
Abstract
Introduction
Intra-articular (IA) corticosteroids, including triamcinolone acetonide (TA), are a recommended treatment for hip osteoarthritis. We compared the safety and systemic exposure of TA extended-release (TA-ER) versus TA crystalline suspension (TAcs) in patients with hip osteoarthritis.
Methods
In this phase 2, randomized, multicenter, open-label, single-dose study (NCT03382262), patients with hip osteoarthritis were randomly assigned 1:1 to receive single IA injections of TA-ER 32 mg or TAcs 40 mg. Safety assessments included treatment-emergent adverse events (TEAEs). Blood samples were collected for pharmacokinetic (PK) analysis up to day 85. PK parameters included area under the concentration–time curve, total body drug clearance, maximum concentration (Cmax), mean residence time, half-life, and time to maximum concentration.
Results
Of 30 patients (TA-ER: n = 15; TAcs: n = 15) randomized and included in the Safety Population, 25 patients were evaluated in the PK Population. TEAEs were reported in four of 15 (26.7%) patients who received TA-ER and in seven of 15 (46.7%) patients who received TAcs. The most common TEAEs included arthralgia and headache. All TEAEs were of grade 1 or 2 in severity. TA-ER produced substantially lower peak plasma TA concentrations compared with TAcs (Cmax geometric mean: 890.4 vs. 5549.4 pg/ml), and these were less variable with TA-ER versus TAcs. Similarly, overall TA systemic exposure was substantially lower for TA-ER versus TAcs, with gradual elimination from systemic circulation through day 85.
Conclusions
Following a single IA injection in the hip, TA-ER was generally well tolerated, with a safety profile comparable to that of TAcs. Systemic TA exposure was markedly lower in TA-ER-treated patients, consistent with the PK profile observed in knee osteoarthritis.
Clinicaltrials.gov identifier: NCT03382262.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Triamcinolone acetonide extended-release (TA-ER) is approved by the US Food and Drug Administration (FDA) for the management of pain associated with knee osteoarthritis (OA). |
The extended-release profile of TA-ER has been confirmed in pharmacokinetic (PK) studies in patients with knee OA; however, the PK profile of TA-ER has not been evaluated in other large joints. |
This phase 2 study was the first conducted comparing the safety and systemic exposure of TA following a single intra-articular (IA) injection of TA-ER or triamcinolone acetonide crystalline suspension (TAcs) into the hip joint in patients with hip OA. |
What was learned from the study? |
In patients with hip OA, a single IA injection of TA-ER into the index hip joint was generally safe and well tolerated, and total systemic exposure to TA was substantially lower with TA-ER compared with TAcs during the study period. |
The PK profile of TA-ER following injection into the hip was consistent with that of single- and bilateral-knee injections in the knee. |
Introduction
The hip joint is the second most common site of osteoarthritis (OA), a debilitating disease associated with pain and limitations in physical function [1,2,3,4,5]. Knee and hip OA were ranked as the 11th highest contributors to global disability in the Global Burden of Disease 2010 study based on years lived with disability [4]. The updated Global Burden of Disease 2017 study highlighted the continued and growing burden of these diseases and estimated the global prevalence of knee and hip OA at 303.1 million cases in 2017, with an age-standardized prevalence of 3.75%, an increase of 9.3% since 1990. In addition to prevalence, OA currently accounts for approximately 7.1% of the burden of years lived with disability, a statistically significant increase of 31.4% since 2007, building on the 63.1% increase between 1990 and 2007. At the regional level, the highest prevalence and incidence rates of OA were in North America, affecting about 32.5 million US adults [6, 7]. The age-standardized prevalence of radiographic hip OA was 19.6% in a 3-year US-based community study of 978 participants [8]. In a study of 26 patients with end-stage hip OA, knee muscle strength, functional performance, and physical activity were substantially reduced compared with healthy individuals (P < 0.01) [2]. Similarly, patients with hip OA experienced significantly more pain and greater limitations in daily activities compared with unaffected individuals in a longitudinal cohort study (P = 0.02 and P = 0.05, respectively; N = 66) [3].
Hip OA symptoms and progression are linked to the presence of pro-inflammatory mediators and inflammation in synovial tissues [9,10,11]. For example, levels of interleukin-6 (IL-6) and interferon-γ–inducible protein-10 (IP10) were significantly associated with Hip Disability and Osteoarthritis Outcome Score pain (P = 0.02 and P = 0.04, respectively). Only IP10 expression was observed in the synovium of patients with hip OA [9]. In addition, pain as assessed by the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) and the Visual Analogue Scale was significantly correlated with synovial fluid IL-6 levels (P < 0.01) and visfatin levels (P < 0.01 and P < 0.05, respectively) in a cross-sectional study of 112 patients with end-stage hip OA [10]. Furthermore, pain severity was associated with mean serum high-sensitivity C-reactive protein levels in 770 patients with advanced knee and hip OA [11].
There are several pharmacologic treatment options for hip OA, with oral nonsteroidal anti-inflammatory drugs (NSAIDS) and intra-articular corticosteroids (IACS) categorized as “strongly recommended” by the American College of Rheumatology/Arthritis Foundation [12]. NSAIDS have been shown to provide moderate pain relief across multiple well-designed clinical trials involving patients with OA. However, oral NSAIDS can be limited, in particular, by the occurrence gastrointestinal toxicities [13]. IACS agents also exert anti-inflammatory effects and have been shown to provide relief from hip OA pain for ≤ 8 weeks post injection [14, 15].
Triamcinolone acetonide extended-release (TA-ER) is a microsphere-based formulation of triamcinolone acetonide approved by the US Food and Drug Administration as an intra-articular (IA) injection for the management of OA pain of the knee [16]. TA-ER has demonstrated efficacy and safety through week 16 following a single injection compared with saline-placebo and through week 12 compared with triamcinolone acetonide crystalline suspension (TAcs) in patients with knee OA [17, 18]. Moreover, in patients with knee OA, TA-ER provided similar improvements in pain scores following a subsequent IA injection administered 12–24 weeks after the initial dose [19].
IA injection of TAcs may result in high peak systemic TA concentrations due to rapid efflux of corticosteroids from the joint [20]. This is of particular concern in patients with diabetes, who can experience elevated blood glucose levels for several days after injection of TAcs [21]. Previous phase 2 pharmacokinetic (PK) studies of TA-ER in patients with knee OA demonstrated its extended-release profile [20, 22]. Quantifiable TA levels were detected in synovial fluid through week 12 following a single IA injection in the knee, and systemic peak TA concentrations were substantially lower with TA-ER compared with TAcs [20]. Single or bilateral IA injection of TA-ER showed a gradual increase in plasma TA that plateaued through 24 h post injection, followed by slow elimination from the systemic circulation [20, 22]. Furthermore, single IA injection of TA-ER in the knee showed minimal blood glucose disruption during the days following injection [21].
Although the PK profile of TA-ER compared with that of TAcs has been well characterized following IA injection of TA-ER or TAcs in patients with knee OA, it has not been evaluated in other large joints. Here, we evaluate the safety and systemic exposure of TA following IA injection of TA-ER and TAcs in a phase 2 study of patients with hip OA.
Methods
Ethics
This study was conducted according to the Helsinki Declaration of 1964, as revised in 2013, and International Committee on Harmonisation Good Clinical Practice guidelines. Governing ethical bodies approved the protocol, and all patients provided written informed consent.
Patient Eligibility and Study Design
This was a phase 2, randomized, open-label, single-dose study (NCT03382262) that enrolled patients aged ≥ 40 years with body mass index (BMI) ≤ 40 kg/m2, symptomatic hip OA based on pain as defined by the American College of Rheumatology clinical/radiographic criteria for hip OA [23], and ≥ 2 of the following: erythrocyte sedimentation rate < 20 mm/h, radiographic femoral or acetabular osteophytes, and radiographic joint space narrowing (superior, axial, and/or medial) [23]. Eligible patients had symptoms consistent with hip OA in the index hip for ≥ 6 months prior to screening, and pain in the index hip joint for > 15 days in the preceding month. Patients were excluded who had systemic inflammatory joint disease; history of infection in the hip joint; IA treatment of any joint with corticosteroids (including TA-ER) or biologic agent within 6 months of screening; IA treatment of the index hip with hyaluronic acid within 6 months of screening; parenteral or oral corticosteroid use within 3 months of screening; inhaled, intranasal, or topical corticosteroid use within 2 weeks of screening; uncontrolled diabetes as indicated by hemoglobin A1c level of > 7.5% (> 59 mmol/mol); or use of any other investigational drug, biologic agent, or device within 3 months.
Patients meeting inclusion criteria were randomly assigned 1:1 to a single IA injection of TA-ER 32 mg or TAcs 40 mg using a central system directly accessed by the study sites and were evaluated for 12 weeks following injection. Analgesic medication use was permitted.
Study Interventions
TA-ER 32 mg was provided as a powder that was reconstituted in diluent containing an isotonic, sterile aqueous sodium chloride solution (0.9% weight/weight [w/w]), carboxymethylcellulose (0.5% w/w), and polysorbate-80 (0.1% w/w) and administered as a 5-ml IA injection. Commercially available TAcs 40 mg (Kenalog®-40; Bristol-Myers Squibb Company, Princeton, NJ, USA; 1 ml) [24] was initially administered as a 1-ml IA injection for comparison with TA-ER. As per the protocol amendment, however, 1 ml was combined with 4 ml normal saline and sodium chloride (0.9%) solution and administered as a 5-ml injection. As per the protocol, the investigator could choose the position of the hip and the approach for injection. A 21-gauge or larger needle was used for injection into the hip joint. The injection was performed with ultrasound guidance. Patients were instructed to avoid strenuous activities or prolonged weight-bearing activities for approximately 24 to 48 h post injection and to maintain stable physical activity throughout the study.
Study Assessments
Safety was assessed over a 12-week period (85 days) based on treatment-emergent adverse events (TEAEs), physical examination, index joint assessments, vital signs, and laboratory evaluations. TEAEs were spontaneously reported by the patient or discovered by the investigator and were defined as any untoward medical occurrence in a patient who received study drug treatment that may or may not have been related to study treatment.
IA injections were administered on day 1. Blood samples were collected at baseline and on days 2, 3, 5, 8, 15, 22, 29, 57, and 85 post injection for PK analysis. On day 1, blood samples were collected at hours 1–6, 8, 10, and 12 post injection. Plasma TA concentrations were assayed using a validated liquid chromatography tandem mass spectrometry method.
Statistical Analysis
All descriptive statistical analyses were performed using SAS® statistical software (Version 9.4).
The Safety Population included all patients who received study drug treatment. TEAEs were coded using the Medical Dictionary for Regulatory Activities (Version 20.1), and concomitant medications were coded using the World Health Organization Drug Dictionary (Version B3; March 2014 edition).
The PK Population included all patients from the Safety Population who received a full dose of study drug, completed PK sampling, and had sufficient plasma concentration measurements for PK analysis. PK plasma TA concentrations were assessed using non-compartmental analysis from individual TA plasma concentration–time profiles using Phoenix WinNonlin® Version 8 (Certara Corporation), and mean concentration profiles were calculated for each treatment arm. Descriptive statistics were calculated by time point for plasma TA concentration levels. PK parameters included area under the concentration–time curve (AUC), total body drug clearance (CL), maximum concentration (Cmax), mean residence time (MRT), half-life (T1/2), and time to maximum concentration (Tmax). A linear trapezoidal rule was applied to calculate observed AUC. AUC parameters were calculated for the following intervals: AUC from time 0 to hour 24 (AUC0–24) and hour 96 (AUC0–96), AUC from time 0 to the last quantifiable concentration (AUC0–last), and AUC from time 0 to infinity post injection (AUC0–∞). Plasma PK data of subjects with extrapolated AUC0-∞ ≥ 25% of predicted value were not included in overall plasma PK parameter summary calculations.
Sample size was based on the PK primary endpoint, and a sample size of 12 in each treatment arm was estimated for analysis. A total sample size of 24 patients achieved approximately 90% power, with 2-sided α = 0.05, to detect a ratio of < 1 of the exposure PK parameter means (TA-ER/TAcs), with a pooled coefficient of variation estimation of 0.68 (PASS 15 Power Analysis and Sample Size Software [2017], NCSS, LLC; Kaysville, UT, USA). The sample size of 24 patients assumed a 10% drop-out rate for providing complete blood samples for PK analysis.
Results
Patient Disposition
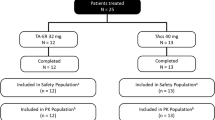
This study was conducted from December 2017 to October 2018 at six US study centers. A total of 30 patients were randomized in the study, received a single IA injection of TA-ER 32 mg (n = 15) or TAcs 40 mg (n = 15) on day 1, and were included in the Safety Population. Two patients discontinued the study due to withdrawal of consent (n = 1; 6.7%; TAcs) and withdrawal by the investigator or sponsor (n = 1; 6.7%; TA-ER 32 mg). A total of 28 (93.3%) patients (TA-ER 32 mg: n = 14; TAcs 40 mg: n = 14) completed the study (Fig. 1). Four unique patients were excluded from PK analysis due to study drug administration issues. Three patients had incomplete administration of the intended TA-ER 32 mg dose, and one patient enrolled and received study medication at two sites (TAcs followed by TA-ER). The PK population for analysis thus comprised 25 patients (TA-ER 32 mg: n = 11; TAcs 40 mg: n = 14).

Patient disposition. PK pharmacokinetic, TAcs triamcinolone acetonide crystalline suspension, TA-ER triamcinolone acetonide extended-release. aOne patient was enrolled in the study at two different study centers and received TAcs at one study center and then received TA-ER 5 days later at another study center, with both study drugs administered in the right hip; this patient is included in both treatment groups. bThe Safety Population included all patients who received any amount of study treatment. The patient noted above who received two injections is included in both treatment groups. cThe PK Population included all patients from the Safety Population who received a full dose of study drug (three patients excluded from TA-ER PK Population because of incomplete dosing), completed PK sampling, and had sufficient plasma concentration measurements for PK analysis. The patient noted above who received two injections (TAcs followed by TA-ER) is excluded from both treatment groups
Baseline Disease Characteristics
Of all participants, 56.7% were male, the mean age (standard deviation [SD]) at study outset was 59.1 (7.3) years, 90% were overweight (BMI 25.0–29.9 kg/m2) or obese (BMI 30.0–39.9 kg/m2), and the mean number of years since initial diagnosis (SD) was 5.1 (4.4) years. There were slightly more males in the TAcs group (10/15 patients; 66.7%) compared with the TA-ER group (7/15 patients; 46.7%). However, baseline demographic characteristics and OA history were well balanced across the TA-ER and TAcs treatment groups for most parameters, except for prior surgery or procedures (0 [0%] vs. 3 patients [20%], respectively) (Table 1).
Safety
TA-ER and TAcs were generally well tolerated. TEAEs were reported in four of 15 (26.7%) patients who received TA-ER and in seven of 15 (46.7%) patients who received TAcs (Table 2). All TEAEs were of grade 1 or 2, and no TEAEs led to study discontinuation.
Commonly occurring TEAEs included arthralgia (TA-ER: n = 1 [6.7%]; TAcs: n = 2 [13.3%]), headache (both n = 1 [6.7%]), upper respiratory tract infection (both n = 1 [6.7%]), musculoskeletal pain (TA-ER: n = 1 [6.7%]; TAcs: n = 0 [0%]), and nasopharyngitis (TA-ER: n = 0 [0%]; TAcs: n = 1 [6.7%]). Index-hip TEAEs occurred in 1 (6.7%) patient who received TA-ER and in three (20.0%) patients who received TAcs. No study drug-related TEAEs occurred in patients who received TA-ER. Furthermore, no clinically relevant changes in vital signs or laboratory parameters were observed.
Pharmacokinetics
Following a single IA injection in the hip, rapid systemic absorption of TA was demonstrated. In TA-ER-treated patients, TA plasma concentrations peaked with a geometric mean (GM) (95% confidence interval [CI]) Cmax of 890.4 (415.9–1906.6) pg/ml at a median tmax of 5.0 h, and the GM MRT (95% CI) was 758.6 (452.6–1271.7) h (Table 3). In contrast, in TAcs-treated patients, TA plasma concentrations peaked with a higher GM (95% CI) Cmax of 5549.4 (3086.0–9979.2) pg/ml, at a median tmax of 4.0 h. The GM MRT (95% CI) for TAcs was 402.2 (191.0–847.2) h and shorter than that observed with TA-ER). Median t1/2 (minimum, maximum) values of 461.5 (342, 828) h and 388.5 (32, 1283) h were reported for TA-ER and TAcs, respectively.
At hour 5 post injection, TA concentrations increased to a GM of 793 pg/ml in the systemic circulation of TA-ER-treated patients compared with a GM of 4673 pg/ml in TAcs-treated patients. Systemic plasma TA concentrations plateaued through hour 72 post injection, and TA was slowly eliminated from systemic circulation through day 85 in the TA-ER group. In contrast, in the TAcs group, systemic TA concentrations remained substantially elevated compared with TA-ER through hour 24 post injection and decreased through day 85 (Fig. 2a–d). A GM AUC0–last of 397,457.3 h·pg/ml and 784,250.1 h·pg/ml was reported for the TA-ER and TAcs groups, respectively. Measurable GM TA plasma concentrations were higher with TA-ER compared with TAcs at day 85 (112 pg/ml vs. 93 pg/ml), with 10 of 11 (90.1%) and 12 of 14 (85.7%) patients contributing PK samples with measurable TA-ER and TAcs concentrations, respectively. However, the duration of measurable levels of TA was comparable for the TA-ER and TAcs groups (1575.7 vs. 1590.5 h). Taken together, total systemic exposure to TA was substantially higher with TAcs compared with TA-ER during the study period.

Plasma TA concentration over time after injection of TA-ER (n = 11) or TAcs (n = 14). a Log-linear GM (95% CI) baseline to day 85; b Linear–linear GM (95% CI) baseline to day 85; c Log-linear GM (95% CI) baseline to hour 24; d Linear–linear GM (95% CI) baseline to hour 24 (PK Population). CI confidence interval, GM geometric mean, PK pharmacokinetic, TA triamcinolone acetonide, TAcs triamcinolone acetonide crystalline suspension, TA-ER triamcinolone acetonide extended-release
Discussion
This is the first study to assess the safety and PK of TA-ER following a single IA injection in the hip joint. In patients with hip OA, this treatment with TA-ER into the index hip joint was generally well tolerated, no new safety signals were identified, and all TEAEs were grade 1 or 2 in severity, consistent with the safety profile of TAcs as well as that of TA-ER following IA injection in the knee [20]. Peak TA plasma concentrations were considerably lower with TA-ER compared with TAcs; similarly, systemic TA exposure was markedly lower with TA-ER compared with TAcs.
The PK profile of TA-ER following a single injection into the hip was consistent with that of single- and bilateral-knee injections that showed early systemic absorption and a plateau in plasma TA concentration through hour 24, followed by gradual systemic elimination [20, 22]. In a phase 2 open-label pharmacokinetics study, GM Cmax of TA was substantially lower in TA-ER-treated patients (32 mg) compared with those given TAcs (40 mg) (966.7 vs. 11,064.7 pg/ml) at a similar time point following a single IA injection in the knee [20]. In another phase 2a open-label study in which patients with knee OA received bilateral knee injections of TA-ER (32 mg) or TAcs (40 mg), mean plasma TA concentrations were lower in the TA-ER group compared with those treated with TAcs (GM Cmax: 2277.7 vs. 7394.7 pg/ml) [22]. In addition, plasma TA concentrations were less variable with TA-ER compared with TAcs at each collection point throughout the present study, which is consistent with findings from the previous phase 2 study comparing single injections of either treatment into one knee [20]. Because systemic exposure of TA was substantially lower with TA-ER compared with TAcs, TA-ER may exhibit an improved safety profile relative to TAcs in the hip. These results are also consistent with findings from the initial phase 2 study assessing the efficacy, safety, and PK of three dose levels of TA-ER and a single dose of TAcs (40 mg) in patients with knee OA [25]. The mean plasma concentration–time profile of TA was markedly lower for TA-ER at the 40 mg dose (approximately 32 mg delivered) compared with TAcs (40 mg dose) (GM Cmax: 707 vs. 13,196 pg/ml, respectively) following a single IA injection in the knee. In the same study, TA-ER provided greater pain relief compared with TAcs up to week 12 post injection as measured by Average Daily Pain scores, with differences observed from weeks 5 to 10 (P < 0.05).
Three occurrences, representing a 20% incidence, of incomplete administration of TA-ER during ultrasound-guided hip injection were observed and were unexpected. Incomplete administration has not been noted in clinical studies involving hundreds of patients receiving TA-ER injections into the knee joint, or in a small study of TA-ER injections into the glenohumeral joint in patients with shoulder OA [26]. The standard anterior approach to the hip joint in patients in a supine position may have contributed to this observation with injection of the TA-ER microsphere suspension. Additional evaluation will be necessary to further assess the potential for incomplete TA-ER administration and identify if adjustments to the injection technique will decrease the incidence of incomplete administration observed in this study.
Limitations of this study include its small sample size and open-label study design. However, it is encouraging that the findings of the present study are consistent with two previous phase 2 studies that assessed the safety and PK of TA-ER in larger patient populations with knee OA (N = 228 and N = 81) [20, 25].
Conclusions
The symptomatic management of hip OA remains an important area of research to reduce symptoms and increase quality of life in patients with OA. IA injection therapies for hip OA are an area of increasing interest. The current study suggests that IA injection of TA-ER 32 mg offers safe treatment in the index hip joint, along with reduced systemic exposure, and present a promising treatment alternative.
References
Turkiewicz A, Petersson IF, Bjork J, et al. Current and future impact of osteoarthritis on health care: a population-based study with projections to year 2032. Osteoarthr Cartil. 2014;22(11):1826–32.
Judd DL, Thomas AC, Dayton MR, Stevens-Lapsley JE. Strength and functional deficits in individuals with hip osteoarthritis compared to healthy, older adults. Disabil Rehabil. 2014;36(4):307–12.
Kumar D, Wyatt C, Chiba K, et al. Anatomic correlates of reduced hip extension during walking in individuals with mild-moderate radiographic hip osteoarthritis. J Orthop Res. 2015;33(4):527–34.
Cross M, Smith E, Hoy D, et al. The global burden of hip and knee osteoarthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis. 2014;73(7):1323–30.
Barbour KE, Lui LY, Nevitt MC, et al. Hip osteoarthritis and the risk of all-cause and disease-specific mortality in older women: a population-based cohort study. Arthritis Rheumatol. 2015;67(7):1798–805.
Safiri S, Kolahi AA, Hoy D, et al. Global, regional and national burden of rheumatoid arthritis 1990–2017: a systematic analysis of the Global Burden of Disease study 2017. Ann Rheum Dis. 2019;78(11):1463–71.
Osteoarthritis Action Alliance. OA prevalence and burden. Chapel Hill, NC: Osteoarthritis Action Alliance. 2021. https://oaaction.unc.edu/oa-module/oa-prevalence-and-burden/.
Kim C, Linsenmeyer KD, Vlad SC, et al. Prevalence of radiographic and symptomatic hip osteoarthritis in an urban United States community: the Framingham osteoarthritis study. Arthritis Rheumatol. 2014;66(11):3013–7.
Ren G, Lutz I, Railton P, et al. Serum and synovial fluid cytokine profiling in hip osteoarthritis: distinct from knee osteoarthritis and correlated with pain. BMC Musculoskelet Disord. 2018;19(1):39.
Bas S, Finckh A, Puskas GJ, et al. Adipokines correlate with pain in lower limb osteoarthritis: different associations in hip and knee. Int Orthop. 2014;38(12):2577–83.
Sturmer T, Brenner H, Koenig W, Gunther KP. Severity and extent of osteoarthritis and low grade systemic inflammation as assessed by high sensitivity C reactive protein. Ann Rheum Dis. 2004;63(2):200–5.
Kolasinski SL, Neogi T, Hochberg MC, et al. 2019 American College of Rheumatology/Arthritis Foundation guideline for the management of osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken). 2020;72(2):149–62.
Katz JN, Arant KR, Loeser RF. Diagnosis and treatment of hip and knee osteoarthritis: a review. JAMA. 2021;325(6):568–78.
Atchia I, Kane D, Reed MR, Isaacs JD, Birrell F. Efficacy of a single ultrasound-guided injection for the treatment of hip osteoarthritis. Ann Rheum Dis. 2011;70(1):110–6.
Lambert RG, Hutchings EJ, Grace MG, Jhangri GS, Conner-Spady B, Maksymowych WP. Steroid injection for osteoarthritis of the hip: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2007;56(7):2278–87.
ZILRETTA® (triamcinolone acetonide extended-release injectable suspension), for intra-articular use. Burlington, MA: Flexion Therapeutics, Inc. 2020. Accessed 1 2020.
Conaghan PG, Hunter DJ, Cohen SB, et al. Effects of a single intra-articular injection of a microsphere formulation of triamcinolone acetonide on knee osteoarthritis pain: a double-blinded, randomized, placebo-controlled, multinational study. J Bone Joint Surg Am. 2018;100(8):666–77.
Conaghan PG, Cohen SB, Berenbaum F, Lufkin J, Johnson JR, Bodick N. Brief report: a phase IIb trial of a novel extended-release microsphere formulation of triamcinolone acetonide for intraarticular injection in knee osteoarthritis. Arthritis Rheumatol. 2018;70(2):204–11.
Spitzer AI, Richmond JC, Kraus VB, et al. Safety and efficacy of repeat administration of triamcinolone acetonide extended-release in osteoarthritis of the knee: a phase 3b, open-label study. Rheumatol Ther. 2019;6(1):109–24.
Kraus VB, Conaghan PG, Aazami HA, et al. Synovial and systemic pharmacokinetics (PK) of triamcinolone acetonide (TA) following intra-articular (IA) injection of an extended-release microsphere-based formulation (FX006) or standard crystalline suspension in patients with knee osteoarthritis (OA). Osteoarthr Cartil. 2018;26(1):34–42.
Russell SJ, Sala R, Conaghan PG, et al. Triamcinolone acetonide extended-release in patients with osteoarthritis and type 2 diabetes: a randomized, phase 2 study. Rheumatol (Oxf). 2018;57(12):2235–41.
Kivitz A, Kwong L, Shlotzhauer T, Lufkin J, Cinar A, Kelley S. A randomized, phase IIa study to assess the systemic exposure of triamcinolone acetonide following injection of extended-release triamcinolone acetonide or traditional triamcinolone acetonide into both knees of patients with bilateral knee osteoarthritis. Ther Adv Musculoskelet Dis. 2019;2019:11.
Altman R, Alarcon G, Appelrouth D, et al. The American College of Rheumatology criteria for the classification and reporting of osteoarthritis of the hip. Arthritis Rheum. 1991;34(5):505–14.
KENALOG®-40 injection (triamcinolone acetonide injectable suspension, USP). Bristol Myers Squibb: Princeton, NJ. 2019. Accessed Apr 2019.
Bodick N, Lufkin J, Willwerth C, et al. An intra-articular, extended-release formulation of triamcinolone acetonide prolongs and amplifies analgesic effect in patients with osteoarthritis of the knee: a randomized clinical trial. J Bone Joint Surg Am. 2015;97(11):877–88.
Hanson P, Kivitz A, Mehra P, et al. Safety and systemic exposure of triamcinolone acetonide following ultrasound-guided intra-articular injection of triamcinolone extended-release or standard triamcinolone acetonide in patients with shoulder osteoarthritis: an open-label, randomized study. Drugs R D. 2021;21(3):285–93.
Acknowledgements
Funding
This study, the Rapid Service Fee, and the Open Access Fee were funded by Flexion Therapeutics, Inc., a wholly owned subsidiary of Pacira BioSciences, Inc. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Medical Writing and/or Editorial Assistance
Professional medical writing and editorial assistance in the preparation of this article was provided by Holly Tomlin, MS, Flexion Therapeutics, Inc., and Stephanie Agbu, PhD, ApotheCom, Yardley, PA, and was funded by Flexion Therapeutics, Inc. The authors thank Dr. Louis Levy of TriWest Research Associates (El Cajon, CA, USA) for his contribution to the study. Flexion Therapeutics, Inc. is a wholly owned subsidiary of Pacira BioSciences, Inc.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
AK and AC assisted in data acquisition, analysis, and interpretation. All authors reviewed and approved the manuscript.
Prior Presentation
This work was previously presented at the Osteoarthritis Research Society International (OARSI) 2019 World Congress, May 3–4, 2019, Toronto, Canada.
Disclosures
Alan Kivitz is a shareholder of Amgen, Gilead, GlaxoSmithKline, Novartis, Pfizer, and Sanofi. He has received consulting fees from AbbVie, Boehringer Ingelheim, Flexion Therapeutics, Gilead, Janssen, Pfizer, Regeneron, Sanofi, and Sun Pharma Advanced Research. He has also served as a paid presenter or speaker for AbbVie, Celgene, Flexion Therapeutics, Genzyme, GlaxoSmithKline, Eli Lilly, Merck, Novartis, Pfizer, Sanofi, and UCB. Purvi Mehra is an employee of ARTEMIS Institute for Clinical Research, a clinical research firm with clients in the pharmaceutical and medical device industries. Peter Hanson is an employee of BioSolutions Clinical Research Center; is a paid consultant for Centrexion Therapeutics and Flexion Therapeutics, Inc.; and has received research support from Centrexion Therapeutics, Flexion Therapeutics, Inc., and Samumed. Louis Kwong has nothing to disclose. Amy Cinar is a former employee of Flexion Therapeutics, Inc. Joelle Lufkin is a former employee of Flexion Therapeutics, Inc. She is an independent contractor in Beverly, MA, USA. Scott Kelley is a former employee of Flexion Therapeutics, Inc. and owns stock in Medtronic. He is now affiliated with Brigham and Women’s Hospital, Boston, MA, USA. Flexion Therapeutics, Inc. is a wholly owned subsidiary of Pacira BioSciences, Inc.
Compliance with Ethics Guidelines
All aspects of this study were reviewed and approved by a central institutional review board (IRB; Schulman Central IRB 4445 Lake Forest Drive, Suite 300 Cincinnati, OH 45242, USA), compiling approvals from each individual study site’s IRB (Altoona Center for Clinical Research, 175 Meadowbrook Ln, Duncansville, PA 16635, USA; IRB date of approval, November 22, 2017; Artemis Institute for Clinical Research - San Diego, 770 Washington St STE 300, San Diego, CA 92103, USA; IRB date of approval, November 22, 2017; TriWest Research Associates, 300 S Pierce St #201, El Cajon, CA 92020, USA; IRB date of approval, November 21, 2017; Rochester Clinical Research, Inc., 500 Helendale Rd, Rochester, NY 14609, USA; IRB date of approval, November 22, 2017; UCLA Medical Center, 1000 W Carson St, Torrance, CA 90502, USA; IRB date of approval, November 28, 2017; BioSolutions Clinical Research Center, 5565 Grossmont Center Dr #253, La Mesa, CA 91942, USA; IRB date of approval, November 21, 2017). All aspects of the study were performed in accordance with the Helsinki Declaration of 1964 and its later amendments. Written informed consent was obtained from all participants before study inclusion.
Data Availability
The datasets generated and/or analyzed within this publication are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Additional information
Drs. Cinar, Lufkin and Kelley were employees of Flexion Therapeutics, Inc. at the time this research was conducted. Flexion Therapeutics, Inc. is a wholly owned subsidiary of Pacira Biosciences, Inc.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kivitz, A., Mehra, P., Hanson, P. et al. A Randomized, Open-Label, Single-Dose Study to Assess Safety and Systemic Exposure of Triamcinolone Acetonide Extended-Release in Patients With Hip Osteoarthritis. Rheumatol Ther 9, 679–691 (2022). https://doi.org/10.1007/s40744-022-00430-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-022-00430-3