
Abstract
Introduction
The objective of this study was to compare a single intra-articular injection of an innovative high and low molecular weight hyaluronic acid formulation (HA-HL) versus placebo in treating moderate-to-severe symptomatic knee osteoarthritis.
Methods
Subjects with primary osteoarthritis knee pain (Kellgren and Lawrence grade 2–3) were randomly assigned to intra-articular HA-HL or placebo in a prospective, double-blind, 24-week study. The primary outcome variable was change from screening to week 24 of a Visual Analogue Scale (VAS) pain score. Secondary outcomes included Lequesne’s algofunctional index, EuroQol 5-Dimension Questionnaire, 5-level version (EQ-5D-5L), Outcome Measures in Arthritis Clinical Trials-Osteoarthritis Research Society International (OMERACT-OARSI) response and rescue medication usage.
Results
In a total of 692 randomized patients, a rapid decrease was observed in mean VAS pain score from baseline to week 1 (26 ± 24 mm in the HA-HL group vs. 23 ± 23 mm in the placebo group); pain intensity continued to decrease during 24 weeks of follow-up, reaching a mean change from baseline of 35 ± 28 mm vs. 32 ± 27 mm at week 24. Mixed model analysis demonstrated statistically significant differences between groups in favor of the HA-HL group at weeks 1, 6, 12, and 24. HA-HL was also more effective than placebo in improving Lequesne’s algofunctional index, OMERACT-OARSI response, and health-related quality of life. The use of rescue medication (paracetamol 500 mg tablets; ≤ 6 per day) was lower in the HA-HL group. Both treatments were similarly well tolerated.
Conclusions
A single intra-articular injection of an innovative high and low molecular weight hyaluronic acid formulation (HA-HL) is effective in providing fast, sustained, and clinically relevant reductions in pain, functional limitation, and health-related quality of life that were apparent at 1 week after the intra-articular injection and maintained throughout the 24-week follow-up in subjects with painful knee osteoarthritis, with a good safety profile.
Trial Registration
ClinicalTrials.gov identifier: NCT03200288.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
The use of intra-articular administration of hyaluronic acid is well established for the relief of joint pain and to improve joint function in knee osteoarthritis. |
We conducted a prospective, double-blind, 24-week study to compare a single intra-articular injection of an innovative high and low molecular weight hyaluronic acid formulation (HA-HL) versus placebo in treating moderate-to-severe symptomatic knee osteoarthritis. |
What was learned from the study? |
A single intra-articular injection of high and low molecular weight hyaluronic acid is more effective than placebo in treating the pain associated with moderate-to-severe knee osteoarthritis. The clinically relevant effects on pain, functionality, and health-related quality of life are quickly achieved and sustained throughout 24 weeks of follow-up. |
This study shows that a single intra-articular injection of an innovative formulation containing high and low molecular weight hyaluronic acid is safe as placebo with a similar incidence of adverse events. |
The single intra-articular administration of this new hyaluronic acid formulation represents a safe, effective, rapid, and sustained therapeutic strategy that opens new perspectives: improved adherence to hyaluronic acid treatments; improved safety because of the reduction in the number of injections; and the achievement of an onset of action similar to corticosteroids, the substitution of which would reduce the risk of steroid-related adverse events. |
Introduction
Osteoarthritis (OA) of the knee is characterized by chronic degeneration of the hyaline cartilage, meniscus, subchondral bone, and consecutive inflammation of the synovial membrane and other tissues [1, 2]. Increasing pain, loss of mobility, and the consequent loss of independence as people get older or as knee OA progresses result in the substantial burden of disability and impaired quality of life associated with the disease [1, 3, 4]. The use of intra-articular (i.a.) viscosupplementation with the non-sulphated glycosaminoglycan hyaluronic acid to relieve joint inflammation and pain and improve joint function in knee OA is well established [5,6,7,8,9,10], and included as a second-line therapeutic modality in current evidence-based treatment guidelines, consensus statements, and decision algorithms [3, 10,11,12,13,14,15,16,17].
A single-injection i.a. administration of a hyaluronic acid derivative able to maintain a level of clinical performance at least similar, if not superior, to that of other already approved products administered as a series of weekly injections, would represent a valuable therapeutic option for subjects with OA, while reducing the potential risks associated with multiple injections. An innovative formulation for i.a. injection consisting of high and low molecular weight hyaluronic acid (HA-HL) has been developed to address potential issues of hyaluronic acid concentration, injection volume, and viscosity. This new device injected into the knee joint rapidly reduces pain and allows for the re-establishment of the physiological and rheological properties of affected joints, with an expected final advantage for the subjects in terms of the risk/benefit ratio [18].
The medical device was granted CE certification in February 2015; it is marketed as Sinovial® HLFootnote 1 (IBSA) in Italy, the Czech Republic, Slovak Republic, Switzerland, Hungary, France, Poland, Albania, and Turkey, and it is currently licensed for the management of knee OA.
The objective of the current study was to compare the safety and efficacy of one i.a. injection of HA-HL with that of one i.a. injection of placebo in treating the primary clinical symptoms of pain and functional capacity, patient global status evaluation, and analgesic consumption, in subjects with symptomatic knee OA.
Methods
Study Design
This was a 24-week, phase 3, multicenter, randomized, double-blind, placebo-controlled, parallel-group study (ClinicalTrials.gov identifier NCT03200288) comparing the clinical performance and safety of a single i.a. injection of HA-HL with a single i.a. injection of placebo (saline) in the treatment of pain in symptomatic knee osteoarthritis.
Participants were recruited among outpatients of public/private/university clinics and/or hospitals in Belgium, Germany, Hungary, Italy, and Poland. The trial protocol is available at https://clinicaltrials.gov/ct2/show/NCT03200288.
The trial protocol was approved by the local Ethical Committee of each participating site and the National Regulatory Authorities of Italy, according to the specific national regulation. The full list of institutional review boards/independent ethics committees that approved this study can be found in the Supplementary Material. The study coordinator was Prof. Jean Yves Reginster, MD, PhD, of the University of Liège, Liège, Belgium. The trial was conducted in accordance with the Declaration of Helsinki and its modifications, the rules of the International Conference on Harmonization (ICH) Good Clinical Practices (GCP), and ISO 14155, the European Union Council Directive 93/42/EEC amended by 2007/47/EC, the MEDDEV 2. 12–1 rev. 6 and amendments, and all relevant local legislation in force on the conduct of clinical investigations with medical devices in OA [19,20,21,22,23,24,25,26,27,28]. All patients provided written informed consent. In addition, particular reference was made to the methodology described in Kirchner et al. [29], a study of a new i.a. HA product already authorized in both Europe and the USA.
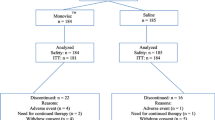
A total of seven visits were performed (Fig. 1). Visits consisted of screening (visit 0), a baseline visit (visit 1), which included randomization, and administration of i.a. HA-HL or placebo. Follow-up visits were performed after 7 days from treatment (week 1; visit 2); and at weeks 6, 12, 18 and 24 (visits 3–6). Visit 6 (week 24) was the final visit and the end of the study. An end of study/early termination visit was performed within 2 weeks from the withdrawal of any subject from the study.

Study design and subject disposition. The baseline visit (randomization) was arranged after the screening visit (visit 0). If a wash-out was not necessary, the patient could be admitted to visit 1 (baseline) on the same day as the screening visit, in which case attention had to be paid that all the procedures/assessments foreseen at the baseline visit were performed. The wash-out period was only needed if patients were taking analgesics/NSAIDs. The per-protocol (PP) population consisted of all patients in the intention-to-treat (ITT) population who did not have any major protocol violations. The PP population consisted of 341 patients in the HA-HL group and 340 in the placebo group; six patients in the HA-HL group and five in the placebo group had major protocol deviations, most commonly intake of a prohibited medication (n = 5) or violation of an exclusion criteria (n = 3)
A period of 24 weeks on-study (including screening and post-treatment follow-up period) was planned for each patient to ensure the long-term evaluation of the treatment. For patients who were taking analgesic/nonsteroidal anti-inflammatory drugs (NSAIDs) at the time of the screening, a wash-out period lasting five half-lives of analgesic/NSAIDs was observed before the baseline visit. During the study, rescue medication (paracetamol 500 mg tablets provided by the sponsor of the study; ≤ 6 per day) was permitted in the case of unbearable pain at the target knee or another condition requiring analgesic therapy. Rescue medication was not permitted during the 24-h preceding each visit. The subjects were required to record daily consumption of rescue medication in a diary and to return totally used, partially used or unused boxes at the next visit. Topical analgesics/NSAIDs, topical corticosteroids, and non-pharmacological therapy to joints other than the target knee were allowed. Adverse events (AEs), changes in concomitant medications, and changes in lifestyle were checked and recorded at each visit. Target knee assessments and global assessments of status were performed, and the level of treatment satisfaction and AEs recorded at each visit.
Patient and Public Involvement
Patients were not involved in the design or conduct of the study, development of outcomes, or dissemination of study results.
Participants
Eligible participants were female and male subjects ≥ 40–80 years of age with primary knee OA according to American College of Rheumatology (ACR) criteria, with Kellgren and Lawrence grade 2–3 radiographic evidence of OA [30], symptoms of at least 3 months in duration, and moderate-to-severe pain at inclusion. Screening pain intensity in the target knee measured by 100 mm Visual Analogue Scale (VAS) was required to be ≥ 40 mm VAS (and ≤ 20 mm in the contralateral knee) and confirmed at randomization after wash-out from analgesics/NSAIDs. Participants had to be willing and able to comply with study procedures, including usage of paracetamol (acetaminophen) as the only analgesic. Key exclusion criteria and prohibited concomitant medications are summarized in Table 1.
Treatments and Randomization
Eligible subjects were randomized to a single i.a. injection of HA-HL or placebo (buffered sterile physiological saline solution). Subjects were assigned sequentially into one of two parallel treatment groups in a balanced 1:1 ratio within each site using a computer-generated randomization list prepared by the Randomisation Manager using validated SAS® software. A strict protocol of data management, confidentiality, and access to the randomization list was followed. In addition to randomization, a number of measures were taken to minimize bias. The patients and the performance evaluator were blinded to treatment group assignment and, in recognition of the different viscosities of HA-HL and the placebo solutions, the physician who performed the injection (injector) was in all instances different from the physician who performed the efficacy and safety assessments (evaluator) and never evaluated the clinical status of patients or study parameters. Injectors used the same gauge of needles for both injections. Both treatments were packaged identically in order to maintain blinding. Target knee assessment was performed and recorded by the evaluator, who was not present at any of the injections and was blinded to the study product administration. The injector always performed the injections but never evaluated the clinical status of the patients or the study parameters.
The medical device was a sterile solution containing 64 mg (32 mg of high and 32 mg of low molecular weight) non-chemically modified hyaluronic acid sodium salt of biofermentation origin per 2 ml of 3.2% buffered solution in a 2.25 ml unit-dose syringe. The reference therapy (placebo) was a sterile buffered physiological solution with the same constituents, pH, volume and osmolality, with the exception of hyaluronic acid. The device manufacturer was IBSA.
Treatment was administered as an i.a. injection at day 1 (baseline visit) into the synovial space of the target knee. The treatments were administered by a physician with knowledge and experience in the assessment and management of subjects with knee OA and performing i.a. injections. The same standardized injection technique for HA-HL and placebo recipients was adopted at each site, according to the injecting physician's usual clinical practice, the subject's condition, and in accordance with the study protocol. Pre-administration of topical or subcutaneous local anesthetic was permitted.
Outcome Measures
The primary outcome variable was defined as the change from screening to visit 6 (week 24) of the VAS pain score calculated as a VAS measure ranging from 0 to 100 mm, where 0 represented no pain and 100 represented unbearable pain. Secondary outcomes were assessed at 1, 6, 12, 18, and 24 weeks and included change from baseline in Lequesne’s algofunctional index; change from baseline in global status assessed by subject using the EuroQol 5-Dimension Questionnaire, 5-level version (EQ-5D-5L); change from baseline in global status assessed by the physician on a 5-point verbal Likert scale, dichotomized to ‘Improvement’ and ‘No change/Worsening’; overall response rate according to Outcome Measures in Arthritis Clinical Trials-Osteoarthritis Research Society International (OMERACT-OARSI) criteria [31]; and rescue medication usage.
Safety outcomes included the incidence and frequency of AEs and change from baseline in vital signs and physical examination parameters. The level of treatment satisfaction was assessed by subject at baseline (within 15 min after the i.a. injection), in 24 ± 1 h after the injection, and at week 1 and week 6. AEs were coded by system organ class (SOC) and preferred term using the Medical Dictionary for Regulatory Activities (MedDRA)™ Version 21.1.
Statistical Methods
The sample size calculation was based on findings from a prior registration study of HA knee injection study (IBSA data on file) that showed a 13-week difference in pain score between treated and placebo groups of 6.39 mm (two-sided p value, 0. 0374). Based on the normal distribution with a two-sided alpha level of 0.05, a power of 0.80 and a 1:1 ratio of treated to placebo patients, a total sample size of 606 patients (303 assigned to treatment and 303 assigned to placebo) was calculated to demonstrate superiority. Considering a dropout rate up to 15%, a sample size of 720 patients was initially proposed. However, enrolment was stopped before reaching the 720 enrolled patients, considering the low dropout rate.
Analysis of the primary efficacy endpoint was performed in the intent-to-treat (ITT) population, and sensitivity analyses were repeated in the per-protocol (PP) population. The VAS knee pain score was analyzed using a mixed model for repeated measures approach, including treatment*time interaction, site, treatment*site interaction, baseline score, visit, and treatment*visit. A first-order autoregressive covariance structure was used. If the treatment*site interaction term was not significant (p > 0.15), it was removed from the model. A set of hierarchical analysesFootnote 2 was derived from slices based on visits to test the effect at each visit. Analyses of available and imputed data were performed separately for each visit on both absolute and change from baseline data.
The safety analyses were performed in the safety population (which coincided with the ITT population). Hypothesis testing was carried out at the two-sided α = 0.05 level when comparing subsequent assessment time points and/or treatments. Descriptive statistics were provided for all variables by treatment group; continuous variables were summarized by using no., mean, median, standard deviation (SD) and range; categorical variables were summarized by using frequency distributions and percentages. The statistical analyses were performed by an independent consultant using SAS® Statistical Analysis Software v 9.2 (SAS Institute, Cary, NC, USA).
Results
The study was conducted in 31 centers in five European countries (one in Belgium, four in Germany, six in Hungary, three in Italy, and 17 in Poland). The first subject was enrolled on the June 29, 2017 and the last subject completed on the October 30, 2018.
Subject Disposition
A total of 708 subjects were screened, and 692 of them were randomized (HA-HL, n = 347; placebo, n = 345) and received a single i.a. injection per-protocol, while 16 subjects were not randomized and were not treated. As the treatment was administered at each of the investigational sites by a qualified professional (unblinded injector), compliance was 100%. Subject disposition for the study is presented in Fig. 1. The ITT and safety populations consisted of 692 subjects (347 in the HA-HL group and 345 in the placebo group). Minor protocol deviations led to some exclusions from the PP population, which consisted of 681 subjects (341 in the HA-HL group and 340 in the placebo group).
Subject demographics at baseline are presented in Table 2. The average age was 63.7 years, and 67% of enrolled subjects were females. There were no statistically significant differences in baseline characteristics between groups, including knee deviation, OA location, Kellgren and Lawrence grade, and ACR criteria. Approximately 60% of subjects in both groups had radiographically confirmed Kellgren and Lawrence grade 2 knee OA, and the remainder grade 3. The mean time since diagnosis of knee OA was 5.0 years in the HA-HL group and 4.8 years in the placebo group (p = 0.656). All patients had knee pain and osteophytes, with the majority of patients experiencing morning stiffness and crepitus (83.5% and 83.0%, respectively).
Overall, 273 subjects (78.7%) in the HA-HL group and 274 (79.4%) in the placebo group had ≥ 1 concomitant medical condition (p = 0.852). The most common conditions included vascular disorders, metabolism and nutrition disorders, and musculoskeletal disorders.
A total of 663 subjects (95.5%), 336 (96.8%) in the HA-HL group and 327 (94.8%) in the placebo group, completed the study and 29 subjects (4.2%), 11 (3.2%) in the HA-HL group and 18 (5.2%) in the placebo group, discontinued the study because of voluntary withdrawal or were lost to follow-up. Major protocol deviations were reported in six subjects (1.7% of ITT) in the HA-HL group and in five (1.5%) in the placebo group, most commonly because of intake of prohibited medication (five patients overall) and violation of an exclusion criterion (three patients overall).
Clinical Outcomes
VAS Pain Score
The mean VAS for pain decreased from baseline to any post-baseline time point in both treatment groups, with the decrease according to the no-imputation method for the ITT population (primary efficacy endpoint) greater in the HA-HL group than with placebo at all post-baseline time points, and significantly greater at any time point with the exception of week 18 (Table 3).
Overall, there was a rapid decrease from baseline to week 1 in mean VAS pain score (26 ± 24 mm in the HA-HL group vs. 23 ± 23 mm in the placebo group); pain intensity continued to decrease during 24 weeks of follow-up, reaching a mean change from baseline of 35 ± 28 mm versus 32 ± 27 mm at week 24.
Analysis of the primary endpoint using a mixed model analysis with site*treatment interaction term on changes in VAS pain score in the ITT population confirmed that the interaction between treatment and site term was maintained (p = 0.001). Mixed model analysis demonstrated statistically significant differences between groups in favor of the HA-HL group at weeks 1, 6, 12, and 24, whereas the difference between groups at week 18 remained borderline significant (difference 3.4 mm, p = 0.051; Table 3 and Fig. 2).

Visual Analogue Scale (VAS) pain reduction using a repeated measures mixed model analysis of covariance that included factors for treatment, baseline, and site (scale of 0-best to 100-worst)
The results of the VAS pain score in the PP population and with the last observation carried forward (LOCF) approach were consistent with those observed with the no imputation method. Further analysis using age, gender, and body mass index (BMI) as terms found that neither age, BMI, nor gender were confounding factors in the response of subjects to HA-HL treatment (data not shown). Similar results and statistically significant between-group differences at all time points were shown when data were analyzed using a multiple imputation model (data not shown), although the 18-week data did not consistently demonstrate statistical superiority of the experimental therapy.
Lequesne’s Algofunctional Index
The extent of decrease from baseline of the mean score of the Lequesne’s algofunctional index (with no imputation) was significantly greater in the HA-HL group than in the placebo group at any post-baseline time point (Table 4).
The results in the ITT population with the LOCF approach were consistent with those seen in the no imputation method, and the mixed model analysis with site*treatment interaction term showed statistically significant effects in favor of the HA-HL for treatment, visit, baseline, site and treatment*site interaction (all p ≤ 0.001), whereas the treatment*visit interaction was not statistically significant (Fig. 3).

Reduction in Lequesne algofunctional Index using a repeated measures mixed model analysis of covariance that included factors for treatment, baseline, and site (scale of 0-best to 24-worst)
Health-Related Quality of Life
All components of the patient-assessed EQ-5D-5L showed improvements over the treatment period in both groups. The extent of the increase from baseline mean EQ-5D-5L general health VAS score was greater in the HA-HL group than in the placebo group at any post-baseline time point, and the comparison between groups showed statistically significant differences, in favor of the HA-HL group, in the distribution of scores of each item of the EQ-5D-5L 5-point verbal Likert scale (mobility problems, self-care problems, usual activities problems, pain and discomfort problems, and anxiety problems) (Fig. 4).

Change in EuroQol 5-Dimension Questionnaire, 5-level version (EQ-5D-5L) over time. *Analysis of covariance model; p value < 0.05. p values from Cochran–Mantel–Haenszel (CMH) test adjusted for baseline value
Investigator’s Assessment of Global Status
The extent of the decrease of the mean score of the investigator’s assessment of global status was slightly greater in the HA-HL group than in the placebo group at any post-baseline time point, and the comparison between groups in the distribution of scores of the investigator’s assessment of global status (very good, good, fair, poor, very poor) showed statistically significant differences at any post-baseline time point in favor of the HA-HL group compared to the placebo group (Fig. 5).

Investigator’s assessment of global status on a scale of 1-very good to 5-very poor
Overall Response Rate
The percentage of patients with success according to OMERACT-OARSI was significantly higher in the HA-HL group than in the placebo group at any post-baseline time point (except at week 18) (Fig. 6).

Overall response rate according to the Outcome Measures in Arthritis Clinical Trials-Osteoarthritis Research Society International (OMERACT-OARSI) criteria over time in the ITT population (N = 692)
Use of Rescue Medication
Approximately 75% of patients in both treatment groups used at least one tablet of rescue paracetamol in the overall 0 to 24-week follow-up period. There was no statistically significant difference between groups in the total amount of rescue medication during weeks 0–24 of the study (mean 84.9 vs. 73.4 tablets in the placebo vs. HA-HL group, p = 0.271), or when evaluated as number of tablets between visits or rescue medication use at any time.
Safety and Tolerability
Overall, both treatments were well tolerated, the safety and tolerability of HA-HL were similar to that of placebo, and there were no changes from baseline in vital signs or physical examination. A total of 845 post-treatment AEs were reported in 187 subjects (53.9%) in the HA-HL group and 703 post-treatment AEs were reported in 180 subjects (52.2%) in the placebo group (p = 0.703) (Table 5). The most commonly reported post-treatment AEs by preferred term were: headache, with 79 subjects (22.8%) in the HA-HL group and 74 (21.4%) in the placebo group; arthralgia, with 52 subjects (15.0%) in the HA-HL group and 51 (14.8%) in the placebo group; and nasopharyngitis, with 40 subjects (11.5%) in the HA-HL group and 39 (11.3%) in the placebo group.
Fifteen post-treatment serious AEs (SAEs) were reported in nine subjects (2.6%) in HA-HL group and ten post-treatment SAEs were reported in nine subjects (2.6%) in the placebo group (none was fatal). The difference between groups was not statistically significant (p = 1.000). None of the SAEs in either of the treatment groups was treatment-related. Eleven post-treatment AEs of severe intensity were reported in seven subjects (2.0%) in HA-HL group and two were reported in two subjects (0.6%) in the placebo group. The difference between groups was not statistically significant (p = 0.177) (Table 5). Of the five treatment-related AEs reported in five subjects (1.4%) in HA-HL group and 13 treatment-related AEs reported in nine subjects (2.6%) in the placebo group (p = 0.296), most consisted of local injection site reactions (injection site pain, rash, bruising, and hypoesthesia).
Subjects in the HA-HL group had a significantly higher level of satisfaction with treatment than those in the placebo group after both 1 week and 6 weeks from treatment. At week 1, 194 patients (55.9%) in the HA-HL group and 171 (49.6%) in the placebo group were very or extremely satisfied with the treatment, whereas 35 patients (10.1%) in the HA-HL group and 47 (13.6%) in the placebo group were not at all or slightly satisfied with the treatment (p = 0.035).
At week 6, 179 patients (52.3%) in the HA-HL group and 161 (47.3%) in the placebo group were very or extremely satisfied with the treatment, whereas 38 patients (11.1%) in the HA-HL group and 55 (16.2%) in the placebo group were not at all or slightly satisfied with the treatment (p = 0.039).
Discussion
An effective conservative pharmacological therapy able to provide pain relief and functional amelioration in knee OA with a high-dose, low-volume single injection would offer advantages over conservative therapies for OA that require multiple injections, including better patient compliance and a reduced risk of AEs compared with NSAIDs [8]. This study showed that a single i.a. knee injection of a high and low molecular weight hyaluronic acid formulation in patients with knee OA provided clinical benefit that was evident from week 1 and sustained for an extended period with excellent tolerability.
The objective of this study was to evaluate the clinical performance and the safety of the hyaluronic acid formulation in the symptomatic treatment of pain due to OA of the knee, as compared to matched placebo. The two treatment groups were well matched for demographic and other baseline characteristics, including baseline VAS pain score, OA history, and radiological assessments. The results of the primary efficacy endpoint for the ITT population, using a mixed model analysis with site*treatment interaction showed that the decrease of VAS for pain score from baseline to any post-baseline time point was significantly greater in the HA-HL group than in the placebo group.
Several studies have investigated the efficacy and safety of the high and low molecular weight hyaluronic acid formulation (Sinovial® HL; HA-HL) in OA. However, the present study involved the largest number of patients. A randomized controlled trial compared three i.a. injections of HA-HL to platelet-rich plasma (PRP; 5.5 ml) in 48 professional soccer players with unilateral pain not responsive to conservative treatment. A good response was observed in both groups at 3, 6, and 12 months, whereas on intergroup analysis, the HA-HL group showed a better outcome at 3 and 6 months of follow-up compared to PRP [18].
The efficacy of two i.a. injections of HA-HL was also compared to two i.a. injections of high molecular weight hyaluronic acid in patients with a BMI > 30 affected by Kellgren and Lawrence grade > 3 knee OA not responsive to conservative treatments. The randomized controlled trial involved 48 patients and showed that treatment with HA-HL provided better outcomes in term of pain reduction and function than high molecular weight HA [32]. Another study of 35 patients suffering from radiologically confirmed symptomatic knee OA reported that two i.a. injections of HA-HL 2 weeks apart provided good outcomes that lasted over 6 months, with reduction of VAS pain, Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) total score, consumption of NSAIDs and improvement of range of motion [33].
The HA-HL formulation has been utilized in hip and hand OA with similar findings of efficacy and safety. Abate and Salini enrolled 20 patients with moderate-to-severe hip OA; each patient received an i.a. ultrasound-guided injection of HA-HL at baseline and after 40 days [34]. VAS, Lequesne index, and Harris Hip score were evaluated at 3 and 6 months. The data collected were retrospectively compared with those obtained in a cohort of 20 patients matched for sex, age, and severity of hip OA, treated with high molecular weight hyaluronic acid. The intra-group comparison showed a significant improvement in clinical and functional outcomes at 3 and 6 months in both cohorts, while the infra-group comparison showed better results in patients treated with HA-HL.
The high and low molecular weight formulation of HA has also been suggested to be more effective than triamcinolone in pain relief and joint function improvement in hand OA, with a rapid and persistent effect, providing a valid alternative to steroids in the management of OA of the trapeziometacarpal joint (TMJ) [35]. In another small study in patients with TMJ OA, the efficacy on pain and function was evident as early as the first month and persisted at 6 months of follow-up [36].
This was the first study to explore the efficacy of a single injection of the new HA-HL formulation.
Hyaluronic acids do not generally have a quick and simple analgesic effect, and their full benefits may not be apparent until at least 4 weeks. Nevertheless, symptomatic relief after a single i.a. injection of HA-HL was rapidly achieved and the statistically significant between-group differences in pain intensity in favor of HA-HL in our study approached or exceeded the 5 mm difference, which is considered clinically relevant for symptomatic slow-acting drugs for OA (SYSADOA) [27], at most time points, and was significantly in favor of HA-HL at all time points, except for week 18, when it reached only borderline significance.
The differences in mean VAS pain reductions between the active treatment and placebo were modest, and may not fully explain the potential benefits of HA-HL in individuals with chronic pain. For example, at the individual level, the decrease in pain intensity from baseline was greater than 30 mm at week 6 and all subsequent time points in the HA-HL group, a decrease of more than 50% from study inclusion. This difference can be considered clinically relevant [37, 38]. Indeed, group mean differences may obscure meaningful improvements for individual patients, and the determination of clinical effectiveness should take into consideration risks and benefits beyond the provision of pain relief [37]. In the case of hyaluronic acid, this includes potentially delaying knee arthroplasty [17, 39]. Furthermore, the effect of placebo in OA, particularly when given by injection, is well known [40], and our control arm, i.a. injection of saline, is in itself known to provide benefit in OA [41,42,43]. However, despite the “placebo response”, the decrease in pain intensity in our study was still greater for patients treated with HA-HL. The issue of what constitutes a proper placebo in randomized controlled trials thus arises. Considering the evidence for the benefits of i.a. saline injection, a sham injection as the control arm may be necessary to reveal the real differences in efficacy for i.a. administration in OA [44].
At all post-treatment time points, the HA-HL treatment group was judged by the investigators to have had a better outcome than the placebo group, mostly due to a higher percentage of patients with a score of good/very good in the assessment of global status. In agreement with the findings for the other efficacy variables, OMERACT-OARSI success scores were more favorable for HA-HL patients than those given placebo at all time points, attaining statistical significance at all but week 18.
Our findings further support the evidence from network meta-analysis of randomized controlled trials that i.a. HA decreases symptomatic pain and/or improves physical function in knee OA [45]. Furthermore, the post hoc analyses of this study show that the effect size for HA-HL for VAS pain intensity was in the range of what is observed with NSAIDs until week 12 [17], and was so until at least week 18 for the Lequesne's Index, supporting a clinically relevant effect size for i.a. HA-HL. In agreement with the data of previous studies, HA-HL was as safe as placebo; there were no treatment-related SAEs and no changes from baseline in vital signs and physical examination. Most of the treatment-related AEs in both treatment groups consisted of local injection site reactions, and there were no discontinuations related to HA-HL.
In health research, the perspective of patients is increasingly considered pivotal to support the development, approval, and reimbursement of health interventions that best meet the needs of patients. This approach has been developed by a panel of experts from the World Health Organization (WHO), the European Society for Clinical and Economic Aspects of Osteoporosis, Osteoarthritis and Musculoskeletal Diseases (ESCEO), the European Medicines Agency (EMA), and the Outcomes Measures in Rheumatology (OMERACT), among others [46]. In our study, subjects given i.a. HA-HL were generally more satisfied with their treatment than those receiving the placebo, principally due to a higher proportion characterizing their response as “extremely” satisfying. This result confirms the use of hyaluronic acid in the management of OA, given the high degree of patients’ satisfaction [47].
Conclusions
In conclusion, the results of the study have shown that a single i.a. injection of an innovative high and low molecular weight hyaluronic acid formulation (HA-HL), was more effective than placebo in treating the pain associated with moderate-to-severe knee OA, resulting in symptomatic relief that was apparent 1 week after the i.a. injection and maintained throughout the 24-week follow-up period. Similar findings were observed for all secondary outcome variables, and both treatments were equally well tolerated, with the incidence of adverse events indistinguishable between groups.
Notes
Other brand name: Intragel® HL.
European Medicines Agency “Points to Consider on Multiplicity Issues in Clinical Trials” (CPMP/EWP/908/99) and FDA fixed sequence method of the “Multiple endpoints in Clinical Trials Guidance for Industry” draft guidance, January 2017.
References
Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. Lancet. 2019;393(10182):1745–59.
Mobasheri A, Batt M. An update on the pathophysiology of osteoarthritis. Ann Phys Rehabil Med. 2016;59(5–6):333–9.
Bannuru RR, Osani MC, Vaysbrot EE, et al. OARSI guidelines for the non-surgical management of knee, hip, and polyarticular osteoarthritis. Osteoarthri Cartil. 2019;27(11):1578–89.
Cross M, Smith E, Hoy D, et al. The global burden of hip and knee osteoarthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis. 2014;73(7):1323–30.
Altman RD. Pharmacological therapies for osteoarthritis of the hand: a review of the evidence. Drugs Aging. 2010;27(9):729–45.
Bruyere O, Cooper C, Arden N, et al. Can we identify patients with high risk of osteoarthritis progression who will respond to treatment? A focus on epidemiology and phenotype of osteoarthritis. Drugs Aging. 2015;32(3):179–87.
Cooper C, Rannou F, Richette P, et al. Use of intraarticular hyaluronic acid in the management of knee osteoarthritis in clinical practice. Arthritis Care Res (Hoboken). 2017;69(9):1287–96.
Maheu E, Bannuru RR, Herrero-Beaumont G, et al. Why we should definitely include intra-articular hyaluronic acid as a therapeutic option in the management of knee osteoarthritis: Results of an extensive critical literature review. Semin Arthritis Rheum. 2019;48(4):563–72.
Maheu E, Rannou F, Reginster JY. Efficacy and safety of hyaluronic acid in the management of osteoarthritis: Evidence from real-life setting trials and surveys. Semin Arthritis Rheum. 2016;45(4 Suppl):S28-33.
Raman R, Henrotin Y, Chevalier X, et al. Decision algorithms for the retreatment with viscosupplementation in patients suffering from knee osteoarthritis: Recommendations from the EUROpean VIScosupplementation COnsensus Group (EUROVISCO). Cartilage. 2018;9(3):263–75.
Bruyere O, Cooper C, Pelletier JP, et al. An algorithm recommendation for the management of knee osteoarthritis in Europe and internationally: a report from a task force of the European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis (ESCEO). Semin Arthritis Rheum. 2014;44(3):253–63.
Fernandes L, Hagen KB, Bijlsma JW, et al. EULAR recommendations for the non-pharmacological core management of hip and knee osteoarthritis. Ann Rheum Dis. 2013;72(7):1125–35.
Hochberg MC, Altman RD, April KT, et al. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken). 2012;64(4):465–74.
Jordan KM, Arden NK, Doherty M, et al. EULAR Recommendations 2003: an evidence based approach to the management of knee osteoarthritis: Report of a Task Force of the Standing Committee for International Clinical Studies Including Therapeutic Trials (ESCISIT). Ann Rheum Dis. 2003;62(12):1145–55.
Rillo O, Riera H, Acosta C, et al. PANLAR consensus recommendations for the management in osteoarthritis of hand, hip, and knee. J Clin Rheumatol. 2016;22(7):345–54.
Ariani A, Manara M, Fioravanti A, et al. The Italian Society for Rheumatology clinical practice guidelines for the diagnosis and management of knee, hip and hand osteoarthritis. Reumatismo. 2019;71(S1):5–21.
Bruyere O, Honvo G, Veronese N, et al. An updated algorithm recommendation for the management of knee osteoarthritis from the European Society for Clinical and Economic Aspects of Osteoporosis, Osteoarthritis and Musculoskeletal Diseases (ESCEO). Semin Arthritis Rheum. 2019;49(3):337–50.
Papalia R, Zampogna B, Russo F, et al. Comparing hybrid hyaluronic acid with PRP in end career athletes with degenerative cartilage lesions of the knee. J Biol Regul Homeost Agents. 2016;30(4 Suppl 1):17–23.
US Department of Health and Human Services, U.S. Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Center for Devices and Radiological Health (CDRH). Guidance for Industry Clinical Development Programs for Drugs, Devices, and Biological Products Intended for the Treatment of Osteoarthritis (OA). Draft Guidance. US Food and Drug Administration; 1999.
Committee for Medicinal Products for Human Use (CHMP). Guideline on Clinical Investigation of Medicinal Products Used in the Treatment of Osteoarthritis (CPMP/EWP/784/97 Rev 1). London: European Medicines Agency; 2010.
Group for the Respect Of Ethics and Excellence in Science (GREES): Osteoarthritis Section. Recommendations for the registration of drugs used in the treatment of osteoarthritis. Ann Rheum Dis. 1996;55(8):552–7.
McAlindon TE, Driban JB, Henrotin Y, et al. OARSI clinical trials recommendations: design, conduct, and reporting of clinical trials for knee osteoarthritis. Osteoarthr Cartil. 2015;23(5):747–60.
Jordan JM, Henrotin Y. Osteoarthritis Research Society international initiative on recommendations for conducting clinical trials in osteoarthritis: overview. Osteoarthr Cartil. 2015;23(5):671–3.
Altman R, Brandt K, Hochberg M, et al. Design and conduct of clinical trials in patients with osteoarthritis: recommendations from a task force of the Osteoarthritis Research Society. Results from a workshop. Osteoarthr Cartil. 1996;4(4):217–43.
Dougados M, Devogelaer J-P, Annefeld M, et al. Recommendations for the registration of drugs used in the treatment of osteoarthritis. Ann Rheum Dis. 1996;55:552–7.
McAlindon TE, Bannuru RR, Sullivan MC, et al. OARSI guidelines for the non-surgical management of knee osteoarthritis. Osteoarthr Cartil. 2014;22(3):363–88.
Reginster JY, Reiter-Niesert S, Bruyere O, et al. Recommendations for an update of the 2010 European regulatory guideline on clinical investigation of medicinal products used in the treatment of osteoarthritis and reflections about related clinically relevant outcomes: expert consensus statement. Osteoarthr Cartil. 2015;23(12):2086–93.
Strand V, Kelman A. Outcome measures in osteoarthritis: randomized controlled trials. Curr Rheumatol Rep. 2004;6(1):20–30.
Kirchner M, Marshall D. A double-blind randomized controlled trial comparing alternate forms of high molecular weight hyaluronan for the treatment of osteoarthritis of the knee. Osteoar Cartil. 2006;14(2):154–62.
Kellgren JH, Lawrence JS. Radiological assessment of osteo-arthrosis. Ann Rheum Dis. 1957;16(4):494–502.
Pham T, van der Heijde D, Altman RD, et al. OMERACT-OARSI initiative: Osteoarthritis Research Society International set of responder criteria for osteoarthritis clinical trials revisited. Osteoarthr Cartil. 2004;12(5):389–99.
Papalia R, Russo F, Torre G, et al. Hybrid hyaluronic acid versus high molecular weight hyaluronic acid for the treatment of osteoarthritis in obese patients. J Biol Regul Homeost Agents. 2017;31(4 Suppl 2):103–9.
Manciameli A. Trattamento della gonartrosi moderata mediante iniezione intrarticolare di acido ialuronico sale sodico [Italian]. Giornale Ital Ortop Traumatol. 2018;44:146–9.
Abate M, Salini V. Efficacy and safety study on a new compound associating low and high molecular weight hyaluronic acid in the treatment of hip osteoarthritis. Int J Immunopathol Pharmacol. 2017;30(1):89–93.
Tenti S, Pascarelli NA, Giannotti S, et al. Can hybrid hyaluronic acid represent a valid approach to treat rizoarthrosis? A retrospective comparative study. BMC Musculoskelet Disord. 2017;18(1):444.
Bartoloni E, Luccioli F, La Paglia GMC, et al. Effect of Sinovial High-Low® injections in trapeziometacarpal osteoarthritis. Clin Exp Rheumatol. 2019;37(1):166.
Dworkin RH, Turk DC, McDermott MP, et al. Interpreting the clinical importance of group differences in chronic pain clinical trials: IMMPACT recommendations. Pain. 2009;146(3):238–44.
Moore RA, Moore OA, Derry S, McQuay HJ. Numbers needed to treat calculated from responder rates give a better indication of efficacy in osteoarthritis trials than mean pain scores. Arthritis Res Ther. 2008;10(2):R39.
Altman R, Lim S, Steen RG, Dasa V. Hyaluronic acid injections are associated with delay of total knee replacement surgery in patients with knee osteoarthritis: evidence from a large U.S. health claims database. PLoS ONE. 2015;10(12):e0145776.
Zhang W, Robertson J, Jones AC, Dieppe PA, Doherty M. The placebo effect and its determinants in osteoarthritis: meta-analysis of randomised controlled trials. Ann Rheum Dis. 2008;67(12):1716–23.
Saltzman BM, Leroux T, Meyer MA, et al. The therapeutic effect of intra-articular normal saline injections for knee osteoarthritis: a meta-analysis of evidence level 1 studies. Am J Sports Med. 2017;45(11):2647–53.
Bar-Or D, Rael LT, Brody EN. Use of saline as a placebo in intra-articular injections in osteoarthritis: potential contributions to nociceptive pain relief. Open Rheumatol J. 2017;11:16–22.
Altman RD, Devji T, Bhandari M, et al. Clinical benefit of intra-articular saline as a comparator in clinical trials of knee osteoarthritis treatments: a systematic review and meta-analysis of randomized trials. Semin Arthritis Rheum. 2016;46(2):151–9.
Migliore A, Bizzi E, Herrero-Beaumont J, et al. The discrepancy between recommendations and clinical practice for viscosupplementation in osteoarthritis: mind the gap! Eur Rev Med Pharmacol Sci. 2015;19(7):1124–9.
Beaudart C, Lengele L, Leclercq V, et al. Symptomatic efficacy of pharmacological treatments for knee osteoarthritis: a systematic review and a network meta-analysis with a 6-month time horizon. Drugs. 2020;80(18):1947–59.
de Wit M, Cooper C, Tugwell P, et al. Practical guidance for engaging patients in health research, treatment guidelines and regulatory processes: results of an expert group meeting organized by the World Health Organization (WHO) and the European Society for Clinical and Economic Aspects of Osteoporosis, Osteoarthritis and Musculoskeletal Diseases (ESCEO). Aging Clin Exp Res. 2019;31(7):905–15.
Conrozier T, Monet M, Lohse A, Raman R. Getting better or getting well? The Patient Acceptable Symptom State (PASS) better predicts patient’s satisfaction than the decrease of pain, in knee osteoarthritis subjects treated with viscosupplementation. Cartilage. 2018;9(4):370–7.
Acknowledgements
Funding
Funding for this study and the journal’s Rapid Service Fee were provided by IBSA Institut Biochimique S.A., Lugano, Switzerland, who also provided funding for the statistical analysis, conducted by Fred Hoeler.
Medical Writing Assistance
We thank Ray Hill, an independent medical writer, who provided medical writing support funded by IBSA Institut Biochimique S.A., Lugano, Switzerland.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
J-YR contributed to study conception and/or design and to the interpretation of study results, AM, J-YR, TB, RP, ZZ, OGK, GG, contributed to the acquisition of study results and all authors contributed to critical revision of the manuscript and gave final approval for submission for publication.
Disclosures
Alberto Migliore reports grants as a consultant from Eli Lilly, Fidia, MSD, Novartis, Pfizer, Roche, Sanofi, IBSA, Jansen and Abiogen for national and international studies and courses. François Rannou reports personal fees from Pierre Fabre, Sanofi-Aventis, Pfizer, Bayer, Expansciences, Thuasne, Grünenthal, Lohmann et Rauscher, and Janssen-Cilag, and grants from PHRC, ANR, H2020, the Fondation Arthritis. Jean-Yves Reginster reports grants and personal fees from IBSA-Genevrier, Mylan, Cniel, and Radius Health, and personal fees from Pierre Fabre, the Dairy Research Council (DRC), Faes Pharma, Rejuvenate Biomed, Samumed, Teva Pharmaceuticals, Theramex, Pfizer, Mithra Pharmaceuticals, Nutricia, Danone, Agnovos. Tomasz Blicharski, Rafal Plebanski, Zbigniew Zegota and Győrfi Gyula have nothing to disclose.
Compliance with Ethics Guidelines
The trial protocol was approved by the local Ethical Committee of each participating site and the National Regulatory Authorities of Italy, according to the specific national regulation. The full list of institutional review boards/independent ethics committees that approved this study can be found in the Supplementary Material. The study coordinator was Prof. Jean Yves Reginster, MD, PhD, of the University of Liège, Liège, Belgium. The trial was conducted in accordance with the Declaration of Helsinki and its modifications, the rules of the International Conference on Harmonization (ICH) Good Clinical Practices (GCP), and ISO 14155, the European Union Council Directive 93/42/EEC amended by 2007/47/EC, the MEDDEV 2. 12–1 rev. 6 and amendments, and all relevant local legislation in force on the conduct of clinical investigations with medical devices in OA [19,20,21,22,23,24,25,26,27,28]. All patients provided written informed consent.
Data Availability
The datasets generated during and/or analyzed during the current study are available on reasonable request. All requests should be made in writing to the corresponding author.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Migliore, A., Blicharski, T., Plebanski, R. et al. Knee Osteoarthritis Pain Management with an Innovative High and Low Molecular Weight Hyaluronic Acid Formulation (HA-HL): A Randomized Clinical Trial. Rheumatol Ther 8, 1617–1636 (2021). https://doi.org/10.1007/s40744-021-00363-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-021-00363-3