
Abstract
Purpose of Review
RNA epigenetic modifications have been identified as novel, dynamic regulators of gene expression, with important impacts on stem cell fate decisions. Here, we examine the functions of RNA modifications, with a focus on N6-methyladenosine (m6A), in hematopoietic stem cells under normal conditions and in malignancy.
Recent Findings
The m6A RNA modification is a critical regulator of hematopoiesis. Disruption of different elements of the m6A machinery can skew the balance of self-renewal and differentiation in normal hematopoietic stem cells. The m6A reader, writer, and eraser proteins are also overexpressed in myeloid leukemia, and disruption of their function impairs leukemogenesis. RNA m6A modification governs important aspects of immune system function, including immune cell development, immune signaling, and recognition of RNA as foreign or self. In hematopoietic stem cells, endogenously derived double-stranded RNA can form in the absence of m6A, inducing deleterious inflammatory pathways which compromise stem cell function.
Summary
The RNA modification m6A exerts a variety of functions in normal hematopoietic stem cells as well as leukemic cells. Pharmacologic modulation of different elements of the m6A machinery provides a promising avenue for ex vivo expansion of hematopoietic stem cells in the transplant setting, as well as for leukemia therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The past half century has witnessed burgeoning discoveries in the mechanisms regulating gene expression, thereby largely expanding the tenets of the central dogma. Post-transcriptional RNA modifications encompass more than 160 different chemical variations on the four canonical ribonucleotides, with diverse impacts on transcript function and regulation [1, 2].
Eukaryotic mRNA is well-known to be modified by a 5′-m7G cap and 3′-polyadenylated tail, which facilitate sufficient output of translational products. In addition, mRNA can be exquisitely decorated by a collection of internal modifications [2]. The modified nucleotide N6-methyladenosine (m6A) was first identified as an abundant internal mRNA modification as early as the mid-1970s, and its principal methyltransferase, methyltransferase like 3 (METTL3), was discovered shortly thereafter [3,4,5]. We now know that mRNA can be modified by over 10 different chemical marks, including m6A, N6,2’-O-dimethyladenosine (m6Am), 5-methylcytosine (m5C), and rarely N1-methyladenosine (m1A). Among these, m6A is the most prevalent internal modification in mammalian cells, accounting for 1–3 adenosines per mRNA transcript on average [6].
A contemporary renaissance in RNA epigenetics was unlocked by the publication of the first transcriptome-wide m6A maps, which were derived by coupling RNA immunoprecipitation with an m6A-specific antibody with next-generation sequencing (MeRIP-seq) [7, 8]. Across the transcriptome, m6A is predominantly distributed throughout the coding region of mRNAs, with enrichment in the 3′-untranslated region (3′UTR) and near-stop codons with an RRACH (R = A/G, H = A/C/U) consensus motif.
Over the past decade, mechanistic studies facilitated by these techniques have implicated the m6A modification in RNA processing, metabolism, and structure. Demonstrated functions include regulation of transcript stability, translational efficiency, micro RNA (miRNA) processing, long non-coding RNA (lncRNA) function, RNA/DNA hybridization, and RNA conformation [9,10,11,12,13]. While some studies have shown that m6A guides RNA splicing by recruiting splicing factors, this remains a point of contention [14, 15].
Corresponding biologic phenotypes have garnered significant interest. Early studies demonstrated that perturbation of the m6A modification exerts remarkable effects on stem cell populations. Constitutive deletion of Mettl3 in mice results in early embryonic lethality with persistence of embryonic stem cells in “ground-state” or “naïve” pluripotency, implicating m6A as a key regulator of self-renewal [16, 17]. This has prompted further investigation into the effects of m6A on other stem cell populations in normal and diseased states.
As a result, emerging literature has begun to describe the role of the m6A machinery in normal hematopoiesis and malignancy. The m6A modification is an essential regulator of hematopoietic stem cell (HSC) self-renewal and differentiation, and various elements of the m6A machinery have been implicated in myeloid malignancies including myelodysplasia (MDS) and acute myeloid leukemia (AML). In this review, we will describe the m6A machinery and its functions within the hematopoietic system.
Overview of the m6A Machinery
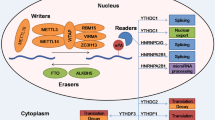
The establishment of transcriptome-wide methods for m6A mapping has facilitated extensive characterization of the armament of m6A reader, writer, and eraser proteins and their impacts on RNA metabolism.
Core components of the m6A methyltransferase complex include METTL3, METTL14, WT1-associated protein (WTAP), and KIAA1429. m6A is deposited co-transcriptionally, guided in part by binding of METTL14 to H3K36me3 [14, 18, 19]. The presence of m6A can in turn regulate other histone modifications and mediate chromatin accessibility [20••]. RNA-binding motif protein 15 (RBM15) and RBM15B participate in the recruitment of the METTL3/14/WTAP complex to a subset of RNAs including the lncRNA Xist, which mediates X chromosome inactivation in an m6A-dependent fashion [11]. METTL5, zinc finger CCHC-type containing 4 (ZCCHC4), and METTL16 are ancillary m6A methyltransferases, which independently install m6A on a few specific target transcripts including the 28S rRNA, spliceosomal U6 snRNA, and S-adenosyl methionine synthetase [21,22,23,24].
The best characterized m6A readers are the YTH domain-containing family of proteins. Among these, YTH N6-methyladenosine RNA-binding protein 1 (YTHDF1) and YTH domain-containig 2 (YTHDC2) predominantly promote translational efficiency, while YTHDF2 accelerates mRNA decay by trafficking target mRNAs to P bodies for degradation [9, 10, 25]. YTHDF3 facilitates the functions of both YTHDF1 and 2 and also enables translation of circular RNAs (circRNAs) [26,27,28,29]. YTHDC1 participates in RNA splicing, circRNA synthesis, Xist-mediated X chromosome inactivation, and global regulation of chromatin accessibility [11, 20, 29, 30]. Additional readers include eukaryotic initiation factor 3 (eIF3), insulin-like growth factor 2-binding protein (IGF2BP) family proteins, and heterogeneous nuclear ribonucleoprotein (hnRNP) family proteins hnRNPA2B1, hnRNPC, and hnRNPG [12, 31,32,33,34,35]. For a description of their functions, we recommend a detailed review by Meyer and Jaffrey [36].
While alkB homolog 5, RNA demethylase (ALKBH5) has been firmly established as an m6A eraser, the second proposed m6A demethylase, fat mass and obesity associated (FTO), has a higher affinity for cap-associated m6Am, and its true substrate remains unclear [37,38,39,40].
Role of the m6A Methyltransferases in Hematopoiesis
METTL3 and METTL14 in Hematopoietic Stem Cells
An important role for m6A modification in homeostatic hematopoiesis was first demonstrated in zebrafish models, as mettl3-deficient embryos die early during development due to profound hematopoietic failure secondary to an arrested endothelial-to-hematopoietic transition (EHT). In zebrafish, this critical cell fate decision marks the emergence of the first hematopoietic stem and progenitor cells from the hemogenic endothelium in the aorta-gonad-mesonephros (AGM). Mettl3-deficient zebrafish fail to navigate this transition, as endothelial identity is maintained in the AGM at the expense of primitive hematopoiesis. This effect is mediated by the stabilization of notch1a transcripts, which are normally targeted for degradation by m6A during the EHT [41••].
In a subsequent study, tissue-specific Cre-Lox models of Mettl3 deletion in endothelial and hematopoietic tissues driven by the Vec and Vav promoters, respectively, was performed. Analysis of mice at E10.5 recapitulated the hematopoietic phenotype in Vec-Cre but not Vav-Cre mice [42]. This led to the suggestion that the observed hematopoietic phenotype in Mettl3 deficient mice is exclusively attributable to defects within the Vec+ endothelial compartment, and not due to effects in hematopoietic tissues. However, this interpretation is limited as analysis was performed prior to the onset of complete Vav expression in hematopoietic tissues at approximately E11.5.
Indeed, in our own analysis, we found that Vav-Cre+-Mettl3fl/fl mice die during embryogenesis due to profound hematopoietic failure, with rare surviving pups exhibiting profound cytopenias and bone marrow aplasia [43••]. Our findings are in keeping with multiple preceding models of Mettl3 deletion in adult mice. Three independent groups characterized Mx1-Cre+-Mettl3fl/fl mice, in which Mettl3 deletion is induced in the hematopoietic tissues of adult mice in response to polyinosinic:polycytidylic acid (pI:pC) treatment. Consistently across these studies, loss of METTL3 results in profound hematopoietic failure, with resultant peripheral blood cytopenias, reduced marrow cellularity, spleen hypertrophy, and extramedullary hematopoiesis [44,45,46].
Despite defective hematopoiesis, both E14.5 Vav-Cre+-Mettl3fl/fl and adult Mx1-Cre+-Mettl3fl/fl mice have significantly expanded bulk HSPC populations, identifiable by the Lin−Sca-1+c-kit+ (LSK) surface markers. This finding suggests an arrest in hematopoietic differentiation in m6A-deficient mice. Within the LSK compartment, there is a further enrichment of LSK subpopulations with immunophenotypes normally characteristic of self-renewing HSCs with long-term reconstitution potential. Despite the increased proportion of phenotypic HSCs in both models, these cells were deficient in functional assays including in vitro colony formation and hematopoietic reconstitution in competitive transplantation assays [43,44,45,46]. Taken together, these findings indicate that loss of METTL3 in the hematopoietic system results in accumulation of phenotypic HSCs that have profoundly reduced hematopoietic potential.
Evidence thus far indicates that loss of METTL3 destabilizes HSC identity rather than reinforcing it. Mx1-Cre+-Mettl3fl/fl phenotypic HSCs showed enhanced metabolic and proliferative activity, exiting quiescence and entering the cell cycle [44, 45]. This is accompanied by diminished expression of hallmark HSC self-renewal genes [44]. Single-cell RNA sequencing of Mx1-Cre+-Mettl3fl/fl HSPCs demonstrated the emergence of two novel “HSC-like” populations, one of which exhibited diminished expression of core HSC self-renewal genes. Comparison with wildtype HSPC subtypes showed that Mettl3−/− HSC populations most closely resembled wildtype multipotent progenitors. Functional analyses via transplant of sorted populations demonstrated that phenotypic Mettl3−/− HSCs engrafted preferentially in the MPP compartment and eventually disappeared in long-term analyses, suggesting limited self-renewal capacity [45••]. Mechanistic studies of this phenotype have centered predominantly on expression of Myc, which is downregulated in Mettl3−/− HSCs. Global MYC downregulation and altered segregation of MYC at the level of individual cell divisions impair the capacity for lineage commitment of HSCs, instead favoring self-renewal via symmetric replication [45, 46].
In comparison with Mx1-Cre+-Mettl3fl/fl mice, Mx1-Cre+-Mettl14fl/fl mice exhibit an attenuated hematopoietic phenotype. HSC frequency is unaltered in primary Mx1-Cre+-Mettl14fl/fl compared with controls. Engraftment potential is diminished in competitive reconstitution assays but to a lesser degree than that of Mettl3−/− cells [47]. METTL3 and METTL14 form a heterodimer, and METTL3 is typically degraded upon loss of METTL14. Independent roles of METTL3 or METTL14 or residual METTL3 function following Mettl14 deletion may account for this difference.
Overall, these findings highlight the functional importance of the m6A methyltransferase complex in the normal hematopoietic system. Both METTL3 and METTL14 are necessary for normal hematopoiesis, and dysregulation of either produces dramatic impairment of HSC function. The above described model systems and their corresponding phenotypes are summarized in Table 1.
METTL3 and METTL14 in Myelopoiesis and Myeloid Malignancies
The m6A methyltransferase subunits METTL3 and METTL14 have been described as important regulators of normal and malignant myelopoiesis. While this has been of interest for the development of therapeutics, observations have varied across experimental systems, and it remains to be seen whether proof of concept experiments will translate to effective clinical therapies.
Initial in vitro experiments in human CD34+ cord blood progenitors, which are enriched for HSPCs, showed that loss of either METTL3 or METTL14 promotes spontaneous myeloid differentiation [48]. Furthermore, METTL3 and METTL14 are overexpressed in multiple subtypes of acute myeloid leukemia and are recurrently identified as essential genes in genome-wide CRISPR-Cas9 dropout screens in experimental models of AML [54••]. Indeed, loss of METTL3 or METTL14 in both murine AML models and human AML cell lines attenuates proliferation, diminishes engraftment, and delays mortality following transplantation into recipient mice. Furthermore, loss of either methyltransferase component results in the acquisition of mature myeloid cell surface markers and mature myeloid morphology [47, 48, 54].
A variety of mechanisms accounting for METTL3-mediated inhibition of myeloid differentiation have been identified. Vu et al. found that METTL3 was essential for expression of the oncogenes MYC and BCL2, as well as suppression of the PI3K/Akt signaling pathway, which normally promotes hematopoietic differentiation [48]. Barbieri et al. found that METTL3 is recruited to transcription start sites by the transcription factor CEBPZ. Promoter-bound METTL3 methylates emerging transcripts, enhancing the translation of key oncogenes such as SP1 by alleviating ribosome stalling [54••]. Weng et al. showed that METTL14 is downregulated by the myeloid transcription factor SPI1 (PU.1), which facilitates differentiation via downregulation of the m6A target genes Myb and Myc [47]. The experimental systems used in these studies and the observed mechanisms of leukemogenesis are summarized in Table 2.
While these data support a role for METTL3 as a regulator of myelopoiesis, results have varied across different models. As described above, loss of METTL3 in mice results in deficient myeloid reconstitution and depletion of myeloid progenitors, which contrasts with the pro-myeloid differentiation phenotype seen in vitro in human CD34+ cells [43,44,45]. Lee et al. further parsed the role of METTL3 in myeloid cells in vivo by performing myeloid-specific deletion of Mettl3 driven by the LysM promoter. These mice showed no quantitative deficits in peripheral blood, bone marrow, or spleen counts, and myeloid lineage cells retained normal phenotype and function [46••]. As such, it will be important to reconcile the results of experiments across different model systems to fully resolve the role of METTL3 in normal and malignant myelopoiesis.
m6A Readers in Normal Hematopoiesis and Malignancy
YTHDF2 Expands Functional Hematopoietic Stem Cells
While perturbation of the m6A writer proteins has allowed for interrogation of the global effects of RNA methylation on hematopoiesis, studying individual reader proteins allows for further mechanistic dissection of these phenotypes. Mx1-Cre+-Ythdf2fl/fl (Ythdf2−/−) mice have a striking hematopoietic phenotype that differs from m6A methyltransferase-deficient mice in important ways.
Multiple groups have examined Mx1-Cre+-Ythdf2fl/fl mice, with concordant results. Phenotypic HSCs are dramatically expanded in Ythdf2−/− marrow, strongly resembling the phenotype seen in Mx1-Cre-Mettl3fl/fl mice. However, whereas Mettl3-deficient mice experience profound cytopenias and hematopoietic failure, Ythdf2−/− HSCs maintain normal tri-lineage hematopoiesis with only minor changes in peripheral blood counts. Colony-forming capacity is preserved, and functional repopulating HSCs are fourfold enriched in Ythdf2−/− marrow by limiting dilution assays [49, 50]. Ythdf2−/− HSCs also expand more readily in response to stressors such as myeloablative 5-fluorouracil treatment and radiation [49]. While Ythdf2−/− marrow engrafts in equal proportions with wildtype marrow in competitive transplantation, Ythdf2−/− cells exhibit advantages in repopulation of the HSC, myeloid, and erythroid compartments with a relative deficit in T cell engraftment (Table 1) [50••].
The enhanced HSC proliferation with preserved repopulating capacity seen in Mx1-Cre+-Ythdf2fl/fl mice is compelling as it points to a viable avenue for the ex vivo expansion of hematopoietic stem cells for transplant applications. Indeed, in preliminary studies, shRNA-mediated knockdown of YTHDF2 in human CD34+ cord blood results in up to 15-fold expansion of HSCs, providing a promising proof of concept for therapeutic applications [49].
YTHDF2 was identified as a putative oncogene based on its overexpression in bulk AML cells, with enriched expression in leukemia-initiating cell (LIC) subpopulations. Interestingly, while loss of YTHDF2 enhances HSC activity in normal cells, Ythdf2 deletion impairs leukemogenesis in murine AML models and human AML cell lines and reduces LIC frequency. The loss of leukemogenic activity following Ythdf2 deletion in AML is attributable to upregulation of TNF receptor 2, which sensitizes leukemic cells to TNF-induced apoptosis (Table 2) [50••]. The preferential disruption of leukemogenesis with preserved normal hematopoiesis seen in Mx1-Cre+-Ythdf2fl/fl mice identifies another promising opportunity for therapeutic intervention.
The m6A Erasers in Hematopoietic Malignancies
Several observations have implicated either gain or loss of function of the m6A erasers in myeloid malignancies. Both FTO and ALKBH5 were found to be overexpressed in various subtypes of AML, launching investigation into their possible role as oncogenes [52, 53, 55]. In contrast, in an analysis of copy number variation in acute myeloid leukemia, loss of ALKBH5 was proposed to correlate with poorer prognosis [56].
FTO and ALKBH5 could also hypothetically be downregulated in the context of isocitrate dehydrogenase 1 and 2 (IDH1/2) mutations in myeloid malignancies, which occur in approximately 20–30% of patients with MDS or AML [57, 58]. IDH1/2 normally catalyzes the conversion of isocitrate to α-ketoglutarate (α-KG), a necessary substrate of the α-KG-dependent dioxygenase family of enzymes, which includes both FTO and ALKBH5. Substitutions in key catalytic site residues endow mutant IDH1/2 with neomorphic activity, resulting in aberrant conversion of α-KG to 2-hydroxyglutarate (2-HG), which competitively inhibits the α-KG-dependent dioxygenases [59,60,61,62]. It therefore stands to reason that inhibition of FTO or ALKBH5 might be important in the pathophysiology of IDH1/2-mutant leukemias. In keeping with this hypothesis, preliminary experiments showed that overexpression of mutant IDH1/2 in cell lines resulted in a 2-HG-dependent increase in the overall amount of m6A modification.
These preliminary observations provided the basis for further investigation of FTO and ALKBH5 in myeloid malignancies.
FTO in Myeloid Malignancies
In patient samples, FTO is overexpressed in AML subtypes bearing particular translocations or mutations, such as the PML-RARA fusion oncogene. Enforced Fto expression correspondingly enhances leukemogenesis in murine AML models bearing these translocations, while Fto knockdown prolongs survival. This effect is mediated by FTO-mediated downregulation of ASB2 and retinoic acid receptor alpha (RARA), two m6A target genes that are typically upregulated during both normal hematopoiesis and ATRA-mediated differentiation therapy in PML-RARA acute myeloid leukemia [55]. This presents a simple model whereby FTO overexpression regulates the expression of key leukemia genes via particular m6A sites.
The proposed role of FTO in IDH1/2-mutant malignancies is more complex. Su et al. broadly assessed the mechanism of IDH1/2 mutations in human leukemias by directly treating an array of human cell lines with 2-HG. Surprisingly, they found that 2-HG largely inhibits cell growth, viability, and leukemic activity in transplant assays in a subset of human leukemia cell lines, while other cell lines are resistant to this effect. At baseline, 2-HG-sensitive cell lines are distinguishable from resistant lines by increased expression of FTO, globally decreased m6A levels, and downstream activation of MYC with suppression of ASB and RARA, in keeping with previous findings. By contrast, 2-HG-resistant cell lines are characterized by decreased levels of 5-hmC, implicating inhibition of the DNA demethylase ten-eleven translocation 2 (TET2) rather than either of the RNA demethylases [39]. To explain the counterintuitive anti-tumor activity of an apparent oncometabolite, they assessed gene expression in TCGA human AML samples bearing IDH1/2 mutations. In doing so, they found that MYC is overexpressed independently of FTO levels in both IDH1/2-mutant patient samples and 2-HG-resistant cell lines, but not in 2-HG-sensitive cells. Pharmacologic inhibition of MYC restores sensitivity to 2-HG in this context, suggesting that MYC overexpression is necessary to protect IDH1/2-mutant cells and 2-HG-resistant cells from the inherent anti-tumor activity of 2-HG (Table 2) [39].
It will be important to determine to what extent this dichotomy between 2-HG-resistant and 2-HG-sensitive leukemias is consistently recreated across a spectrum of patient samples. In this regard, patient-derived xenograft models may be of interest. Furthermore, there are several established murine models of IDH1/2 mutations which may allow for further mechanistic validation in vivo.
ALKBH5 in Normal Hematopoiesis and Myeloid Malignancies
While a single study showed that loss of ALKBH5 copy number in AML is associated with inferior prognosis, reanalysis of the same patient data by two independent groups ultimately showed a relatively low rate of ALKBH5 deletions. Furthermore, reanalysis showed that ALKBH5 is in fact overexpressed in these AML patient samples, with increased expression correlating to diminished survival [52, 53, 56].
Additional experiments demonstrated that ALKBH5 is preferentially overexpressed within the leukemia-initiating cell compartment and in post-relapse patient samples. ALKBH5 was shown to be necessary for AML cell proliferation and survival in vitro, as well as for leukemogenic activity in vivo. By contrast, ALKBH5 appeared dispensable for normal hematopoiesis, as Alkbh5-deficient mice maintain normal proportions of HSPCs with preserved peripheral blood counts and repopulating potential in competitive transplant assays (Table 1) [52, 53]. This again exposes a leukemia-specific dependence on a component of the m6A machinery which may represent a therapeutic vulnerability.
Preferential expression of ALKBH5 in leukemic cells is facilitated by widespread alterations in chromatin accessibility. The ALKBH5 locus is preferentially depleted of the repressive histone mark H3K9me3 in leukemic cells, with a corresponding enrichment of the H3K9 demethylase KDM4C. Loss of KDM4C concordantly impairs AML cell proliferation and clonogenic capacity, closely resembling ALKBH5 depletion phenotypes [52]. At the level of downstream targets, depletion of ALKBH5 results in downregulation of the pro-proliferative receptor tyrosine kinase AXL, whose transcripts display increased m6A levels after the deletion of ALKBH5 [52]. TACC3 is another m6A-modified transcript whose stability is dependent on ALKBH5 expression. Loss of TACC3 function also resembles loss of ALKBH5, resulting in diminished leukemic activity in vitro and in vivo (Table 2) [53]. These findings highlight an integrated effect of epigenetic chromatin and RNA modifications in myeloid malignancy.
RNA Modification and Immune Function
Recently, m6A RNA modification has been found to play a significant role in immune regulation, broadening our understanding of the function of m6A in the hematopoietic system.
While m6A modification of endogenous RNAs impacts immune cell function and development in several contexts, many studies have also investigated how m6A modification of exogenous viral RNAs modulates the host immune response during infection [63]. The roles of m6A in the immune response have been summarized in depth in a recent review by Shulman and Stern-Ginossar [64]. Here, we will highlight a few relevant studies which offer a general overview of m6A function in immune regulation and how this might globally impact hematopoiesis.
m6A in “Self” Versus “Non-self” Recognition
In certain contexts, the m6A modification has been hypothesized to function in distinguishing RNAs as “self” versus “non-self.” Viral single-stranded or double-stranded RNAs (dsRNAs) are well-established pathogen-associated molecular patterns (PAMPs), which are typically identified as non-self during the innate immune response by pattern recognition receptors (PRRs) such as the DExD/H-box helicase 58 (DDX58, also known as RIG-I) or interferon induced with helicase C domain 1 (IFIH1, also known as MDA5). PRRs in turn activate immune signaling pathways including interferon signaling to coordinate the antiviral response.
Using human metapneumovirus (HMPV) as a model, Lu et al. show that depletion of m6A from viral RNA facilitates their detection by PRRs. Loss of m6A consequently enhances HMPV immunogenicity and attenuates infectivity [65]. In theory, manipulation of m6A could therefore facilitate adjuvant or vaccine development. Chen et al. similarly showed that m6A mediates recognition of circRNAs as “self” in mammalian cells. While m6A-modified circRNA is non-immunogenic, unmodified circRNA activates RIG-I, mitochondrial antiviral signaling (MAVS) protein filamentation, and interferon signaling to generate antigen-specific T and B cell responses [66••].
In our recent paper, we found that loss of m6A in Vav-Cre+-Mettl3fl/fl mice results in the formation of endogenously derived double-stranded RNAs. These dsRNA species are likely detected as “non-self” by PRRs, activating dsRNA response pathways including MDA5-RIG-I, PKR-eIF2a, and 2′-5′-oligoadenylate synthetase-ribonuclease L (OAS-RNaseL). This cell-intrinsic innate immune response contributes to the hematopoietic failure phenotype observed in these mice and can be partially rescued by knockdown or deletion of downstream immune mediators [43••]. Of note, these findings were found in Vav-Cre mice but not described in Mx1-Cre-mediated models of Mettl3 deletion; the latter depend on induction of Cre expression with the dsRNA mimetic pI:pC, which would mask any dsRNA response phenotype in comparisons between control and experimental mice. In keeping with our findings, previous studies have shown that the incorporation of m6A or other modified nucleotides m5C, m5U, s2U, or Ψ in RNA suppresses their detection by Toll-like receptors or RIG-I [67, 68].
The proposed effect of m6A modification on dsRNA formation and recognition resonates with the adenosine-to-inosine (A-to-I) editing function of the adenosine deaminase acting on RNA (ADAR). Interestingly, ADAR1 editing activity prevents the formation of endogenously derived dsRNA and consequent MDA5 activation, and ADAR1 activity is negatively correlated with m6A deposition transcriptome wide [69, 70]. Notably, Adar1-deficient embryos die of hematopoietic failure and exhibit an expansion of functionally defective phenotypic HSCs that closely mimics the phenotype of Mettl3−/− hematopoiesis [71]. It is therefore reasonable to hypothesize that both m6A modification and A-to-I editing prevent dsRNA formation and thereby mediate a common phenotype in hematopoietic stem cells—this possibility will need to be investigated further.
The dsRNA response can play an important role in malignancy, as Ishizuka et al. found that loss of ADAR1 in tumor cells results in a sensitization of cells to immunotherapy, overcoming resistance to checkpoint blockade. This effect is mediated in part by the formation of endogenous dsRNAs which triggers a deleterious immune response [72]. In this way, suppression of endogenous dsRNA formation can be considered a mechanism of immune evasion in cancers. It will be of interest to determine whether perturbation of m6A could similarly promote dsRNA formation and anti-tumor effects in solid or hematologic malignancies.
m6A in Immune Cell Signaling and Development
The m6A modification has also been shown to modulate the development and function of immune cells. Li et al. showed that m6A modification on mRNAs controls T cell homeostasis by regulation of Socs mRNA stability in CD4-Cre-Mettl3fl/fl mice [73]. The m6A modification level is increased during dendritic cell (DC) maturation and is required for DC maturation during an immune response to lipopolysaccharide (LPS) in a YTHDF1-dependent manner [74]. However, as discussed above, Lee et al. have shown that m6A RNA modification is not required for the maintenance or function of mature myeloid cells in Lyzm-Cre-Mettl3fl/fl mice [46••]. These findings demonstrate the importance of m6A for immune cell development and function and further illustrate the context dependence of m6A function in hematopoiesis and the immune response.
Other RNA Modifications in Hematopoiesis
In this review, we have focused on the role of m6A in hematopoiesis as it is the most common mRNA modification and its function has been the most extensively characterized. However, additional RNA modifications have also been found to influence hematopoiesis. While the tumor suppressor TET2 is known to suppress leukemogenesis by hydroxylating DNA 5-mC to 5hmC, TET2 was also recently found to promote myelopoiesis in the context of infection by demethylating RNA 5-mC [75]. Several tRNA modifications impact hematopoiesis as well. Pseudouridylation of tRNA governs HSC commitment and influences disease pathophysiology in MDS, and the m1A modification modulates erythropoiesis [76, 77]. DNMT2 mediates methylation of cytosine on tRNAAsp and is essential for hematopoiesis [78]. In general, tRNA metabolism can influence HSPC quiescence and other aspects of hematopoiesis, and it is possible that these functions are dependent on specific modifications [79, 80].
Future Directions
The many studies presented here demonstrate the essential function of m6A RNA modification in the hematopoietic system. Nevertheless, many questions remain regarding the relationship between m6A and malignant hematopoiesis.
First, it is notable that all components of the m6A machinery—writers, readers, and erasers—have now been found to be overexpressed in various subtypes of AML. For instance, METTL3, METTL14, YTHDF2, ALKBH5, and FTO have all been reported to be upregulated in MLL-rearranged leukemias, and perturbation of any of these proteins disrupts leukemogenicity. It therefore appears that a model whereby leukemia is promoted via simple gain or loss of total m6A content is unlikely. Instead, global upregulation of the m6A machinery in leukemia might be indicative of a generalized state of increased RNA metabolism and turnover. Under these conditions, compromising any element of the m6A machinery might be deleterious. Alternatively, each individual component of the m6A machinery may govern distinct leukemogenic mechanisms. It will be important to determine which of these regulators ultimately represent the best therapeutic target.
Other elements of the m6A machinery are likely to be mechanistically relevant in malignancy. The methyltransferase adapter RBM15 is a component of the fusion oncogene RBM15-MKL in acute megakaryoblastic leukemia [81]. Interestingly, Mettl3 deletion results in the accumulation of megakaryocytic progenitors in mice, directly implicating m6A in the regulation of this lineage [46••]. It remains to be seen whether RBM15-MKL promotes leukemogenesis via an m6A-mediated mechanism. WTAP has been described as an oncogene in AML, but this function has not yet been tied directly to m6A. Interestingly, the m6A-dependent lncRNA Xist is essential for normal hematopoiesis and functions as a tumor suppressor in hematologic malignancies [82]. It is possible that the role of m6A as a regulator of HSC cell fate decisions is partially mediated by effects on Xist or its reader YTHDC1.
Conclusions
The RNA modification m6A affects RNA function through a variety of mechanisms. Beyond direct effects on transcript stability and translational efficiency, the m6A modification can influence transcript conformation and recognition by immune regulators and interact with other epigenetic modifiers to regulate global chromatin structure. Further investigation will be necessary to determine the extent to which these different effects contribute to the regulation of hematopoiesis and leukemogenesis. It is clear that RNA modifications offers an avenue for the development of therapeutics for the ex vivo expansion of hematopoietic stem cells as well for the treatment of myeloid malignancies.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46(D1):D303–D7. https://doi.org/10.1093/nar/gkx1030.
Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169(7):1187–200. https://doi.org/10.1016/j.cell.2017.05.045.
Perry RP, Kelley DE. Existence of methylated messenger RNA in mouse L cells. Cell. 1974;1(1):37–42. https://doi.org/10.1016/0092-8674(74)90153-6.
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71(10):3971–5.
Adams JM, Cory S. Modified nucleosides and bizarre 5′-termini in mouse myeloma mRNA. Nature. 1975;255(5503):28–33. https://doi.org/10.1038/255028a0.
Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20(10):608–24. https://doi.org/10.1038/s41580-019-0168-5.
• Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–6. https://doi.org/10.1038/nature11112One of two initial papers describing the transcriptome-wide distribution of m6A using immunoprecipitation coupled with RNA sequencing.
• Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell. 2012;149(7):1635–46. https://doi.org/10.1016/j.cell.2012.05.003One of two initial papers describing the transcriptome-wide distribution of m6A using immunoprecipitation coupled with RNA sequencing.
• Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–20. https://doi.org/10.1038/nature12730Demonstrates the generalized function of the reader YTHDF1 in transcript degradation.
• Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–99. https://doi.org/10.1016/j.cell.2015.05.014Demonstrates the function of the reader YTHDF2 in promoting translation efficiency, and characterizes the integrated effects of both YTHDF1 & YTHDF2 on gene expression.
Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537(7620):369–73. https://doi.org/10.1038/nature19342.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518(7540):560–4. https://doi.org/10.1038/nature14234.
Abakir A, Giles TC, Cristini A, Foster JM, Dai N, Starczak M, et al. N(6)-methyladenosine regulates the stability of RNA:DNA hybrids in human cells. Nat Genet. 2020;52(1):48–55. https://doi.org/10.1038/s41588-019-0549-x.
Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vagbo CB, Geula S, et al. m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017;31(10):990–1006. https://doi.org/10.1101/gad.301036.117.
Rosa-Mercado NA, Withers JB, Steitz JA. Settling the m(6)A debate: methylation of mature mRNA is not dynamic but accelerates turnover. Genes Dev. 2017;31(10):957–8. https://doi.org/10.1101/gad.302695.117.
•• Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15(6):707–19. https://doi.org/10.1016/j.stem.2014.09.019Demonstrates the role of m6A as a critical regulator of self-renewal in ESCs.
•• Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science. 2015;347(6225):1002–6. https://doi.org/10.1126/science.1261417Further characterizes Mettl3−/− ESCs, highlighting reinforced ‘naïve’ pluripotency.
Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, et al. Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature. 2019;567(7748):414–9. https://doi.org/10.1038/s41586-019-1016-7.
Knuckles P, Carl SH, Musheev M, Niehrs C, Wenger A, Buhler M. RNA fate determination through cotranscriptional adenosine methylation and microprocessor binding. Nat Struct Mol Biol. 2017;24(7):561–9. https://doi.org/10.1038/nsmb.3419.
• Liu J, Dou X, Chen C, Chen C, Liu C, Xu MM, et al. N (6)-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science. 2020;367(6477):580–6. https://doi.org/10.1126/science.aay6018Demonstrates the influence of m6A methylation on nascent chromosome-associated RNA transcripts on the expression and recruitment of epigenetic modifiers to chromatin, with consequent effects on chromatin accessibility. This reflects the integrated roles of RNA and chromatin epigenetic modifications in the regulation of gene expression.
van Tran N, Ernst FGM, Hawley BR, Zorbas C, Ulryck N, Hackert P, et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019;47(15):7719–33. https://doi.org/10.1093/nar/gkz619.
Mendel M, Chen KM, Homolka D, Gos P, Pandey RR, McCarthy AA, et al. Methylation of structured RNA by the m(6)A writer METTL16 is essential for mouse embryonic development. Mol Cell. 2018;71(6):986–1000 e11. https://doi.org/10.1016/j.molcel.2018.08.004.
Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, et al. The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell. 2017;169(5):824–35 e14. https://doi.org/10.1016/j.cell.2017.05.003.
Ma H, Wang X, Cai J, Dai Q, Natchiar SK, Lv R, et al. N(6-)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol. 2019;15(1):88–94. https://doi.org/10.1038/s41589-018-0184-3.
Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27(9):1115–27. https://doi.org/10.1038/cr.2017.99.
Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, et al. Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res. 2017;27(3):444–7. https://doi.org/10.1038/cr.2017.10.
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27(3):315–28. https://doi.org/10.1038/cr.2017.15.
Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, et al. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 2017;27(5):626–41. https://doi.org/10.1038/cr.2017.31.
Di Timoteo G, Dattilo D, Centron-Broco A, Colantoni A, Guarnacci M, Rossi F, et al. Modulation of circRNA metabolism by m(6)A modification. Cell Rep. 2020;31(6):107641. https://doi.org/10.1016/j.celrep.2020.107641.
Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61(4):507–19. https://doi.org/10.1016/j.molcel.2016.01.012.
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5' UTR m(6)A promotes cap-independent translation. Cell. 2015;163(4):999–1010. https://doi.org/10.1016/j.cell.2015.10.012.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285–95. https://doi.org/10.1038/s41556-018-0045-z.
Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. 2015;162(6):1299–308. https://doi.org/10.1016/j.cell.2015.08.011.
Alarcon CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519(7544):482–5. https://doi.org/10.1038/nature14281.
Zhou KI, Shi H, Lyu R, Wylder AC, Matuszek Z, Pan JN, et al. Regulation of co-transcriptional pre-mRNA splicing by m(6)A through the low-complexity protein hnRNPG. Mol Cell. 2019;76(1):70–81 e9. https://doi.org/10.1016/j.molcel.2019.07.005.
Meyer KD, Jaffrey SR. Rethinking m(6)A readers, writers, and erasers. Annu Rev Cell Dev Biol. 2017;33:319–42. https://doi.org/10.1146/annurev-cellbio-100616-060758.
Zheng GQ, Dahl JA, Niu YM, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29. https://doi.org/10.1016/j.molcel.2012.10.015.
Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature. 2017;541(7637):371–5. https://doi.org/10.1038/nature21022.
Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. 2018;172(1–2):90–105 e23. https://doi.org/10.1016/j.cell.2017.11.031.
Akichika S, Hirano S, Shichino Y, Suzuki T, Nishimasu H, Ishitani R, et al. Cap-specific terminal N (6)-methylation of RNA by an RNA polymerase II-associated methyltransferase. Science. 2019;363(6423). https://doi.org/10.1126/science.aav0080.
•• Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma D, et al. m6A modulates haematopoietic stem and progenitor cell specification. Nature. 2017;549(7671):273–6. https://doi.org/10.1038/nature23883The first study to demonstrate a hematopoietic phenotype caused by deletion of the m6A methyltransferase.
Lv J, Zhang Y, Gao S, Zhang C, Chen Y, Li W, et al. Endothelial-specific m(6)A modulates mouse hematopoietic stem and progenitor cell development via notch signaling. Cell Res. 2018;28(2):249–52. https://doi.org/10.1038/cr.2017.143.
•• Gao Y, Vasic R, Song Y, Teng R, Liu C, Gbyli R, et al. m(6)A Modification prevents formation of endogenous double-stranded RNAs and deleterious innate immune responses during hematopoietic development. Immunity. 2020;52:1–15. https://doi.org/10.1016/j.immuni.2020.05.003In this work, we show that loss of m6A in HSCs results in the formation of endogenously derived dsRNA which mediate a deleterious inflammatory response resulting in deficient hematopoiesis.
Yao QJ, Sang L, Lin M, Yin X, Dong W, Gong Y, et al. Mettl3-Mettl14 methyltransferase complex regulates the quiescence of adult hematopoietic stem cells. Cell Res. 2018;28(9):952–4. https://doi.org/10.1038/s41422-018-0062-2.
•• Cheng Y, Luo H, Izzo F, Pickering BF, Nguyen D, Myers R, et al. m(6)A RNA methylation maintains hematopoietic stem cell identity and symmetric commitment. Cell Rep. 2019;28(7):1703–16 e6. https://doi.org/10.1016/j.celrep.2019.07.032Provides a detailed single-cell RNA-seq analysis of Mettl3−/− HSCs, demonstrating destabilized HSC identity and impaired function in transplant assays.
•• Lee H, Bao S, Qian Y, Geula S, Leslie J, Zhang C, et al. Stage-specific requirement for Mettl3-dependent m(6)A mRNA methylation during haematopoietic stem cell differentiation. Nat Cell Biol. 2019;21(6):700–9. https://doi.org/10.1038/s41556-019-0318-1Characterizes the effects of tissue-specific Mettl3 deletion in both HSCs and mature myeloid cells.
Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes Leukemogenesis via mRNA m(6)A modification. Cell Stem Cell. 2018;22(2):191–205 e9. https://doi.org/10.1016/j.stem.2017.11.016.
Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23(11):1369–76. https://doi.org/10.1038/nm.4416.
Li Z, Qian P, Shao W, Shi H, He XC, Gogol M, et al. Suppression of m(6)A reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res. 2018;28(9):904–17. https://doi.org/10.1038/s41422-018-0072-0.
•• Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I, et al. Targeting the RNA m(6)A reader YTHDF2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell. 2019;25(1):137–48 e6. https://doi.org/10.1016/j.stem.2019.03.021Characterizes the selective dependence of myeloid leukemia on YTHDF2 and characterizes the enhanced self-renewal and preserved repopulating capacity of Ythdf2−/− HSCs.
Wang H, Zuo H, Liu J, Wen F, Gao Y, Zhu X, et al. Loss of YTHDF2-mediated m(6)A-dependent mRNA clearance facilitates hematopoietic stem cell regeneration. Cell Res. 2018;28(10):1035–8. https://doi.org/10.1038/s41422-018-0082-y.
Wang J, Li Y, Wang P, Han G, Zhang T, Chang J, et al. Leukemogenic chromatin alterations promote AML leukemia stem cells via a KDM4C-ALKBH5-AXL signaling axis. Cell Stem Cell. 2020;27:81–97.e8. https://doi.org/10.1016/j.stem.2020.04.001.
Shen C, Sheng Y, Zhu AC, Robinson S, Jiang X, Dong L, et al. RNA demethylase ALKBH5 selectively promotes tumorigenesis and cancer stem cell self-renewal in acute myeloid leukemia. Cell Stem Cell. 2020;27:64–80.e9. https://doi.org/10.1016/j.stem.2020.04.009.
•• Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature. 2017;552(7683):126–31. https://doi.org/10.1038/nature24678Demonstrates recruitment of METTL3 to promoters, and disease relevance of this mechanism in the context of myeloid leukemias.
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell. 2017;31(1):127–41. https://doi.org/10.1016/j.ccell.2016.11.017.
Kwok CT, Marshall AD, Rasko JE, Wong JJ. Genetic alterations of m(6)A regulators predict poorer survival in acute myeloid leukemia. J Hematol Oncol. 2017;10(1):39. https://doi.org/10.1186/s13045-017-0410-6.
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–66. https://doi.org/10.1056/NEJMoa0903840.
Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol. 2010;28(14):2348–55. https://doi.org/10.1200/JCO.2009.27.3730.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–67. https://doi.org/10.1016/j.ccr.2010.11.015.
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–44. https://doi.org/10.1038/nature08617.
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225–34. https://doi.org/10.1016/j.ccr.2010.01.020.
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–8. https://doi.org/10.1038/nature10860.
Williams GD, Gokhale NS, Horner SM. Regulation of viral infection by the RNA modification N6-methyladenosine. Annu Rev Virol. 2019;6(1):235–53. https://doi.org/10.1146/annurev-virology-092818-015559.
Shulman Z, Stern-Ginossar N. The RNA modification N(6)-methyladenosine as a novel regulator of the immune system. Nat Immunol. 2020;21(5):501–12. https://doi.org/10.1038/s41590-020-0650-4.
Lu M, Zhang Z, Xue M, Zhao BS, Harder O, Li A, et al. N(6)-methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat Microbiol. 2020;5(4):584–98. https://doi.org/10.1038/s41564-019-0653-9.
•• Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L, et al. N6-methyladenosine modification controls circular RNA immunity. Mol Cell. 2019. https://doi.org/10.1016/j.molcel.2019.07.016Demonstrates the role of m6A in mediating detection of double-stranded circRNA as “self” or foreign.
Kariko K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23(2):165–75. https://doi.org/10.1016/j.immuni.2005.06.008.
Durbin AF, Wang C, Marcotrigiano J, Gehrke L. RNAs containing modified nucleotides fail to trigger RIG-I conformational changes for innate immune signaling. mBio. 2016;7(5). https://doi.org/10.1128/mBio.00833-16.
Xiang JF, Yang Q, Liu CX, Wu M, Chen LL, Yang L. N(6)-methyladenosines modulate A-to-I RNA editing. Mol Cell. 2018;69(1):126–35 e6. https://doi.org/10.1016/j.molcel.2017.12.006.
Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015;349(6252):1115–20. https://doi.org/10.1126/science.aac7049.
Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol. 2009;10(1):109–15. https://doi.org/10.1038/ni.1680.
Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature. 2019;565(7737):43–8. https://doi.org/10.1038/s41586-018-0768-9.
Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, et al. m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. 2017;548(7667):338–42. https://doi.org/10.1038/nature23450.
Wang H, Hu X, Huang M, Liu J, Gu Y, Ma L, et al. Mettl3-mediated mRNA m(6)A methylation promotes dendritic cell activation. Nat Commun. 2019;10(1):1898. https://doi.org/10.1038/s41467-019-09903-6.
Shen Q, Zhang Q, Shi Y, Shi Q, Jiang Y, Gu Y, et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature. 2018;554(7690):123–7. https://doi.org/10.1038/nature25434.
Murakami S, Suzuki T, Yokoyama W, Yagi S, Matsumura K, Nakajima Y, et al. Nucleomethylin deficiency impairs embryonic erythropoiesis. J Biochem. 2018;163(5):413–23. https://doi.org/10.1093/jb/mvx086.
Guzzi N, Ciesla M, Ngoc PCT, Lang S, Arora S, Dimitriou M, et al. Pseudouridylation of tRNA-derived fragments steers translational control in stem cells. Cell. 2018;173(5):1204–16 e26. https://doi.org/10.1016/j.cell.2018.03.008.
Tuorto F, Herbst F, Alerasool N, Bender S, Popp O, Federico G, et al. The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. Embo J. 2015;34(18):2350–62. https://doi.org/10.15252/embj.201591382.
Goncalves KA, Silberstein L, Li S, Severe N, Hu MG, Yang H, et al. Angiogenin promotes hematopoietic regeneration by dichotomously regulating quiescence of stem and progenitor cells. Cell. 2016;166(4):894–906. https://doi.org/10.1016/j.cell.2016.06.042.
Kanaji T, Vo MN, Kanaji S, Zarpellon A, Shapiro R, Morodomi Y, et al. Tyrosyl-tRNA synthetase stimulates thrombopoietin-independent hematopoiesis accelerating recovery from thrombocytopenia. Proc Natl Acad Sci U S A. 2018;115(35):E8228–E35. https://doi.org/10.1073/pnas.1807000115.
de Rooij JD, Branstetter C, Ma J, Li Y, Walsh MP, Cheng J, et al. Pediatric non-down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet. 2017;49(3):451–6. https://doi.org/10.1038/ng.3772.
Yildirim E, Kirby JE, Brown DE, Mercier FE, Sadreyev RI, Scadden DT, et al. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell. 2013;152(4):727–42. https://doi.org/10.1016/j.cell.2013.01.034.
Acknowledgments
We apologize to our colleagues whose original work could not be cited due to space limitations.
Funding
This study was supported in part by the State of Connecticut under the Regenerative Medicine Research Fund (to S.H.; its contents are solely the responsibility of the authors and do not necessarily represent the official views of the State of Connecticut or Connecticut Innovations), the NIH/NIDDK R01DK102792 (to S.H.), the NCI R01CA222518 (to S.H.), the NIH/NIDDK U54DK106857 (Yale Cooperative Center of Excellence in Hematology; S.H.), and The Frederick A. Deluca Foundation (to S.H.). Y.G. was supported by the James Hudson Brown-Alexander Brown Coxe Postdoctoral Fellowships. R.V. was supported by the American Society of Hematology Physician Scientist Career Development Award and the William U. Gardner Memorial Student Research Fellowship. C.L. was supported by the Exchange Undergraduate Scholarship of Wuhan University, China.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Classical Signalling Pathways
Rights and permissions
About this article

Cite this article
Vasic, R., Gao, Y., Liu, C. et al. The Role of RNA Epigenetic Modification in Normal and Malignant Hematopoiesis. Curr Stem Cell Rep 6, 144–155 (2020). https://doi.org/10.1007/s40778-020-00178-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-020-00178-y