] vs. ≥65 years [9.4%]), a diagnosis other than MM or ML (other 24.7% vs. MM 10.6% and ML 11.1%), and the presence of complications (yes 15.9% vs. no 9.4%) were associated with the incidence of ADRs. As a variety of diseases were classified as ‘other’, including AL amyloidosis, neuroblastoma, POEMS syndrome, plasma cell leukemia, medulloblastoma, and Ewing’s sarcoma, which are generally accepted as having a poorer prognosis than MM and ML, this finding was not unexpected. Further, it is likely that pediatric patients were also included in this subgroup, and our analysis recognized such patients as having a high incidence of ADRs.
The incidence of ADRs was comparable between patients with and without renal impairment, those with and without hepatic dysfunction, and elderly (aged ≥65 years) versus non-elderly patients (aged <65 years). Given this lack of difference, the lack of safety concerns in these populations suggests that no dosage adjustments are required in these patients. ADRs of special interest occurred infrequently and tended to occur either during or in the week following plerixafor administration; therefore, particularly close monitoring for leukocytosis and thrombocytopenia during this time period is recommended. No instances of tumor cell recruitment were observed in this large post-marketing study.
In terms of effectiveness, the proportion of patients achieving the target CD34+ cell yield of ≥2 × 106 cells/kg within ≤4 days of treatment in our study was acceptably high (71.1%) but was lower than rates reported in the global phase III and domestic phase II clinical studies [39,40,41,42]. In the phase II study conducted by Ri et al. [39], 100% of patients in the plerixafor plus G-CSF arm and 85.7% of patients in the G-CSF alone arm had a minimum CD34+ collection yield of ≥2 × 106 CD34+ cells/kg. Of these, 71.4% had a collection yield of ≥6 × 106 CD34+ cells/kg within ≤2 days of apheresis compared with no patients receiving G-CSF alone. Similar results were observed in the subsequent phase II study conducted in 32 Japanese patients undergoing SCT for NHL [40]. In this study, 93.8% of patients in the plerixafor plus G-CSF arm and 31.3% of patients in the G-CSF arm had a minimum CD34+ collection yield of ≥2 × 106 CD34+ cells/kg, and 56.3% had a CD34+ collection target of ≥5 × 106 CD34+ cells/kg within ≤4 days of apheresis compared with 6.3% of patients in the G-CSF arm. However, it is important to note that these studies excluded patients for whom previous mobilization attempts had failed, who had received chemotherapy in the previous 2 weeks, or who had an ECOG PS of >1 [39, 40]. In contrast, 21.3% of patients in our study had undergone prior HSC collection. When examining effectiveness by HSC collection status, a smaller proportion (43.8%) of patients who had undergone prior HSC collection had a CD34+ cell yield of ≥2 × 106 cells/kg within ≤4 days compared with patients who had not undergone prior HSC collection (78.6%). Nevertheless, this result was still clinically significant when considering the difficult-to-mobilize population in the current study compared with the phase II and III studies, which excluded patients for whom previous HSC collection attempts had failed or who had an ECOG PS of ≥2. Although the effectiveness of plerixafor in this study was lower than the rate reported in the Japanese pre-approval clinical trials [39, 40], the effectiveness rate (71.1%) was considered sufficient, particularly given the patient population. Patients in our study tended to be heavily pretreated, and many had undergone prior HSC collection, which negatively affected results.
When effectiveness results were examined by baseline CD34+ cell count, baseline CD34+ counts >2 cells/μL were significantly associated with the target CD34+ cell yield of ≥2 × 106 cells/kg within ≤4 days of treatment in our study. Over 80% of patients with a baseline CD34+ count >2 had a target CD34+ cell yield of ≥2 × 106 cells/kg within ≤4 days of treatment, with a trend towards an increasing proportion of patients with a target CD34+ yield as the baseline CD34+ cell count increased. Our findings paralleled those of other studies, which reported higher rates of effectiveness with increasing baseline CD34+ counts. In a retrospective analysis of data from two Japanese university hospitals over 8 years, 100% of patients with solid tumors had a CD34+ yield of 2.0 × 106/kg when the CD34+ pre-count was ≥10 cells/μL [41]. Similarly, the proportion of patients with MM with a CD34+ yield of 2.0 × 106/kg increased with increasing pre-count levels, from 80.0% of patients with ML with a CD34+ pre-count of 20.0─24.9 cells/μL and 88.9% of patients with MM and a CD34+ pre-count level of 25.0─29.9/μL, to 100% of patients with ML and MM and a pre-count level of ≥30 cells/μL, respectively [41]. Taken together, the findings reported in our study and others that a higher baseline CD34+ cell count is associated with a target CD34+ yield suggests that earlier initiation of plerixafor should be considered prior to repeated mobilization failures and myelosuppressive chemotherapy regimens. Using this approach, 95─100% of patients should theoretically have a minimum CD34+ yield of ≥2 × 106 cells/kg in clinical practice, based on the results observed in other studies [19, 41]. A lower rate of effectiveness was reported in patients with ML than in patients with MM, suggesting that the negative impact of prior chemotherapy on effectiveness was substantial. In most instances, HSC collection in patients with ML occurs after pretreatment with high-dose chemotherapy [42], which is consistent with the findings reported in the current study.
Limitations were inherent to post-marketing studies and included the noninterventional, observational nature of the study and incomplete or missing data. As this was not a controlled study, results should be interpreted with caution. Further, no data on previous stem cell transplantation were available, and the proportion of patients for whom this was a second transplantation was unclear. It is also possible that apheresis may have been stopped when patients reached 1.9 × 106 cells/kg during the first apheresis rather than continuing to the second apheresis, which may have led to the results being an underestimation. Technical variations in the method used to perform CD34+ measurements at different laboratory facilities may have also influenced the results. In addition, several different statistical methods were employed, which may have increased the likelihood of false-negative results. As such, all P values reported in the current analysis were nominal and should be interpreted with caution. That being said, the probability of false positives was likely low because determination of P value signals was made by medical consideration. As previously mentioned, underreporting of ADRs may also be more common in post-marketing studies because of the potential for investigator bias when reporting. Nevertheless, the study population represented a large cohort of Japanese patients, particularly compared with the Japanese phase II studies, and also reflected the heterogeneous population typically seen in clinical practice.
In conclusion, the results of this large post-marketing surveillance study confirmed the safety and effectiveness of plerixafor for stem cell mobilization prior to A-SCT in Japanese patients. Overall, plerixafor was well-tolerated, and ADRs occurred in a relatively low percentage of patients compared with previous global clinical trials and the phase II Japanese studies. Further, no new safety concerns were raised, and the safety profile of plerixafor was consistent with its known safety profile. The findings reported in the current study add to the wealth of data supporting the safety and effectiveness of plerixafor for stem cell mobilization in patients undergoing A-SCT and provide real-world evidence detailing its safety and effectiveness in myriad indications. Our results support the rationale for early mobilization with plerixafor prior to repeated mobilization failure and pretreatment with intensive myelosuppressive therapies for optimal outcomes.
References
Attal M, Harousseau JL, Stoppa AM, Sotto JJ, Fuzibet JG, Rossi JF, et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. New Engl J Med. 1996;335(2):91–7. https://doi.org/10.1056/nejm199607113350204.
Child JA, Morgan GJ, Davies FE, Owen RG, Bell SE, Hawkins K, et al. High-dose chemotherapy with hematopoietic stem-cell rescue for multiple myeloma. New Engl J Med. 2003;348(19):1875–83. https://doi.org/10.1056/NEJMoa022340.
Linch DC, Winfield D, Goldstone AH, Moir D, Hancock B, McMillan A, et al. Dose intensification with autologous bone-marrow transplantation in relapsed and resistant Hodgkin’s disease: results of a BNLI randomised trial. Lancet. 1993;341(8852):1051–4. https://doi.org/10.1016/0140-6736(93)92411-l.
Schmitz N, Pfistner B, Sextro M, Sieber M, Carella AM, Haenel M, et al. Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haemopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin’s disease: a randomised trial. Lancet. 2002;359(9323):2065–71. https://doi.org/10.1016/s0140-6736(02)08938-9.
Philip T, Guglielmi C, Hagenbeek A, Somers R, Van der Lelie H, Bron D, et al. Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non-Hodgkin’s lymphoma. New Engl J Med. 1995;333(23):1540–5. https://doi.org/10.1056/nejm199512073332305.
Burman J, Tolf A, Hägglund H, Askmark H. Autologous haematopoietic stem cell transplantation for neurological diseases. J Neurol Neurosurg Psychiatry. 2018;89(2):147–55. https://doi.org/10.1136/jnnp-2017-316271.
Brierley CK, Castilla-Llorente C, Labopin M, Badoglio M, Rovira M, Ricart E, et al. Autologous haematopoietic stem cell transplantation for Crohn’s disease: a retrospective survey of long-term outcomes from the European Society for Blood and Marrow Transplantation. J Crohns Colitis. 2018;12(9):1097–103. https://doi.org/10.1093/ecco-jcc/jjy069.
Passweg JR, Baldomero H, Bader P, Bonini C, Cesaro S, Dreger P, et al. Hematopoietic stem cell transplantation in Europe 2014: more than 40,000 transplants annually. Bone Marrow Transplant. 2016;51(6):786–92. https://doi.org/10.1038/bmt.2016.20.
Anderlini P, Rizzo JD, Nugent ML, Schmitz N, Champlin RE, Horowitz MM. Peripheral blood stem cell donation: an analysis from the International Bone Marrow Transplant Registry (IBMTR) and European Group for Blood and Marrow Transplant (EBMT) databases. Bone Marrow Transplant. 2001;27(7):689–92. https://doi.org/10.1038/sj.bmt.1702875.
Haas R, Murea S. The role of granulocyte colony-stimulating factor in mobilization and transplantation of peripheral blood progenitor and stem cells. Cytokines Mol Ther. 1995;1(4):249–70.
Douglas KW, Gilleece M, Hayden P, Hunter H, Johnson PRE, Kallmeyer C, et al. UK consensus statement on the use of plerixafor to facilitate autologous peripheral blood stem cell collection to support high-dose chemoradiotherapy for patients with malignancy. J Clin Apheresis. 2018;33(1):46–59. https://doi.org/10.1002/jca.21563.
Tarella C, Di Nicola M, Caracciolo D, Zallio F, Cuttica A, Omedè P, et al. High-dose ara-C with autologous peripheral blood progenitor cell support induces a marked progenitor cell mobilization: an indication for patients at risk for low mobilization. Bone Marrow Transplant. 2002;30(11):725–32. https://doi.org/10.1038/sj.bmt.1703729.
Perry AR, Watts MJ, Peniket AJ, Goldstone AH, Linch DC. Progenitor cell yields are frequently poor in patients with histologically indolent lymphomas especially when mobilized within 6 months of previous chemotherapy. Bone Marrow Transplant. 1998;21(12):1201–5. https://doi.org/10.1038/sj.bmt.1701267.
Pavone V, Gaudio F, Console G, Vitolo U, Iacopino P, Guarini A, et al. Poor mobilization is an independent prognostic factor in patients with malignant lymphomas treated by peripheral blood stem cell transplantation. Bone Marrow Transplant. 2006;37(8):719–24. https://doi.org/10.1038/sj.bmt.1705298.
DiPersio JF. New hope for mobilization failures … again. Biol Blood Marrow Transplant. 2012;18(2):159–60. https://doi.org/10.1016/j.bbmt.2011.12.578.
Morgan SJ, Seymour JF, Grigg A, Matthews JP, Prince HM, Wolf MM, et al. Predictive factors for successful stem cell mobilization in patients with indolent lymphoproliferative disorders previously treated with fludarabine. Leukemia. 2004;18(5):1034–8. https://doi.org/10.1038/sj.leu.2403326.
Pusic I, Jiang SY, Landua S, Uy GL, Rettig MP, Cashen AF, et al. Impact of mobilization and remobilization strategies on achieving sufficient stem cell yields for autologous transplantation. Biol Blood Marrow Transplant. 2008;14(9):1045–56. https://doi.org/10.1016/j.bbmt.2008.07.004.
Moreb JS, Byrne M, Shugarman I, Zou F, Xiong S, May WS, et al. Poor peripheral blood stem cell mobilization affects long-term outcomes in multiple myeloma patients undergoing autologous stem cell transplantation. J Clin Apheresis. 2018;33(1):29–37. https://doi.org/10.1002/jca.21556.
Russell N, Douglas K, Ho AD, Mohty M, Carlson K, Ossenkoppele GJ, et al. Plerixafor and granulocyte colony-stimulating factor for first-line steady-state autologous peripheral blood stem cell mobilization in lymphoma and multiple myeloma: results of the prospective PREDICT trial. Haematologica. 2013;98(2):172–8. https://doi.org/10.3324/haematol.2012.071456.
Mohammadi S, Malek Mohammadi A, Nikbakht M, Norooznezhad AH, Alimoghaddam K, Ghavamzadeh A. Optimizing stem cells mobilization strategies to ameliorate patient outcomes: a review of guidelines and recommendations. Int J Hematol-Oncol Stem Cell Res. 2017;11(1):78–88.
Stiff PJ, Micallef I, Nademanee AP, Stadtmauer EA, Maziarz RT, Bolwell BJ, et al. Transplanted CD34(+) cell dose is associated with long-term platelet count recovery following autologous peripheral blood stem cell transplant in patients with non-Hodgkin lymphoma or multiple myeloma. Biol Blood Marrow Transplant. 2011;17(8):1146–53. https://doi.org/10.1016/j.bbmt.2010.11.021.
Bensinger W, Appelbaum F, Rowley S, Storb R, Sanders J, Lilleby K, et al. Factors that influence collection and engraftment of autologous peripheral-blood stem cells. J Clin Oncol. 1995;13(10):2547–55. https://doi.org/10.1200/jco.1995.13.10.2547.
Ketterer N, Salles G, Raba M, Espinouse D, Sonet A, Tremisi P, et al. High CD34(+) cell counts decrease hematologic toxicity of autologous peripheral blood progenitor cell transplantation. Blood. 1998;91(9):3148–55.
Bolwell BJ, Pohlman B, Rybicki L, Sobecks R, Dean R, Curtis J, et al. Patients mobilizing large numbers of CD34+ cells (‘super mobilizers’) have improved survival in autologous stem cell transplantation for lymphoid malignancies. Bone Marrow Transplant. 2007;40(5):437–41. https://doi.org/10.1038/sj.bmt.1705763.
Singh AK, Savani BN, Albert PS, Barrett AJ. Efficacy of CD34+ stem cell dose in patients undergoing allogeneic peripheral blood stem cell transplantation after total body irradiation. Biol Blood Marrow Transplant. 2007;13(3):339–44. https://doi.org/10.1016/j.bbmt.2006.10.029.
Schulman KA, Birch R, Zhen B, Pania N, Weaver CH. Effect of CD34(+) cell dose on resource utilization in patients after high-dose chemotherapy with peripheral-blood stem-cell support. J Clin Oncol. 1999;17(4):1227. https://doi.org/10.1200/jco.1999.17.4.1227.
To LB, Haylock DN, Simmons PJ, Juttner CA. The biology and clinical uses of blood stem cells. Blood. 1997;89(7):2233–58. https://doi.org/10.1182/blood.V89.7.2233.
Fruehauf S, Klaus J, Huesing J, Veldwijk MR, Buss EC, Topaly J, et al. Efficient mobilization of peripheral blood stem cells following CAD chemotherapy and a single dose of pegylated G-CSF in patients with multiple myeloma. Bone Marrow Transplant. 2007;39(12):743–50. https://doi.org/10.1038/sj.bmt.1705675.
Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12(2):121–7. https://doi.org/10.1016/s1074-7613(00)80165-x.
Walenkamp AME, Lapa C, Herrmann K, Wester HJ. CXCR4 ligands: the next big hit? J Nuclear Med. 2017;58(Suppl 2):77S-82S. https://doi.org/10.2967/jnumed.116.186874.
Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23(1):43–52. https://doi.org/10.1038/leu.2008.299.
Fricker SP. Physiology and pharmacology of plerixafor. Transfus Med Hemother. 2013;40(4):237–45. https://doi.org/10.1159/000354132.
Pitchford SC, Furze RC, Jones CP, Wengner AM, Rankin SM. Differential mobilization of subsets of progenitor cells from the bone marrow. Cell Stem Cell. 2009;4(1):62–72. https://doi.org/10.1016/j.stem.2008.10.017.
Mozobil [prescribing information]. Sanofi Genzyme, 2019. http://products.sanofi.us/Mozobil/mozobil.html. Accessed 8 Jul 2020.
European Medicines Agency. CHMP Summary of Product Characteristics: Mozobil®. https://www.ema.europa.eu/en/documents/product-information/mozobil-epar-product-information_en.pdf. Accessed 8 Jul 2020.
Karres D, Ali S, van Hennik PB, Straus S, Josephson F, Thole G, et al. EMA recommendation for the pediatric indications of plerixafor (Mozobil) to enhance mobilization of hematopoietic stem cells for collection and subsequent autologous transplantation in children with lymphoma or malignant solid tumors. Oncologist. 2020;25(6):e976–81. https://doi.org/10.1634/theoncologist.2019-0898.
DiPersio JF, Stadtmauer EA, Nademanee A, Micallef IN, Stiff PJ, Kaufman JL, et al. Plerixafor and G-CSF versus placebo and G-CSF to mobilize hematopoietic stem cells for autologous stem cell transplantation in patients with multiple myeloma. Blood. 2009;113(23):5720–6. https://doi.org/10.1182/blood-2008-08-174946.
DiPersio JF, Micallef IN, Stiff PJ, Bolwell BJ, Maziarz RT, Jacobsen E, et al. Phase III prospective randomized double-blind placebo-controlled trial of plerixafor plus granulocyte colony-stimulating factor compared with placebo plus granulocyte colony-stimulating factor for autologous stem-cell mobilization and transplantation for patients with non-Hodgkin’s lymphoma. J Clin Oncol. 2009;27(28):4767–73. https://doi.org/10.1200/jco.2008.20.7209.
Ri M, Matsue K, Sunami K, Shimazaki C, Hayashi A, Sunaga Y, et al. Efficacy and safety of plerixafor for the mobilization/collection of peripheral hematopoietic stem cells for autologous transplantation in Japanese patients with multiple myeloma. Int J Hematol. 2017;106(4):562–72. https://doi.org/10.1007/s12185-017-2255-8.
Matsue K, Kumagai K, Sugiura I, Ishikawa T, Igarashi T, Sato T, et al. Plerixafor for mobilization and collection of haematopoietic stem cells for autologous transplantation in Japanese patients with non-Hodgkin lymphoma: a randomized phase 2 study. Int J Hematol. 2018;108(5):524–34. https://doi.org/10.1007/s12185-018-2505-4.
Nakamura Y, Okubo M, Furuta Y, Tokida M, Ichikawa K, Ohsaka A. Impact of CD34+ pre-counting and plerixafor on autologous peripheral blood stem cell collection in Japanese university hospitals in eight years. Transfus Apher Sci. 2019;58(6): 102664. https://doi.org/10.1016/j.transci.2019.10.006.
LaCasce AS. Treating Hodgkin lymphoma in the new millennium: relapsed and refractory disease. Hematol Oncol. 2019;37(S1):87–91. https://doi.org/10.1002/hon.2589.
Acknowledgements
The authors thank all clinicians for their involvement and contribution to the study. The authors also thank Jordana Campbell, BSc, CMPP of inScience Communications, Springer Healthcare, for writing the outline and the first draft of the manuscript. This medical writing assistance was funded by Sanofi K.K.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Sanofi K.K.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosure of potential conflicts of interest
Nobuhiro Tsukada has received payment for lectures, including service on speaker’s bureaus, from Takeda Pharmaceutical Company Limited and Sanofi K.K. Momoko Nishikori has received grants from Eisai Co., Ltd. and Sumitomo Dainippon Pharma Co., Ltd. and fees for participation in review activities such as data monitoring boards from Sanofi K.K. Hiroaki Goto has received fees for participation in review activities such as data monitoring boards from Amgen K.K., Novartis Pharma K.K., Nippon Shinyaku Co. Ltd., and Bayer Yakuhin Ltd.; has received payment for writing or reviewing manuscripts and delivering lectures, including service on speaker’s bureaus, from Amgen K.K. Rie Kanamori and Takashi Seto are employees of Sanofi K.K. Satoshi Nishina is an employee of Sanofi K.K. and holds stocks/stock options in this company. Shinsuke Iida has received grants, or has grants pending on behalf of Sanofi K.K., Takeda Pharmaceutical Company Limited, Ono Pharmaceutical Co. Ltd., Bristol-Myers Squibb K.K., Celgene Corporation, Janssen Pharmaceutical K.K., Daiichi Sankyo Co. Ltd., Chugai Pharmaceutical Co. Ltd., Kyowa Kirin Co. Ltd., GlaxoSmithKline, and Abbvie Inc.; and has received consulting fees or honorarium from Sanofi K.K., Takeda Pharmaceutical Company Limited, Ono Pharmaceutical Co. Ltd., Bristol-Myers Squibb K.K., Celgene Corporation, Janssen Pharmaceutical K.K., Daiichi Sankyo Co. Ltd.
Availability of data and material
Qualified researchers may request access to patient-level data and related documents (including, for example, the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan, and dataset specifications). Patient-level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi’s data-sharing criteria, eligible studies, and process for requesting access can be found at https://www.clinicalstudydatarequest.com.
Author contributions
SN and TS contributed to study conception/design, data acquisition and data analysis. All authors contributed to data interpretation and writing the manuscript and approved the final draft for submission.
Ethics approval
This study was conducted in accordance with the Japanese regulatory requirements of good post-marketing study practice, and the protocol of this study was approved by the Ministry of Health, Labour and Welfare of the Japanese government. The need for ethics approval and informed consent was waived as this study was mandated by the Japanese regulatory authorities in accordance with the Law for Ensuring the Quality, Efficacy, and Safety of Drugs and Medical Devices (Pharmaceutical and Medical Device Act).
Research involving human participants and/or animals
The study was conducted in accordance with the Japanese regulatory requirements of good post-marketing study practice.
Consent to participate
As this was a mandatory post-marketing study, informed consent from individual patients was not required.
Consent for publication
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Tsukada, N., Nishikori, M., Goto, H. et al. Safety and Effectiveness of Plerixafor for Peripheral Blood Stem Cell Mobilization in Autologous Stem Cell Transplantation: Results of a Post-Marketing Surveillance Study. Drugs - Real World Outcomes 9, 63–78 (2022). https://doi.org/10.1007/s40801-021-00276-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40801-021-00276-1