
Abstract
Introduction
Upadacitinib (UPA), a selective, reversible, oral Janus kinase (JAK)-1 inhibitor, was approved in 2019 in Canada for the treatment of adults with moderately to severely active rheumatoid arthritis (RA). This phase 4 prospective study aimed to characterise the effectiveness of UPA in the real-world population of patients with RA.
Methods
Adults with RA who initiated treatment with once daily UPA (15 mg) and enrolled in the Canadian Real-Life post-marketing Observational Study assessing the Effectiveness of UPadacitinib for treating rheumatoid arthritis (CLOSE-UP) and who completed a 6-month assessment as of 28 February 2023 were included. The primary endpoint of the CLOSE-UP study is the proportion of patients achieving a Disease Activity Score-28 Joint Count C-reactive protein (DAS28-CRP) < 2.6 at 6 months. Data was collected at routine visits. Data analysed and summarised descriptively for the overall interim population and for subgroups based on prior therapy included remission or low disease activity, patient-reported outcomes (PROs), and adverse events.
Results
A total of 392 patients were included in the interim analysis. Overall, 63.5% (191/301) of patients achieved a DAS28-CRP score < 2.6 at month 6, with similar rates observed for all subgroups analysed according to prior therapy including those with prior JAK inhibitor exposure (range 57.4–71.0%), and in patients who received UPA monotherapy (71.6% [48/67]). Early (month 3) and sustained improvements up to 6 months were observed for all PROs. The safety profile was consistent with previous reports.
Conclusion
Real-world improvements in disease activity and PROs in response to UPA treatment were consistent with clinical trial data across a range of Canadian patients with prior therapy exposure and with UPA monotherapy, with an overall favourable benefit–risk profile.
Trial Registration
NCT04574492.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
The safety and efficacy of upadacitinib for the treatment of adult patients with moderate-to-severe rheumatoid arthritis were previously demonstrated in a comprehensive phase 3 clinical development program. |
The generalisability of the data from the pivotal phase 3 program to clinical practice remained to be determined. |
The phase 4 Canadian Real-Life post-marketing Observational Study assessing the Effectiveness of UPadacitinib for treating rheumatoid arthritis (CLOSE-UP) was undertaken to address this knowledge gap. |
What was learned from the study? |
This interim analysis of the ongoing CLOSE-UP study confirmed the real-world safety and effectiveness of upadacitinib in a broad range of Canadians living with rheumatoid arthritis, with similar outcomes observed regardless of prior treatment (including Janus kinase inhibitors) and for upadacitinib monotherapy. |
Introduction
Treat-to-target is standard of care for rheumatoid arthritis (RA). Treatment goals include remission, or, when remission is not attainable, low disease activity (LDA) [1, 2]—outcomes that are associated with better health-related quality of life and functioning, low disability, and minimisation of radiologic damage [3, 4]. Early diagnosis, treatment with disease-modifying antirheumatic drugs (DMARDs), and therapy intensification and escalation are the cornerstones to achieving these goals. Despite aggressive clinical management, including the availability of numerous pharmacological options (conventional synthetic DMARDs [csDMARDs], tumour necrosis factor [TNF] and non-TNF biologic DMARDs [bDMARDs], and targeted synthetic DMARDs [tsDMARDs]), many patients with RA do not reach or maintain recommended treatment targets [3, 4].
Upadacitinib (UPA), a selective, reversible, oral Janus kinase (JAK)-1 inhibitor, was approved in 2019 in Canada for the treatment of adults with moderately to severely active disease at a daily dose of 15 mg, either alone or in combination with methotrexate (MTX) or other csDMARDs. Regulatory approval was based on five pivotal phase 3 clinical trials [5,6,7,8,9] that demonstrated the safety and efficacy of once daily treatment in a broad spectrum of patients including those who were naïve to MTX, had an inadequate response to MTX and/or other csDMARDs, and who had an inadequate response to or were intolerant to one or more bDMARDs.
Although randomised controlled trials (RCTs) are the gold standard for assessing the safety and efficacy of potential new therapies, their results may not be generalisable to clinical practice. Rigorous and non-standardised eligibility criteria may limit external validity, and systematic differences between the characteristics of patients enrolled in RCTs and the real-world population may lead to disparities between efficacy observed in RCTs and real-world effectiveness. Furthermore, RCTs may exclude patient populations who might nevertheless benefit from treatment with an investigational therapy. For example, patients with prior exposure to another tsDMARD (e.g. JAK inhibitor) were excluded from the phase 3 UPA trials. The safety and efficacy of UPA in this patient population has not been formally prospectively studied.
Given these unknowns, a phase 4 study (Canadian Real-Life post-marketing Observational Study assessing the Effectiveness of UPadacitinib for treating rheumatoid arthritis [CLOSE-UP]) evaluating the effectiveness of UPA, including in patients previously treated with tsDMARDS, was undertaken. We performed an interim analysis that included all patients in the CLOSE-UP study who had completed (or discontinued the study prior to) their 6-month assessment to characterise the effectiveness and safety of UPA in the real-world setting.
Methods
Study Design and Patient Population
CLOSE-UP (NCT04574492) is an ongoing, fully enrolled (N = 414), prospective cohort, multicentre (39 sites), open-label study in Canadian patients with RA. Participating investigators follow routine clinical care. UPA is prescribed according to the Canadian product monograph, and adjustment of other treatments, including csDMARDs and corticosteroids is at the discretion of the treating investigator. Enrolled patients are followed for 24 months. Discontinuation of UPA is not a criterion for early withdrawal. The primary endpoint of the study is the proportion of patients who achieve a Disease Activity Score-28 Joint Count C-reactive protein (DAS28-CRP) [10] < 2.6 at 6 months following initiation of UPA therapy.
This interim analysis included all patients who received at least one dose of UPA and completed their 6-month assessment, or who discontinued treatment prior to their 6-month assessment as of 28 February 2023 (N = 392; see Fig. S1 in the Supplementary Material). Eligible patients included those aged ≥ 18 with moderate to severe RA based on the investigator’s diagnosis and for whom treatment with UPA was initiated independent from study participation and prior to determination of patient eligibility. Additional eligibility criteria included prior treatment with a csDMARD (methotrexate, hydroxychloroquine, sulfasalazine, leflunomide, chloroquine, gold salts) and the ability to provide informed consent. Patients with prior treatment with a csDMARD further included those who had no prior treatment with either a bDMARD (etanercept, adalimumab, certolizumab, golimumab, infliximab, rituximab, tocilizumab, sarilumab, abatacept, anakinra) or tsDMARD (tofacitinib, baricitinib), subsequently referred to as the “bio-naïve” subgroup), those with no prior treatment with a tsDMARD and prior treatment with no more than two bDMARDs (“bio-experienced” subgroup), and those with prior treatment with a tsDMARD and no more than one bDMARD (“prior tsDMARD experienced” subgroup). Patients diagnosed with a rheumatic disease other than RA or with juvenile RA, those currently participating in, or with prior exposure to a tsDMARD in a clinical trial, those who received a bDMARD subsequent to treatment with a tsDMARD, and those with any condition that prohibited study participation or obscured treatment assessment based on the opinion of the investigator were ineligible.
Patients were enrolled consecutively. Enrolment was monitored to balance region and immediate previous DMARD therapy.
Ethical Approval
The study is conducted in accordance with the Declaration of Helsinki and International Conference on Harmonisation Guidelines for Good Clinical Practice. Institutional review board/independent ethics committee approval was obtained for all participating centres (Table S1 in the Supplementary Material). All patients provided informed consent to participate in the study.
Assessments and Outcomes
Data were collected from patients’ charts, source documents, physician assessments, and self-administered patient questionnaires during routine visits at baseline, and months 3 and 6. Given the observational nature of this ongoing study, no study-specific visits are required. Failure to observe the data collection schedule was not a protocol violation.
Clinical assessments included swollen (66-joint) and tender (68-joint) joint count (SJC66 and TJC68), and a physician’s global assessment of disease activity (measured on a 100-mm visual analogue scale [VAS]) at baseline, month 3, and month 6. Patient-reported assessments at the same time points included pain (measured on a 100-mm VAS), fatigue (based on the Functional Assessment of Chronic Illness Therapy-Fatigue [FACIT-F]) [11], functioning (based on the Health Assessment Questionnaire-Disability Index [HAQ-DI]) [12], duration and severity (measured on a 100-mm VAS) of joint stiffness, and a patient assessment of global disease activity (PtGA; measured on a 100-mm VAS).
Clinical outcomes included the proportion of patients achieving DAS28-CRP < 2.6 and ≤ 3.2, clinical remission defined by a Clinical Disease Activity Index (CDAI) [13] score ≤ 2.8 or Simplified Disease Activity Index (SDAI) [14] score ≤ 3.3, and LDA defined by CDAI ≤ 10 or SDAI ≤ 11. Remission based on the original American College of Rheumatology (ACR)-European League Against Rheumatism (EULAR) Boolean definition (TJC ≤ 1 based on 28 joints, SJC ≤ 1 based on 28 joints, CRP concentration ≤ 1 mg/dL, and PtGA ≤ 1 on a 0–10 scale) and the ACR-EULAR Boolean 2.0 definition (modified to include a PtGA ≤ 2) was also determined [15, 16].
Serum C-reactive protein (CRP) concentration was recorded at every visit, as was erythrocyte sedimentation rate if performed as a routine assessment.
Adverse events, adverse events of special interest (herpes zoster, major adverse cardiovascular events, venous thromboembolism, infections, serious infections, malignancy [including non-melanoma skin cancer], death), unusual failure in efficacy, and pregnancy were recorded at all visits.
Statistical Analyses
The efficacy and safety populations included patients who received at least one dose of UPA and had a month 6 assessment or who discontinued before month 6. Baseline characteristics and adverse events (coded in Medical Dictionary for Regulatory Activities version 23.1) in the overall interim analysis population and in subgroups according to prior treatment were summarised descriptively. Adverse events of special interest were assessed separately.
Summary statistics were used to describe continuous endpoints, and counts and percentages were used for categorical endpoints, which were also analysed according to exposure to prior treatment and whether UPA was received as monotherapy versus with concomitant csDMARDs. Data were presented as observed. No formal statistical testing was performed. Missing data were not imputed.
Results
Baseline Characteristics and Demographics
As of 28 February 2023, 356 (90.8%) patients had completed their 6-month assessment and 36 (9.2%) had discontinued the CLOSE-UP study prior to this assessment (Fig. S1 in the Supplementary Material). Of these 392 patients, 186 (47.4%) were bio-naïve, 164 (41.8%) were bio-experienced (105 [64.0%] one prior bDMARD and 59 [36.0%] two prior bDMARDs), and 42 (10.7%) were tsDMARD- experienced (28 [66.7%] no prior bDMARDs and 14 [33.3%] one prior bDMARD). Discontinuation of prior bDMARD or tsDMARD in the bio-experienced and tsDMARD-experienced subgroups was due primarily to efficacy-related reasons (no, insufficient, or loss of, clinical response); 130/157 [82.8%] and 33/37 [89.2%], respectively (Table 1).
Most patients in the interim analysis population were female (307 [78.3%]). Mean (standard deviation) age was 60.4 (11.5) years, with 325 (82.9%) of the patients ≥ 50 years of age and 146 (37.2%) ≥ 65 years of age. Mean disease duration was 9.98 (9.41) years. Mean baseline scores on all clinical measures of RA disease activity were consistent with at least moderate (mean DAS28-CRP score 4.39 [0.96]) or high (mean CDAI score 29.34 [11.43]) activity in the overall population as well as in the subgroups analysed according to prior therapy exposure. Mean TJC68 and SJC66 were 14.4 (10.1) and 12.8 (7.7). Approximately half (195 [49.7%]) of the patients were on concomitant (continued or newly received at or after initiation of UPA) MTX, and one-quarter (99 [25.2%]) initiated UPA as monotherapy. The proportion of patients with cardiovascular risk factors at baseline was 67.8% (253/373).
Clinical Outcomes
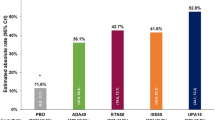
The proportion of patients in the overall population who achieved DAS28-CRP < 2.6 (primary endpoint of the study) at month 6 was 63.5% (191/301), with similar rates observed for this outcome at month 6 in all subgroups analysed according to prior therapy exposure (range 57.4–71.0%) (Fig. 1i). The proportion of patients achieving a DAS28-CRP ≤ 3.2 at month 6 in the overall population was 77.1% (232/301), again, with similar rates observed for all subgroups analysed according to prior therapy exposure (range 74.6–80.6%). Importantly, greater than 50% of patients overall (54.3% [164/302]) and in all subgroups analysed according to prior therapy exposure (range 51.6–56.7%) had already achieved DAS28-CRP < 2.6, and greater than 60% of patients overall (67.5% [204/302]) and in all subgroups analysed according to prior therapy exposure (range 63.6–71.3%) had already achieved a DAS28-CRP ≤ 3.2 by their month 3 assessment. Mean DAS28-CRP in the overall population decreased 1.99 (1.47) points (n = 295) from baseline to month 6, with similar decreases observed in all subgroups analysed according to prior therapy exposure (Table S2).

Proportion of patients in the overall interim analysis CLOSE-UP population and in subgroups of patients based on exposure to prior therapy achieving (i) DAS28-CRP < 2.6 (solid bars) or DAS28-CRP ≤ 3.2 (shaded bars), or remission (solid bars) or low disease activity (shaded bars) defined according to the (ii) CDAI, (iii) SDAI, and (iv) ACR-EULAR Boolean 2.0 criteria. Abbreviations: ACR-EULAR, American College of Rheumatology-European League Against Rheumatism; CDAI, Clinical Disease Activity Index; DAS28-CRP, Disease Activity Score-28 Joint Count C-reactive protein; SDAI, Simplified Disease Activity Index; tsDMARD, targeted synthetic disease-modifying antirheumatic drugs
The proportions of patients in the overall population who achieved CDAI-defined remission at months 3 and 6 were 20.9% (70/335) and 29.0% (92/317) and who achieved CDAI-defined remission or LDA at the same time points were 55.5% (186/335) and 66.6% (211/317) (Fig. 1ii). Generally similar rates for these outcomes were also observed at months 3 and 6 for the subgroups analysed according to prior therapy; CDAI-defined remission rates ranged from 13.2% to 22.6% at month 3 and from 25.7% to 30.2% at month 6, and CDAI-defined remission or LDA rates ranged from 51.9% to 59.1% at month 3 and from 60.5% to 71.2% at month 6. Mean CDAI scores in the overall population decreased 19.76 (14.44) points (29.34 [11.43] to 9.63 [10.48]) from baseline to month 6, with similar decreases observed in all subgroups analysed according to prior therapy exposure (Table S2). Outcomes for the SDAI were comparable to the CDAI (Fig. 1iii).
The proportions of patients in the overall population who achieved remission according to the former ACR-EULAR Boolean definition (PtGA ≤ 1) at months 3 and 6 were 9.9% (36/364) and 17.2% (61/355), whereas the proportions meeting remission according to the ACR-EULAR Boolean 2.0 definition (PtGA ≤ 2) at the same time points were 15.1% (55/364) and 24.2% (86/355) (Fig. 1iv).
Swollen (66) and Tender Joint (68) Counts and Physician Global Assessment
Improvements from baseline were observed at months 3 and 6 in the mean TJC68 and SJC66 and the mean physician global assessment of disease activity score for the overall population and all subgroups analysed according to prior therapy exposure (Table 2). The mean TJC68 decreased at months 3 and 6 in the overall population from 14.4 (10.10) at baseline to 5.8 (8.3) and 4.7 (8.8), respectively, as did the mean SJC66 (from 12.8 [7.7] at baseline to 4.7 [6.0] and 3.8 [6.3]). Mean scores on the physician global assessment of disease activity also decreased at months 3 and 6 in the overall population from 58.79 (20.50) to 21.95 (21.77) and 17.97 (19.79), respectively.
Patient-Reported Outcomes
Improvements in mean pain, FACIT-F, and HAQ-DI scores, as well as in the mean duration and severity of morning stiffness and mean PtGA were observed as early as month 3 and were generally maintained at month 6 in the overall population and all subgroups analysed according to prior therapy exposure (Table 3).
A total of 46.2% (160/346) and 50.7% (171/337) of patients in the overall population reported a 50% reduction in their pain score at months 3 and 6. A minimal clinically important difference (MCID) in patient-reported fatigue (defined as a FACIT-F subscale change ≥ 3.56) [17] was achieved in 60.7% (210/346) and 61.4% (204/332) of patients at months 3 and 6, respectively, and an MCID in patient-reported functioning (HAQ-DI change ≥ -0.22) [18] was achieved in 60.5% (207/342) and 62.6% (206/329) of patients at the same time points.
Efficacy in Patients Treated with UPA Monotherapy
Baseline characteristics of patients who initiated UPA as monotherapy (N = 99) are shown in Table S3 in the Supplementary Material and were generally comparable with the subgroup of patients treated with the combination of UPA and csDMARDs, and the overall population. Although no formal analysis was performed to evaluate the statistical significance of this observation, a higher proportion of patients in the UPA monotherapy subgroup had received prior treatment with a bDMARD or tsDMARD compared to patients treated with the combination of UPA and csDMARDs or the overall population (74.7% [74/99], 45.1% (132/293), and 52.6% (206/392), respectively).
The proportion of patients who achieved remission or LDA at both months 3 and 6 was similar in the UPA monotherapy subgroup compared to patients receiving UPA and csDMARDs (Fig. 2). The proportion of patients who achieved remission increased generally from months 3 to 6, but a particularly notable increase in remission rates was observed for patients in the UPA monotherapy subgroup for CDAI-defined (Fig. 2ii) and ACR-EULAR Boolean-defined (Fig. 2iv) remission compared with patients treated with UPA and csDMARDs.

Proportion of patients in the subgroups of patients treated with UPA monotherapy and a combination of UPA and csDMARDs achieving (i) DAS28-CRP < 2.6 (solid bars) or DAS28-CRP ≤ 3.2 (shaded bars), remission (solid bars) or low disease activity (shaded bars) defined according to the (ii) CDAI, (iii) SDAI, (iv) ACR-EULAR Boolean 2.0 criteria, or (v) a 50% reduction in pain score, or minimal clinically important difference in FACIT-F or HAQ-DI scores from baseline. Abbreviations: ACR-EULAR, American College of Rheumatology-European League Against Rheumatism; CDAI, Clinical Disease Activity Index; csDMARD, conventional synthetic disease-modifying antirheumatic drugs; DAS28-CRP, Disease Activity Score-28 Joint Count C-reactive protein; FACIT-F, Functional Assessment of Chronic Illness Therapy-Fatigue; HAQ-DI, Health Assessment Questionnaire-Disability Index; SDAI, Simplified Disease Activity Index; UPA, upadacitinib
The proportions of patients treated with UPA monotherapy and those treated with UPA in combination with csDMARDs who achieved a 50% reduction in pain, or an MCID in FACIT-F and HAQ-DI scores from baseline at months 3 and 6 are shown in Fig. 2v and are comparable to the proportions observed in the overall population described above.
Safety
Treatment-emergent adverse events (TEAEs) were reported in 50.3% (197/392) of patients in the interim analysis population, and the overall rates were comparable amongst the subgroups of patients analysed according to prior therapy (Table 4). Serious TEAEs occurred in fewer than 5% (19/392) of patients, and TEAE leading to UPA discontinuation occurred in 11.5% (45/392) of patients. Three deaths (0.8%) were reported, due to a previously diagnosed parietal occipital brain mass (n = 1; reported by the investigator as not possibly related to treatment); pneumonia (n = 1; reported as possibly related to treatment); and respiratory failure subsequent to COVID-19 infection (n = 1; reported as not possibly related to treatment). Infections were reported in 25.3% (99/392) of patients, with serious infections reported in 1.5% (6/392) of patients. COVID-19 infections occurred in 12.2% (48/392) of patients. Ten (2.6%) patients developed herpes zoster (5 of whom were not vaccinated at the baseline assessment), 5 (1.3%) patients developed a malignancy (4 basal cell carcinomas and 1 melanoma), and myocardial infarction occurred in 2 (0.5%) patients. Two patients with no prior history of venous thromboembolism experienced one event each of pulmonary embolism.
Discussion
The safety and efficacy of UPA for the treatment of adult patients with moderate-to-severe RA were previously demonstrated in a comprehensive phase 3 program. The effectiveness of UPA in the real-world setting has previously only been reported in abstracts describing data from retrospective analyses of registry data, and from a German prospective post-marketing study [19,20,21,22,23]. The generalisability of the data from the pivotal phase 3 program to clinical practice remained to be determined. This interim analysis of the ongoing observational CLOSE-UP study confirms the effectiveness of UPA in this setting and provides the first prospective data in patients with RA previously treated with other approved tsDMARDs.
The treat-to-target strategy for patients with RA recommends targeting remission, with LDA being an alternative goal in cases where remission is not attainable [1, 2]. Numerous composite measures of disease activity are available to assess these outcomes in both clinical trials and practice. We used multiple validated measures (DAS28-CRP, CDAI, SDAI, ACR-EULAR Boolean remission) whose scores are correlated with disease progression and impaired functioning [13, 16], and cut points that quantitate disease activity and guide clinical decision-making, to assess effectiveness of UPA in this interim analysis. Outcomes in response to initiation of UPA treatment were observed to be similarly independent of prior therapy (including patients with prior exposure to other tsDMARDs) at all time points assessed. It is noteworthy that these outcomes were achieved in patients who had discontinued prior b/tsDMARD therapy for predominantly efficacy-related reasons (163/194; 84.0%). Additionally, although the number of patients with prior tsDMARD (e.g. JAK inhibitor) exposure included in this interim analysis is relatively small (N = 42), data for this subgroup of patients suggest that unlike biologic therapies, prior exposure to a JAK inhibitor may not portend diminished outcomes compared to patients without prior exposure when treatment with UPA is initiated.
The interim analysis also included a relatively large proportion (25%) of patients who initiated treatment with UPA monotherapy. Although current guidelines recommend concurrent treatment with MTX, as many as 30% of patients with RA receive treatment without MTX [24,25,26]. Similar outcomes were attained with UPA monotherapy at both month 3 and month 6 compared with patients receiving UPA in combination with a csDMARD, which may support this approach in patients with tolerability issues, contraindications to treatment with csDMARDs, or for those with comorbidities.
A considerable proportion of patients in this interim analysis achieved remission defined according to ACR-EULAR Boolean 2.0 criteria [16]. The proportion of patients in clinical practice achieving the former criteria [15] in US and German populations has been reported to be as low as 5–7% [27, 28]. This observation is also notable given that 9.9% (36/364) and 17.2% (61/355) of patients in the CLOSE-UP interim population met the former ACR-EULAR Boolean remission criteria at months 3 and 6 (Fig. S2 in the Supplementary Material), proportions that are approximately twice the historical estimates for the US and German clinical practice populations referenced above, and which are consistent with those reported in the UPA phase 3 active comparator trials (but in a broader patient population compared to those studies) [6, 29].
Although not directly comparable because of differences in analytical methods (e.g. non-responder imputation in phase 3 RCTs versus “as observed” analysis in CLOSE-UP) and patient populations, real-world remission and LDA rates observed in the interim-analysis of the CLOSE-UP study appeared to be consistent with those observed in the SELECT-NEXT and SELECT-BEYOND phase 3 RCTs [5, 7].
Outcome domains considered as essential to patient perceived-remission include pain, mobility, physical function, independence, and fatigue [30]. Substantial improvements in the relevant measures of these outcomes (pain VAS, FACIT-F, HAQ-DI) occurred by month 3, were sustained at month 6, and were highly similar in all subgroups independent of prior therapy, and when UPA was administered as monotherapy or in combination with csDMARDs. These data are concordant with clinical improvements observed in the CLOSE-UP study, and with patient-reported data from the UPA phase 3 clinical development program.
One strength of this interim analysis is that it represents the first comprehensive report of the real-world effectiveness of UPA in adult patients with RA. The CLOSE-UP study is fully enrolled, with final 2-year analyses anticipated in 2025. We additionally provide the first prospective efficacy and safety data for UPA in patients with prior JAK inhibitor exposure and real-world data that further support the efficacy of UPA monotherapy. Study limitations should be acknowledged. This is an interim analysis of an ongoing trial; however, our analyses include data for 94.7% (392/414) of all patients enrolled in the CLOSE-UP study, and inconsistencies in outcomes for the complete dataset are not anticipated. The benefits of UPA treatment on structural damage and/or radiographic progression have been demonstrated in RCTs [9, 31]; however, these outcomes were not evaluated in the CLOSE-UP study. Although prior or current smoking status did not appear to influence treatment outcomes in this analysis (data not shown), future research on the influence of smoking and other patient- and/or disease-related characteristics to predict outcomes of UPA treatment would be of interest. As treatment with corticosteroids and other csDMARDs was at the discretion of the investigators, we did not formally assess whether patients in this analysis tapered or discontinued these therapies. These data would also be of interest for future research. Although the low rate of TEAEs, particularly adverse events of interest such as venous thromboembolism, observed in this interim analysis is encouraging, safety data beyond 6 months are needed to confirm the long-term benefit–risk profile of UPA in the real-world setting.
Conclusion
This interim analysis of the CLOSE-UP study confirms the real-world effectiveness of UPA in a broad range of Canadians living with RA. In addition to affirming the potential for comparable clinical and patient-reported outcomes in clinical trials and routine clinical practice, data from this analysis suggests similar effectiveness regardless of prior treatment and for UPA monotherapy. No new safety signals were identified and longer-term data from patients with complete follow-up are expected.
Data Availability
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (e.g. protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the USA and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://vivli.org/ourmember/abbvie/ then select “Home”.
References
Smolen JS, Landewe RBM, Bergstra SA, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann Rheum Dis. 2023;82(1):3–18.
Fraenkel L, Bathon JM, England BR, et al. 2021 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Care Res (Hoboken). 2021;73(7):924–39.
Aletaha D, Smolen JS. Diagnosis and management of rheumatoid arthritis: a review. JAMA. 2018;320(13):1360–72.
Scott IC, Ibrahim F, Panayi G, et al. The frequency of remission and low disease activity in patients with rheumatoid arthritis, and their ability to identify people with low disability and normal quality of life. Semin Arthritis Rheum. 2019;49(1):20–6.
Burmester GR, Kremer JM, Van den Bosch F, et al. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease-modifying anti-rheumatic drugs (SELECT-NEXT): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2018;391(10139):2503–12.
Fleischmann R, Pangan AL, Song IH, et al. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III, double-blind, randomized controlled trial. Arthritis Rheumatol. 2019;71(11):1788–800.
Genovese MC, Fleischmann R, Combe B, et al. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease-modifying anti-rheumatic drugs (SELECT-BEYOND): a double-blind, randomised controlled phase 3 trial. Lancet. 2018;391(10139):2513–24.
Smolen JS, Pangan AL, Emery P, et al. Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT-MONOTHERAPY): a randomised, placebo-controlled, double-blind phase 3 study. Lancet. 2019;393(10188):2303–11.
van Vollenhoven R, Takeuchi T, Pangan AL, et al. Efficacy and safety of upadacitinib monotherapy in methotrexate-naive patients with moderately-to-severely active rheumatoid arthritis (SELECT-EARLY): a multicenter, multi-country, randomized, double-blind, active comparator-controlled trial. Arthritis Rheumatol. 2020;72(10):1607–20.
Fransen J, van Riel PL. The disease activity score and the EULAR response criteria. Rheum Dis Clin North Am. 2009;35(4):745–57.
Cella D, Yount S, Sorensen M, Chartash E, Sengupta N, Grober J. Validation of the functional assessment of chronic illness therapy fatigue scale relative to other instrumentation in patients with rheumatoid arthritis. J Rheumatol. 2005;32(5):811–9.
Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum. 1980;23(2):137–45.
Aletaha D, Nell VP, Stamm T, et al. Acute phase reactants add little to composite disease activity indices for rheumatoid arthritis: validation of a clinical activity score. Arthritis Res Ther. 2005;7(4):R796-806.
Smolen JS, Breedveld FC, Schiff MH, et al. A simplified disease activity index for rheumatoid arthritis for use in clinical practice. Rheumatology (Oxford). 2003;42(2):244–57.
Felson DT, Smolen JS, Wells G, et al. American College of Rheumatology/European League Against Rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Arthritis Rheum. 2011;63(3):573–86.
Studenic P, Aletaha D, de Wit M, et al. American College of Rheumatology/EULAR remission criteria for rheumatoid arthritis: 2022 revision. Ann Rheum Dis. 2023;82(1):74–80.
Keystone E, Burmester GR, Furie R, et al. Improvement in patient-reported outcomes in a rituximab trial in patients with severe rheumatoid arthritis refractory to anti-tumor necrosis factor therapy. Arthritis Rheum. 2008;59(6):785–93.
Wells GA, Tugwell P, Kraag GR, Baker PR, Groh J, Redelmeier DA. Minimum important difference between patients with rheumatoid arthritis: the patient’s perspective. J Rheumatol. 1993;20(3):557–60.
Witte T, Kiltz U, Haas F, et al. Effectiveness of upadacitinib in patients with rheumatoid arthritis in German real-world practice: interim results from a post-marketing observational study. Arthritis Rheumatol. 2021;73:578–81.
Kremer JM, Tundia N, McLean R, Blachley T, Maniccia A, Pappas DA. Characteristics and 6-month outcomes among real-world patients with rheumatoid arthritis initiating upadacitinib: analysis from the corrona registry. Ann Rheum Dis. 2021;80:446.
Gibofsky A, Dhillon B, Pearson ME, et al. Treatment effectiveness of upadacitinib at 3 months in US patients with rheumatoid arthritis from the united rheumatology normalized integrated community evidence (NICE[TM]) real-world data. Ann Rheum Dis. 2021;80:575–6.
Bergman M, Tundia N, Bryant A, et al. POS0436 patient characteristics and outcomesin patients with rheumatoid arthritis treated with upadacitinib: the OM1 RA registry. Ann Rheum Dis. 2021;80:446–7.
Choquette D, Bombardier C, Cesta A, Coupal L. Real-world effectiveness, safety profile, and persistence of upadacitinib. A prototype for collaboration among rheumatology registries in Canada. The RHUMADATA-OBRI partnership. J Rheumatol. 2023;50:75.
Emery P, Pope JE, Kruger K, et al. Efficacy of monotherapy with biologics and JAK inhibitors for the treatment of rheumatoid arthritis: a systematic review. Adv Ther. 2018;35(10):1535–63.
Emery P, Sebba A, Huizinga TW. Biologic and oral disease-modifying antirheumatic drug monotherapy in rheumatoid arthritis. Ann Rheum Dis. 2013;72(12):1897–904.
Bessette L, Florica B, Fournier PA, et al. Canadian retrospective chart review evaluating concomitant methotrexate de-escalation patterns in RA patients treated with biologic or targeted synthetic DMARDS. Ann Rheum Dis. 2022;81:388–9.
Thiele K, Huscher D, Bischoff S, et al. Performance of the 2011 ACR/EULAR preliminary remission criteria compared with DAS28 remission in unselected patients with rheumatoid arthritis. Ann Rheum Dis. 2013;72(7):1194–9.
Shahouri SH, Michaud K, Mikuls TR, et al. Remission of rheumatoid arthritis in clinical practice: application of the American College of Rheumatology/European League Against Rheumatism 2011 remission criteria. Arthritis Rheum. 2011;63(11):3204–15.
Rubbert-Roth A, Enejosa J, Pangan AL, et al. Trial of upadacitinib or abatacept in rheumatoid arthritis. N Engl J Med. 2020;383(16):1511–21.
van Tuyl LH, Sadlonova M, Hewlett S, et al. The patient perspective on absence of disease activity in rheumatoid arthritis: a survey to identify key domains of patient-perceived remission. Ann Rheum Dis. 2017;76(5):855–61.
Fleischmann RM, Genovese MC, Enejosa JV, et al. Safety and effectiveness of upadacitinib or adalimumab plus methotrexate in patients with rheumatoid arthritis over 48 weeks with switch to alternate therapy in patients with insufficient response. Ann Rheum Dis. 2019;78(11):1454–62.
Acknowledgements
The trial investigators and the sponsor are grateful to the patients for their time and participation in this study, and for their contributions to this research that aid in the understanding of RA and its treatment.
Medical Writing, Editorial, and Other Assistance.
Dr. Ekatherina Stoyanova, AbbVie Corp, was instrumental in study protocol design and approval. Editorial and writing assistance was provided by Dr. Lisa M. Shackelton of Alimentiv Inc. Support for this assistance was funded by AbbVie Corp.
Authorship.
All authors whose names appear on the submission have: Made substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data; Drafted the work or revised it critically for important intellectual content; Approved the version to be published; and Agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy of integrity of any part of the work are appropriately investigated and resolved.
Funding
AbbVie Corp (St-Laurent, Québec, Canada) funded this study and participated in the study design, research, analysis, data collection, interpretation of data, reviewing, and approval of the publication. The Rapid Service fee was also funded by AbbVie Corp.
Author information
Authors and Affiliations
Contributions
LB contributed to the study conception and design. LB, JC, AC, LL, NR, P-AF, DL, TG, and DH contributed to data analysis and interpretation, and in critical review and revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
Louis Bessette reports consultancies, research grants, and speaker fees from BMS, Janssen, UCB, AbbVie, Pfizer, Sanofi, Lilly, and Novartis; research grants and speaker fees from Sandoz, Fresnius Kabi, Teva, Organon, and JAMP Pharma; and research grants from Gilead. Jonathan Chan reports consultancy and/or speaker fees and research grants from AbbVie, Pfizer, and UCB, and consultancy and/or speaker fees from Amgen, Celgene, Janssen, Novartis, Roche, Gilead, Eli Lilly, Merck, Fresenius Kabi, and Sandoz. Andrew Chow reports consultancies, research grants, and speaker fees from Abbvie, Amgen, AstraZeneca, BioJamp, Celltrion, Eli Lilly, Fresenius Kabi, Janssen, Novartis, Pfizer, UCB. Larissa Lisnevskaia has no conflicts to report. Nicolas Richard reports consultancies and/or speaker fees from Abbvie, Amgen, AstraZeneca, Eli Lilly, Janssen, Novartis, Pfizer and UCB. Pierre-Andre Fournier, Dalinda Liazoghli and Tanya Girard are employees of AbbVie Corporation. Derek Haaland reports honouraria from Abbvie, Amgen, AstraZeneca, Bristol-Myers Squibb, Eli Lilly, GlaxoSmithKline, Janssen, Merck, Novartis, Pfizer, Roche, Sanofi-Genzyme, Takeda, UCB; advisory board or speakers’ bureau membership for Abbvie, Amgen, AstraZeneca, Bristol-Myers Squibb, Eli Lilly, GlaxoSmithKline, Janssen, Novartis, Pfizer, Roche, Sanofi Genzyme, Takeda; research funding from Abbvie, Adiga Life Sciences, Amgen, AstraZeneca, Bristol-Myers Squibb, Can-Fite Biopharma, Celgene, Eli Lilly, Gilead, GlaxoSmithKline, Janssen, Novartis, Pfizer, Regeneron, Sanofi-Genzyme, and UCB.
Ethical Approval
The study is conducted in accordance with the Declaration of Helsinki and International Conference on Harmonisation Guidelines for Good Clinical Practice. Institutional review board/independent ethics committee approval was obtained for all participating centres (Table S1 in the Supplementary Material). All patients provided informed consent to participate in the study.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bessette, L., Chan, J., Chow, A. et al. Real-World Effectiveness of Upadacitinib for Treatment of Rheumatoid Arthritis in Canadian Patients: Interim Results from the Prospective Observational CLOSE-UP Study. Rheumatol Ther 11, 563–582 (2024). https://doi.org/10.1007/s40744-024-00651-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-024-00651-8