
Opinion statement
There is compelling evidence that interleukin (IL)-23/IL-17 axis plays a critical role in the pathogenesis of inflammatory spondyloarthritides including ankylosing spondylitis, reactive arthritis, psoriatic arthritis, and inflammatory bowel disease-associated arthritis. IL-17 is a family of cytokines. Of the six members of IL-17, IL-17A and IL-17F are the most closely related proinflammatory cytokines and effector molecules. A distinct subset of CD4+ T helper (Th) cells producing IL-17A and IL-17F is designated Th17 cells. Many cytokines are involved in Th17 cell differentiation and therefore are therapeutic targets. IL-6 in combination with transforming growth factor (TGF)-β mediates initiation of Th17 cell differentiation. IL-23 is not required for the initial differentiation of Th17 cells, but is indispensable for stabilizing differentiated Th17 clones in vivo. Clinical trials are undergoing to assess therapeutic efficacy of several monoclonal antibodies against these cytokines to suppress Th17 cells. Ustekinumab, a monoclonal antibody against p40 subunit which is shared by IL-12 and IL-23, has been approved to treat psoriasis and psoriatic arthritis. The therapeutic effect of ustekinumab is thought to be via inhibition of Th17 cells. Monoclonal antibodies to IL-17A and IL-17F and their receptors are also being evaluated in clinical trials. Retinoic acid-related orphan nuclear receptor (ROR)-γt is the master transcription factor for Th17 cells and hence a target. Recently, small molecules blocking ROR-γt activity have been identified and are potentially to be developed as drugs to inhibit Th17 cells. Small interfering RNA (siRNA) and small hairpin RNA (shRNA) targeting Rorc gene can be specifically delivered into CD4+ T cells to suppress IL-17 production for even more precise inhibition of Th17 cells. Technology advancement shows promise for these nucleotide-based therapeutics to be developed for clinical use.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
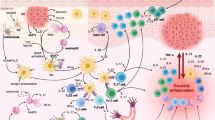
The discovery of interleukin (IL)-23 and subsequent identification of T helper (Th) type 17 cells have drastically changed the paradigm of the pathogenesis of T cell-mediated inflammatory diseases [1, 2]. We now understand that IL-23, produced by macrophages and dendritic cells, exerts its activity mainly via induction of IL-17 by T cells [3], and hence, the cytokine cascade is referred to as IL-23/IL-17 axis or pathway. Activities attributed to IL-12 and Th1 cells in immune-mediated inflammatory process have now been unveiled to be largely contributed by IL-23 and Th17 cells [4]. Evidence from animal models and human studies indicates that IL-23/IL-17 axis is important in mediating inflammation in spondyloarthritides [5•]. There are multiple key points in the IL-23/IL-17 axis that are targets for therapeutic intervention (Fig. 1). Clinical trials targeting this pathway have been exploited with promising results. Current clinical trials are focused on utilizing monoclonal antibodies against upstream cytokines which mediate Th17 cell differentiation, Th17 cytokines, and IL-17 receptors [6]. Transcription factors for the differentiation and development of Th17 cells are potential targets as well, but developing specific agents have been a hurdle until recently. Monoclonal antibodies are not suitable for this purpose due to their inability to access intracellular targets. Fortunately, alternative agents are able to fulfill this task. For instance, small molecules act on the Th17 master transcription factor, retinoic acid-related orphan nuclear receptor (ROR)-γt has been identified and are potentially to be developed as drugs [7, 8]. RNA interference (RNAi)-based technology has advanced to be able to target Rorc gene in CD4+ T cells [9•]. In prospect, these agents are to be explored for therapy in IL-23/IL-17-mediated inflammatory diseases including spondyloarthritides. This brief overview of IL-23/IL-17 axis and its regulatory factors will be focused on providing rationale for potential therapeutic targets. Clinical trials with individual agents in particular diseases will not be discussed in details since they will be covered in other articles of this series.

Multiple targets to block IL-23/IL-17 axis. TGF-β transforming growth factor-β, IL interleukin, TNF tumor necrosis factor, STAT signal transducers and activators of transcription protein, ROR-γt retinoic acid orphan nuclear receptor-γt. IL-23 is a heterodimeric cytokine composed of a p40 subunit that is shared with IL-12 and the p19 subunit; IL-23R, signaling IL-23 receptor, is composed of IL-23R and IL-12Rβ1; both IL-17A and IL-17F form homodimers to signal; IL-17A and IL-17F also form IL-17 A/F heterodimers. IL-17 receptor consists of five members, IL-17RA–RE. IL-17A and IL-17F signal through IL-17RA-RC complex. Act1, cytosolic adaptor protein, a key mediator for IL-17A- and IL-17F-mediated signaling. Names of therapeutic agents in bold are FDA-approved for clinical use.
IL-23/IL-17 axis
IL-17—an important family of cytokines for host defense and inflammation
IL-17 is a family of cytokines comprising six members, IL-17A, IL-17B, IL-17C, IL-17D, IL-17E (also called IL-25), and IL-17F. IL-17A is the prototype of the IL-17 family and often referred to as IL-17. Among these IL-17 family members, IL-17A and IL-17F are the most closely related in structure and function and best characterized. IL-17A and IL-17F bind to the same receptor complex. Thus, these two molecules are likely to have similar biological activities [10, 11]. Furthermore, IL-17A and IL-17F can both be secreted as disulfide-linked homodimers or heterodimers to give rise to three forms of IL-17, IL-17A/A, IL-17A/F, and IL-17F/F, with similar but potentially different biological activities. Indeed, IL-17A/A seems to have the most potent function in tissue inflammation, followed by IL-17A/F [12]. IL-17A and IL-17F are potent effector molecules affecting a broad range of hematopoietic and non-hematopoietic cells to protect the host against invading microbial pathogens. IL-17A and IL-17F promote the recruitment activation and subsequent migration of neutrophils [10, 13], inducing anti-microbial peptides and acute-phase protein production. Studies using gene knockout mice or blocking IL-17 signaling have demonstrated that both IL-17A and IL-17F play a critical role in host defense against a variety of pathogens. These include Salmonella enterica [14], Staphylococcus aureus [15, 16], Streptococcus pneumoniae [15, 16], Listeria monocytogenes [17], Helicobacter pylori [18], Citrobacter rodentium [15, 16], Candida albicans [19], herpes simplex virus [20], and Trypanosoma cruzi [21]. In humans, gene mutations affecting IL-17 production or IL-17 signaling cause increased susceptibility to infection of S. aureus, S. pneumoniae, and C. albicans [22].
In addition, being potent granulocyte attractants, both IL-17A and IL-17F act on a broad range of cell types to induce the expression of inflammatory cytokines including tumor necrosis factor (TNF), IL-1β, IL-6, IL-21, IL-23, granulocyte macrophage-colony stimulating factor, and granulocyte growth factor; chemokines including CXCL1, CXCL5, CXCL8, CXCL10, CCL2, and CCL7; and matrix metalloproteinases (MMP1, MMP3, and MMP13) by activating innate and tissue resident cells, such as fibroblasts and epithelial cells [6, 10]. IL-17A also induces the expression of intercellular cell adhesion molecule 1 (ICAM-1) in keratinocytes as well as inducible nitric oxides and cyclooxygenase-2 in chondrocytes [13]. These properties seem to be mainly associated with inflammation. It must be noted that a wide range of IL-17A and IL-17F activities can be both beneficial to the host in immunity against infections as well as detrimental in the context of immune-mediated inflammation when the IL-23/IL-17 pathway is dysregulated.
Th17—a distinct T helper cell subset
The identification of the IL-17 family of cytokines and the finding that IL-23 mediates the expansion of IL-17-producing T cells led to the discovery of a new subset of T helper cells which is designated Th17 cells. Th17 cells, first described in mice, are the major source of IL-17 in many types of adaptive immunity [23, 24]. Th17 cells produce IL-17A, IL-17F, IL-22, IL-21, and TNF. Similar to Th1 and Th2 cells, Th17 cells require specific cytokines and transcription factors for their differentiation. Transforming growth factor (TGF)-β1 is a regulatory cytokine with a pivotal role in controlling T cell homeostasis and differentiation [6]. Independent studies demonstrated that a combination of the proinflammatory cytokines IL-6 and TGF-β induces in vitro differentiation of highly purified naïve CD4+ T cells into IL-17-producing cells [25, 26]. During in vivo differentiation, a more complex network of cytokines, including IL-23, IL-21, and IL-1β, acts to promote or maintain Th17 development in addition to TGF-β1 and IL-6. During the initial Th17 development, IL-6-induced IL-21 acts as a positive amplification loop to enforce Th17 differentiation [27]. IL-21 was subsequently shown to be able to replace IL-6 at least in vitro. In the absence of IL-6, IL-21 together with TGF-β was shown to inhibit development of inducible T regulatory cells (iTreg) and to promote the differentiation of Th17 cells [27]. However, the in vivo relevance of IL-21 in the induction and function of Th17 remains controversial. It has been reported that the absence of IL-21 or IL-21R has no significant difference on the development of Th17 cells [28]. The most important cytokine involved in Th17 differentiation is IL-23. IL-23 is a heterodimeric cytokine composed of a unique p19 subunit and p40 subunit which is shared with IL-12. It has become clear that many of the previous associations of IL-12 and Th1 cells with autoimmunity can be explained by the requirement for IL-23 [1]. IL-23 was first found to preferentially induce IL-17 production by activated CD4+ T cells [29]. During Th17 cell differentiation, IL-23 is not required in the initial steps driving the differentiation of naïve CD4+ T cells into IL-17-producing cells, but it plays a critical role in stabilizing the phenotypic features of the Th17 lineage. Without IL-23, activated CD4+ T cells in the presence of IL-6 plus TGF-β can produce high amounts of IL-17, but cannot fully develop into pathogenic Th17 cells and acquire bystander regulatory properties mediated by IL-10 production [30]. Thus, IL-23 is essential for pathogenic Th17 cells to fully differentiate and exhibit effector function [30].
Th17 cell differentiation is finely controlled at transcription level by a dynamic regulatory network consisting of positive and negative regulators [31••, 32, 33]. Here, we focus the discussion on ROR-γt and signal transducers and activators of transcription (STAT)-3 in the context of therapeutic intervention. ROR-γt has been identified as the master transcription factor for Th17 cell differentiation. ROR-γt is an isoform of ROR-γ from a splice variant of the Rorc gene that results from initiation by a distinct promoter within the full length of Rorc gene [34–36]. Unlike ROR-γ, ROR-γt is exclusively expressed in lymphoid cells and is critical for T cell development and lymphoid organogenesis [35, 36]. ROR-γt promotes IL-17 expression by directly binding the promoter region of Il17 gene at multiple sites [33, 37, 38]. Rorc gene knockout mice have severely impaired Th17 cell differentiation and are protected from development of experimental autoimmune diseases [37]. Rorc-deficient CD4+ T cells are not responsive to IL-23 for IL-17 production, indicating that IL-23 exerts its effect on Th17 cells via ROR-γt [37]. Conversely, overexpression of ROR-γt promotes IL-17 expression. Furthermore, ROR-γt is required for fully differentiated Th17 cells to maintain their Th17 status [37]. STAT-3 also plays a critical role in Th17 differentiation. Deletion of STAT-3 in T cells abrogates Th17 differentiation [33]. Patients with hyper IgE syndrome due to dominant negative mutations of STAT-3 have severely impaired ability to produce Th17 cells [39]. STAT-3 binds and regulates many other transcription factors that participate in Th17 differentiation including ROR-γt. However, overexpression of STAT-3 in Rorc knockout cells does not restore IL-17 production, suggesting that STAT-3 requires ROR-γt to act on the Il17 locus to drive IL-17 production [33].
IL-17 produced by non-Th17 cells—importance in host defense
In addition to Th17 cells, a wide variety of immune cells including CD8+ T cells, γ/δT cells, natural killer T (NKT) cells, innate lymphoid cells, neutrophils, monocytes, and eosinophils can produce IL-17A and IL-17F under various conditions [40]. It is known that IL-17A and IL-17F produced by these immune cells are also IL-23-dependent. These non-Th17 cells produced IL-17 that are important for host defense but may not participate in immune-mediated inflammation. For example, in animal models, γ/δ T cells and IL-17 produced by them are essential for host defense in S. aureus skin infection [41], whereas IL-17-producing γ/δ T cells are not required for development of arthritis [42]. Furthermore, recently identified IL-17-producing innate lymphoid cells may be important in host defense rather than causing immune-mediated inflammation [43].
IL-17 receptors
The IL-17 family of cytokines mediates its biological functions via surface receptors on target cells. The IL-17 receptor (IL-17R) family contains five members, namely IL-17RA, IL-17RB, IL-17RC, IL-17RD, and IL-17RE [44–46]. All of these receptors have one transmembrane domain and a large intracellular C terminus. Functional receptors form heterodimers with IL-17RA as a common subunit. IL-17RA binds both IL-17A as well as IL-17F but requires the IL-17RC subunit to induce responses to IL-17A and IL-17F [47]. IL-17RA is highly expressed on hematopoietic cells, but also minor on osteoblasts, fibroblasts, endothelial cells, and epithelial cells. In humans, IL-17RA can form a heterodimer with IL-17RC that binds human IL-17A and IL-17F. In contrast to IL-17RA, IL-17RC is expressed only at low levels on hematopoietic cells, but it is highly expressed on non-hematopoietic cells [45, 48]. IL-17 activates several downstream signaling pathways including NFκB, MARP, and C/EBP pathways to induce gene expression of antibacterial peptides, proinflammatory chemokines and cytokines, and MMPs. Signaling through IL-17RA and IL-17RC is mediated by a master cytosolic adaptor molecule, Act1, which is recruited upon binding of IL-17 [45, 48]. Act1 is therefore a potential target to block IL-17 activity.
Role of IL-23/IL-17 axis in spondyloarthritis and psoriatic arthritis
The importance of IL-23/IL-17 axis in the pathogenesis spondyloarthritides is firmly supported by data from animal models, genetic studies, and translational studies. Readers are directed to recent excellent reviews for details of discussion [5•, 49]. Here, we highlight a few points pertinent to therapeutic consideration. In animal models, HLA-B27 overexpression in rats is sufficient to lead to activation of IL-23/IL-17 in the gut and joints. This may be related to the tendency of misfolding of B27 molecule [50, 51]. Moreover, systemic overexpression of IL-23 can result in tissue inflammation including enthesitis which may be caused by increased local production of IL-17A and Il-17F [5•, 49, 52]. Genetic variants identified to influence IL-23 production or signaling and Th17 differentiation include Il-23r, Card9, Il-12B, Il-16r, Il-27, Tyk2, and Stat3 [52, 53]. Increased levels of IL-23 and IL-17 as well as Th17 cells are found in patients with ankylosing spondylitis, psoriatic arthritis, and reactive arthritis. Importantly, both IL-23 and IL-17 are overexpressed in the inflamed synovial tissue [54].
Treatment
The introduction of anti-tumor necrosis factor (TNF) has revolutionized the armamentarium of therapeutic options to treat various autoimmune conditions including inflammatory spondyloarthritides, but there is still a great unmet need for alternative treatments in patients who have contraindications, are intolerant, fail to respond, or become resistant to TNF inhibitors [49]. Various novel therapies to target IL-23/IL-17 pathway are currently under development. IL-23/IL-17 inhibition may yield different outcomes depending on targets. As shown in Fig. 1, therapeutic strategies can be directed to any key point along the IL-23/IL-17 pathway to arrive to the ultimate outcome—inhibition of IL-17-mediated effects, i.e., all these therapeutic interventions aim at either reduction of IL-17 production or inhibition of IL-17 signaling. Current therapeutic options have been developed to inhibit the Th17 differentiation by blocking the cytokines that are upstream of Th17 pathway, e.g., IL-6 and IL-23. This strategy has been exclusively utilizing monoclonal antibodies to block cytokine activity. It has been shown that TNF inhibitors in responsive patients lead to reduction of Th17 cytokines [55]. In theory, targeting IL-1 will also affect Th17 development (Fig. 1). However, these are beyond the scope of discussion of this review. Here, we focus on agents of recently developed or under development in blockade of IL-17 and IL-23. Active research has been undergoing to suppress Th17 cells by targeting the master transcription factor, ROR-γt. It has been demonstrated that small molecules interfering with ROR-γt protein interaction with its target genes and RNAi inhibiting Rorc gene expression can suppress Th17 cells. There is no clinical trial currently ongoing using these small molecules or RNAi, but they are highly likely to be translatable for clinical development.
Blocking activities of cytokines in the upstream of Th17 pathway
Blocking IL-23
Given its critical role in the development of Th17 cells, naturally, IL-23 is an attractive target. Several monoclonal antibodies are being developed to neutralize IL-23 activity.
Ustekinumab is a fully human IgG1 monoclonal antibody which binds to the shared p40 subunit of IL-12 and IL-23, preventing it from binding to the IL-12Rβ1 receptor protein on the surface of immune cell and thereby inhibiting the bioactivity of IL-12 and IL-23 [56]. This antibody was initially developed with intention to block IL-12 before the discovery of IL-23. This antibody is interfering with the Th1 and Th17 pathways and also keratinocyte activation. Ustekinumab is approved for treatment of moderate to severe plaque psoriasis, but at the same time, it is the first anti-IL-12/23 monoclonal approved for the treatment of active psoriatic arthritis in cases of inadequate response to DMARDs or alternative to anti-TNF [56]. Reassuring effect was also noticed in patients with Crohn’s disease. Ustekinumab was also tested in an open-label single-arm trial in ankylosing spondylitis with 65 % achieving ASAS 40 at week 24 [57].
Therapy with monoclonal antibodies against p40 shared subunit of IL-12 and IL-23 causes a downregulation of p40 and IL-23p19, but not of IL-12p35 in psoriatic skin lesions, suggesting that interference with IL-23 rather than IL-12 activity is responsible for this clinical improvement [58]. This is in concordance with results of studies in murine experimental autoimmune encephalomyelitis and collagen-induced arthritis which have indicated that IL-23 rather than IL-12 is responsible for the chronic inflammation [59].
Briakinumab is a fully human antibody which works through the same mechanism as ustekinumab. It has been similarly efficient in moderate to severe psoriasis as anti-TNF and ustekinumab and superior to methotrexate [60]. It was not tested yet in spondyloarthritis. Unfortunately, it had to be withdrawn from the market in the USA and the European Union out of major safety concerns related to major cardiac events in treatment group [61].
Antibodies against p19 subunit (specifically blocking IL-23)
Various IL-23 p19-specific drugs are currently undergoing clinical trials. Specifically targeting the p19 subunit of IL-23 may provide selective inhibition of IL-23 and Th17 cells to increase safety by preventing inhibition of IL-12-mediated Th1 response and protection against intracellular pathogens [49]. Besides preservation of Th1 immunity, it has been stipulated that the presence of Th1 cytokines may actually help inhibit the Th17 cells. Thus, it has been hypothesized that targeting the p19 subunit of IL-23 may be as effective as and safer than blocking p40, the shared subunit of IL-12 and IL-23. Tildrakizumab and guselkumab are monoclonal antibodies targeting the p19 subunit of IL-23. Tildrakizumab is in early phase 3 clinical trials, and guselkumab was studied in a recently completed phase 2 study for psoriasis, but either is not yet tested in spondyloarthritis [49, 62]. Guselkumab responders showed significant reductions in psoriasis gene expression and serum IL-17A levels from baseline at week 1 (P = 0.031) and week 12 (P = 0.0015), and no changes were observed in the placebo group [63].
Blocking IL-6 and IL-6R
In vitro studies have demonstrated the important role of IL-6 in the initiation of Th17 cell differentiation and that IL-6 at least partially mediates inflammation in ankylosing spondylitis; blockade of activity seemed like a reasonable next therapeutic attempt.
Tocilizumab is a humanized monoclonal antibody against IL-6Rα approved for the management of rheumatoid arthritis. Despite previous case reports, case series, and pilot studies reporting successful use of tocilizumab in spondyloarthritis and psoriatic arthritis, recently conducted BUILDER-1 and BUILDER-2 prospective randomized, placebo-controlled trials showed lack of efficacy in TNF inhibitor-naïve and TNF inhibitor-refractory ankylosing spondylitis. Even though the IL-6 signaling blockade was effective, which has been demonstrated by the reduction of acute-phase reactants, there was no significant clinical improvement [64]. More so, sarilumab is the first fully human monoclonal antibody against IL-6Rα which demonstrated similar lack of efficacy in TNF inhibitor-naïve ankylosing spondylitis patients. Despite a statistically significant reduction in highly sensitive C-reactive protein, there was no clinical or imaging improvement [65•]. This translates into substantial differences in the mechanisms underlying rheumatoid arthritis and spondyloarthritis and need for more understanding of the intrinsic inflammatory and immune-mediated pathways [64].
Clazakizumab is a humanized anti-IL-6 monoclonal antibody which not only had promising results in patients with moderate to severe rheumatoid arthritis but also was recently found to be effective in psoriatic arthritis in a phase 2b clinical trial with most clear response in musculoskeletal disease manifestations (arthritis, enthesitis, and dactylitis) [66], but it has not been tested in ankylosing spondylitis yet. More data needs to be accumulated regarding its efficacy.
This poses the obvious question why anti-IL-6R antibodies do not work but anti-IL-6 do? There is no research data to provide any explanation. IL-6 signals through membrane-bound IL-16R and soluble IL-6R to exert anti-inflammatory and proinflammatory activities, respectively. Acute-phase response is actually anti-inflammatory activities mediated by IL-6 via cell membrane-bound IL-6R [67] which may require less anti-IL-6R antibodies to block. At the site of inflammation, IL-6 concentration is likely to largely exceed concentrations of soluble IL-6R where it requires a higher concentration of anti-IL-6R antibodies to block. In contrast, anti-IL-6 antibodies might have an advantage being directly neutralizing IL-6 at the site of inflammation. In addition, detailed studies of effects by anti-IL-6R and anti-IL-6 antibodies on Th17 cells and Th17 cytokines will be likely to offer further explanations.
Blocking Th17 cytokines
Among the Th17 cytokines, IL-17A has been the prime target of several clinical studies.
Two antibodies neutralizing IL-17A, secukinumab and ixekizumab, are being studied in plaque psoriasis and psoriatic arthritis, while secukinumab is also being studied and showing promising results in ankylosing spondylitis. Both compounds proved to have a therapeutic effect in phase 2 trials in rheumatoid arthritis, supporting the role of IL-17A in the pathogenesis [61]. Secukinumab is a fully human anti-IL-17A monoclonal antibody of the IgG1қ isotype which also proved efficacious in the treatment of patients with autoimmune uveitis. In contrast, the secukinumab trial in inflammatory bowel diseases had negative results, rather supporting the protective function of IL-17A in the gut, as observed in animal models of colitis [68–70].
Ixekizumab is a humanized, hinge-modified IgG4 monoclonal antibody against IL-17A. The most frequent adverse events reported in phase 2 clinical trials with IL-17 blockade include worsening psoriasis, nasopharyngitis, upper respiratory infection, headache, and injection site erythema. Larger and longer trials are required to realize the full clinical extent of IL-17 blockade [71].
IL-22 is one of the cytokines produced by Th17 cells and has been implicated in the pathogenesis of psoriasis. IL-22 is pathogenic in psoriasis being involved in keratinocyte proliferation, and elevated levels in the blood have been correlated with disease activity [72]. Fezakinumab is a humanized monoclonal antibody targeting IL-22, in phase 1 trials for moderate to severe psoriasis, not yet studied in psoriatic arthritis. More questions need to be answered; as compared with IL-17A which is known to co-stimulate osteoclast, IL-22 promotes stromal repair and also protects against colitis [49, 73].
Blocking IL-17R—antibody against IL-17RA
IL-17RA and IL-17RC form a heterodimer to signal both IL-17A and IL-17F. Brodalumab is a fully human IgG2 monoclonal antibody against IL-17RA, blocking the effects of IL-17A, IL-17A/F heterodimer, IL-17F, and IL-17E (IL-25). The broader target of brodalumab, while increasing the potential benefit, also poses a higher risk for more adverse effects. IL-17E plays a role in Th2-type inflammatory diseases, dealing with parasitic infections and allergic reactions, such as asthma and atopic dermatitis, and promotes eosinophilia in mice, and blocking its receptor may interfere with those mechanisms [74]. More data and longer time is required to fully appreciate the pros and cons in blocking the IL-17 receptor versus the ligand.
Phase 1 and phase 2 clinical trials show a favorable safety and tolerability profile and also clinical and histological improvement in moderate to severe psoriasis [74]. In a phase 2, randomized, double-blind, placebo-controlled trial in patients with psoriatic arthritis, brodalumab significantly improved response rates in psoriasis, arthritis, and physical function in patients previously naïve or on biologics [75].
Targeting ROR-γt to suppress Th17 cells
As discussed above, it is highly desirable to target pathogenic Th17 cells to avoid impairing the defense effect of IL-17 which are also produced by many immune cells. Recently identified small molecules and RNAi are shown to specifically target ROR-γt and are potentially to be developed as drugs to block Th17 cells.
Small molecules
Through unbiased and candidate screens, several small molecules have been identified to be able to specifically block ROR-γt activity [7, 8]. The cardiac glycoside digoxin and its derivatives bind to ROR-γt ligand binding domain to inhibit its co-activator binding and inhibit Th17 cell differentiation without affecting other T cell lineages [76]. Similarly, by decreasing co-activator recruitment, SR1001 antagonize ROR-α and ROR-γ, and the modified molecule, SR2211, more specifically inhibits ROR-γ and ROR-γt [77, 78]. Ursolic acid and smf azole-type fungicides were identified as ROR-γt or ROR-α/ROR-γt inhibitors, respectively. All these small molecules have been shown to be effective to delay the onset and reduce the severity of experimental autoimmune encephalomyelitis [7]. Moreover, digoxin can suppress adjuvant-induced arthritis. More recently, three other compounds were identified through screening a library of ∼2 million of small molecule compounds to inhibit ROR-γt activity and suppress effector cytokines of Th17 cells. Interestingly, these compounds are shown to act in a different mechanism [79••]. While TMP920 abrogated ROR-γ occupancy of target sequences, TMP778 and GSK805 changed ROR-γt binding to Gata3 and led increased Gata3 mRNA and protein expression. TMP920, TMP778, and GSK805 are all shown to be able to attenuate experimental autoimmune encephalomyelitis and imiquimod-induced skin inflammation [79••, 80].
RNA interference
RNAi-mediated gene silencing holds great promise for manipulating T cells for developing potential T cell-targeted therapeutics. However, efficient delivery of small interfering RNA (siRNA) into primary T cells represents a major hurdle to the wide use of RNAi technology [81]. Retroviral vectors are effective methods to deliver siRNA into T cells [82, 83] but are not suitable for therapy. Recently, aptamer-mediated delivery of therapeutic siRNA and small hair pain RNA (shRNA) has been investigated in animal models of viral infection and tumor. Aptamers are single-stranded oligonucleotides with high affinity and specificity to the target molecules and are often referred to as chemical antibodies [84, 85]. We have generated a CD4-specific aptamer which is linked to shRNA against Rorc. This CD4 aptamer-RORC shRNA chimera was specifically taken up by CD4+ T cells and silenced ROR-γt expression and suppressed Th17 cells [9•]. By substituting the shRNA, the aptamer-shRNA chimera can be developed to target other transcription factors such as STAT-3 that is involved in Th17 cell differentiation. This technology holds great potential for further development of therapeutics to target Th17 cells.
Targeting Act1 to block IL-17 signaling
The adaptor protein, Act1, is critical for IL-17-IL-17RA/RC signaling [45, 48]. Studies using gene knockout mice proved that Act1 is required for the development of several IL-17-mediated inflammatory disease models including collagen-induced arthritis [86], imiquimod-induced psoriasis [87], autoimmune encephalomyelitis- and dextran sodium sulfate-induced colitis [88], cuprizone-induced demyelination [89], lupus nephritis [90], and IL-25 (IL-17E)-induced asthma [91]. All these data suggest that Act1 is a target for blocking IL-17-mediated effects in effector cells. For its intracellular location, small molecules and RNAi-based agents are the best choice to develop for targeting Act1 activity.
Conclusion
IL-23/IL-17 axis is an important component of the immune system in host defense against microbial pathogens, but can mediate inflammation seen in a variety of immune-mediated inflammatory conditions when the pathway is dysregulated. Several lines of evidence indicate that IL-23/IL-17 axis plays a critical role in the pathogenesis of spondyloarthritides. Therefore, blocking IL-23/IL-17 axis is an attractive strategy for developing therapeutics. Many factors are involved in the development and operation of IL-23/IL-17 axis and hence are therapeutic targets. Monoclonal antibodies targeting specific cytokines dominate the current clinical development of agents with success in blocking IL-23 activity and early but promising results in blocking IL-17A activity. Small molecule agents have been identified to be specific in interference of ROR-γt binding to its target genes, are effective in attenuation of animal models of IL-23/IL-17-mediated diseases, and are promising to translate into clinical development as drugs. CD4+ T cell-directed delivery of siRNA or shRNA targeting Rorc gene represents latest advancement in specific targeting of Th17 cells and will be further validated for usefulness in vivo therapy. It is anticipated that small molecules and RNAi agents will draw more interest for developing drugs to treat spondyloarthritides because of the relatively cheaper production of these agents.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8.
McKenzie BS, Kastelein RA, Cua DJ. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 2006;27:17–23.
Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, et al. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity. 2010;33:279–88.
Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40.
Smith JA, Colbert RA. Review: the interleukin-23/interleukin-17 axis in spondyloarthritis pathogenesis: Th17 and beyond. Arthritis Rheumatol. 2014;66:231–41. This article critically analyzes current evidence that IL-23/IL-17 is important in the pathogenesis of spondyloarthropathy.
Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14:585–600.
Huh JR, Littman DR. Small molecule inhibitors of RORgammat: targeting Th17 cells and other applications. Eur J Immunol. 2012;42:2232–7.
Lee JS, Cua DJ. The emerging landscape of RORgammat biology. Immunity. 2014;40:451–2.
Song P, Chou YK, Zhang X, Meza-Romero R, Yomogida K, Benedek G, et al. CD4 aptamer-RORgammat shRNA chimera inhibits IL-17 synthesis by human CD4(+) T cells. Biochem Biophys Res Commun. 2014;452:1040–5. This is the first report that a non-viral-based delivery system is able to efficiently deliver small hairpin RNA into primary T cells to inhibit Th17 cells. This technology has the potential to translate for clinical use.
Reynolds JM, Angkasekwinai P, Dong C. IL-17 family member cytokines: regulation and function in innate immunity. Cytokine Growth Factor Rev. 2010;21:413–23.
Miossec P. Interleukin-17 and Th17 cells: from adult to juvenile arthritis—now it is serious! Arthritis Rheum. 2011;63:2168–71.
Liang SC, Long AJ, Bennett F, Whitters MJ, Karim R, Collins M, et al. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J Immunol. 2007;179:7791–9.
Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. 2011;34:149–62.
Mayuzumi H, Inagaki-Ohara K, Uyttenhove C, Okamoto Y, Matsuzaki G. Interleukin-17A is required to suppress invasion of Salmonella enterica serovar Typhimurium to enteric mucosa. Immunology. 2010;131:377–85.
Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–81.
Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–19.
Xu S, Han Y, Xu X, Bao Y, Zhang M, Cao X. IL-17A-producing gammadeltaT cells promote CTL responses against Listeria monocytogenes infection by enhancing dendritic cell cross-presentation. J Immunol. 2010;185:5879–87.
Shi Y, Liu XF, Zhuang Y, Zhang JY, Liu T, Yin Z, et al. Helicobacter pylori-induced Th17 responses modulate Th1 cell responses, benefit bacterial growth, and contribute to pathology in mice. J Immunol. 2010;184:5121–9.
Conti HR, Gaffen SL. Host responses to Candida albicans: Th17 cells and mucosal candidiasis. Microbes Infect/Inst Pasteur. 2010;12:518–27.
Suryawanshi A, Veiga-Parga T, Rajasagi NK, Reddy PB, Sehrawat S, Sharma S, et al. Role of IL-17 and Th17 cells in herpes simplex virus-induced corneal immunopathology. J Immunol. 2011;187:1919–30.
Miyazaki Y, Hamano S, Wang S, Shimanoe Y, Iwakura Y, Yoshida H. IL-17 is necessary for host protection against acute-phase Trypanosoma cruzi infection. J Immunol. 2010;185:1150–7.
Milner JD, Sandler NG, Douek DC. Th17 cells, Job’s syndrome and HIV: opportunities for bacterial and fungal infections. Curr Opin HIV AIDS. 2010;5:179–83.
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41.
Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32.
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8.
Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–89.
Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–7.
Hirota K, Martin B, Veldhoen M. Development, regulation and functional capacities of Th17 cells. Semin Immunopathol. 2010;32:3–16.
Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–4.
McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–7.
Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature. 2013;496:461–8. This study identifies and validates 39 regulatory factors (both positive and negative) in a dynamic network to regulate and control Th17 cell differentiation. This regulatory network provides novel drug targets.
Hirahara K, Ghoreschi K, Laurence A, Yang XP, Kanno Y, O’Shea JJ. Signal transduction pathways and transcriptional regulation in Th17 cell differentiation. Cytokine Growth Factor Rev. 2010;21:425–34.
Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol. 2007;19:409–17.
He YW, Beers C, Deftos ML, Ojala EW, Forbush KA, Bevan MJ. Down-regulation of the orphan nuclear receptor ROR gamma t is essential for T lymphocyte maturation. J Immunol. 2000;164:5668–74.
Eberl G, Littman DR. The role of the nuclear hormone receptor RORgammat in the development of lymph nodes and Peyer’s patches. Immunol Rev. 2003;195:81–90.
He YW, Deftos ML, Ojala EW, Bevan MJ. RORgamma t, a novel isoform of an orphan receptor, negatively regulates Fas ligand expression and IL-2 production in T cells. Immunity. 1998;9:797–806.
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33.
Chu CQ, Mello A, Gulko P, Elkon KB. RORγt overexpression predisposes to increased susceptibility and severity of experimental arthritis. Arthritis Rheum. 2008;58:S936.
Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 2008;205:1551–7.
Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10:479–89.
Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J Clin Invest. 2010;120:1762–73.
Pollinger B, Junt T, Metzler B, Walker UA, Tyndall A, Allard C, et al. Th17 cells, not IL-17+ gammadelta T cells, drive arthritic bone destruction in mice and humans. J Immunol. 2011;186:2602–12.
Sutton CE, Mielke LA, Mills KH. IL-17-producing gammadelta T cells and innate lymphoid cells. Eur J Immunol. 2012;42:2221–31.
Ramani K, Pawaria S, Maers K, Huppler AR, Gaffen SL, Biswas PS. An essential role of interleukin-17 receptor signaling in the development of autoimmune glomerulonephritis. J Leukoc Biol. 2014;96:463–72.
Gu C, Wu L, Li X. IL-17 family: cytokines, receptors and signaling. Cytokine. 2013;64:477–85.
Song X, Qian Y. The activation and regulation of IL-17 receptor mediated signaling. Cytokine. 2013;62:175–82.
Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, et al. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol. 2006;177:36–9.
Song X, Qian Y. IL-17 family cytokines mediated signaling in the pathogenesis of inflammatory diseases. Cell Signal. 2013;25:2335–47.
Yeremenko N, Paramarta JE, Baeten D. The interleukin-23/interleukin-17 immune axis as a promising new target in the treatment of spondyloarthritis. Curr Opin Rheumatol. 2014;26:361–70.
Colbert RA, DeLay ML, Klenk EI, Layh-Schmitt G. From HLA-B27 to spondyloarthritis: a journey through the ER. Immunol Rev. 2010;233:181–202.
DeLay ML, Turner MJ, Klenk EI, Smith JA, Sowders DP, Colbert RA. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheum. 2009;60:2633–43.
Eiris N, Gonzalez-Lara L, Santos-Juanes J, Queiro R, Coto E, Coto-Segura P. Genetic variation at IL12B, IL23R and IL23A is associated with psoriasis severity, psoriatic arthritis and type 2 diabetes mellitus. J Dermatol Sci. 2014;75:167–72.
Coffre M, Roumier M, Rybczynska M, Sechet E, Law HK, Gossec L, et al. Combinatorial control of Th17 and Th1 cell functions by genetic variations in genes associated with the interleukin-23 signaling pathway in spondyloarthritis. Arthritis Rheum. 2013;65:1510–21.
Cauli A, Mathieu A. Th17 and interleukin 23 in the pathogenesis of psoriatic arthritis and spondyloarthritis. J Rheumatol Suppl. 2012;89:15–8.
Zaba LC, Cardinale I, Gilleaudeau P, Sullivan-Whalen M, Suarez-Farinas M, Fuentes-Duculan J, et al. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med. 2007;204:3183–94.
McKeage K. Ustekinumab: a review of its use in psoriatic arthritis. Drugs. 2014;74:1029–39.
Poddubnyy D, Hermann KG, Callhoff J, Listing J, Sieper J. Ustekinumab for the treatment of patients with active ankylosing spondylitis: results of a 28-week, prospective, open-label, proof-of-concept study (TOPAS). Ann Rheum Dis. 2014;73:817–23.
Levin AA, Gottlieb AB. Specific targeting of interleukin-23p19 as effective treatment for psoriasis. J Am Acad Dermatol. 2014;70:555–61.
Hoeve MA, Savage ND, de Boer T, Langenberg DM, de Waal MR, Ottenhoff TH, et al. Divergent effects of IL-12 and IL-23 on the production of IL-17 by human T cells. Eur J Immunol. 2006;36:661–70.
Traczewski P, Rudnicka L. Briakinumab for the treatment of plaque psoriasis. BioDrugs Clin Immunother Biopharma Gene Ther. 2012;26:9–20.
Gordon KB, Langley RG, Gottlieb AB, Papp KA, Krueger GG, Strober BE, et al. A phase III, randomized, controlled trial of the fully human IL-12/23 mAb briakinumab in moderate-to-severe psoriasis. J Invest Dermatol. 2012;132:304–14.
Hu C, Wasfi Y, Zhuang Y, Zhou H. Information contributed by meta-analysis in exposure-response modeling: application to phase 2 dose selection of guselkumab in patients with moderate-to-severe psoriasis. J Pharmacokinet Pharmacodyn. 2014;41:239–50.
Sofen H, Smith S, Matheson RT, Leonardi CL, Calderon C, Brodmerkel C, et al. Guselkumab (an IL-23-specific mAb) demonstrates clinical and molecular response in patients with moderate-to-severe psoriasis. J Allergy Clin Immunol. 2014;133:1032–40.
Sieper J, Porter-Brown B, Thompson L, Harari O, Dougados M. Assessment of short-term symptomatic efficacy of tocilizumab in ankylosing spondylitis: results of randomised, placebo-controlled trials. Ann Rheum Dis. 2014;73:95–100. This article describe negative results that two monoclonal antibodies (tocilizumab and sarilumab) against IL-6Rα failed to show improvement of clinical outcomes in patients with ankylosing spondylitis in spite of effect on the reduction of acute phase reaction, although both antibodies are efficacious for rheumatoid arthritis.
Sieper J, Braun J, Kay J, Badalamenti S, Radin AR, Jiao L, et al. Sarilumab for the treatment of ankylosing spondylitis: results of a phase II, randomised, double-blind, placebo-controlled study (ALIGN). Ann Rheum Dis. 2014. This article describe negative results that two monoclonal antibodies (tocilizumab and sarilumab) against IL-6Rα failed to show improvement of clinical outcomes in patients with ankylosing spondylitis in spite of effect on the reduction of acute phase reaction, although both antibodies are efficacious for rheumatoid arthritis.
Mease PJ, Gottlieb AB, Berman A, Drescher E, Xing J, Banerjee S, et al. A phase IIb, randomized, double-blind, placebo-controlled, dose-ranging, multicenter study to evaluate the efficacy and safety of clazakizumab, an anti-IL-6 monoclonal antibody, in adults with active psoriatic arthritis. Arthritis Rheumatol. 2014;66:S423.
Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci. 2012;8:1237–47.
Gisondi P, Dalle Vedove C, Girolomoni G. Efficacy and safety of secukinumab in chronic plaque psoriasis and psoriatic arthritis therapy. Dermatol Ther. 2014;4:1–9.
Patel DD, Lee DM, Kolbinger F, Antoni C. Effect of IL-17A blockade with secukinumab in autoimmune diseases. Ann Rheum Dis. 2013;72 Suppl 2:ii116–23.
Ogawa A, Andoh A, Araki Y, Bamba T, Fujiyama Y. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin Immunol. 2004;110:55–62.
Saeki H, Nakagawa H, Ishii T, Morisaki Y, Aoki T, Berclaz PY, et al. Efficacy and safety of open-label ixekizumab treatment in Japanese patients with moderate-to-severe plaque psoriasis, erythrodermic psoriasis and generalized pustular psoriasis. J Eur Acad Dermatol Venereol: JEADV. 2014.
Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005;174:3695–702.
Patel M, Day A, Warren RB, Menter A. Emerging therapies for the treatment of psoriasis. Dermatol Ther. 2012;2:16.
Coimbra S, Figueiredo A, Santos-Silva A. Brodalumab: an evidence-based review of its potential in the treatment of moderate-to-severe psoriasis. Core Evid. 2014;9:89–97.
Mease PJ, Genovese MC, Greenwald MW, Ritchlin CT, Beaulieu AD, Deodhar A, et al. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N Engl J Med. 2014;370:2295–306.
Huh JR, Leung MW, Huang P, Ryan DA, Krout MR, Malapaka RR, et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORgammat activity. Nature. 2011;472:486–90.
Solt LA, Kumar N, Nuhant P, Wang Y, Lauer JL, Liu J, et al. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature. 2011;472:491–4.
Kumar N, Lyda B, Chang MR, Lauer JL, Solt LA, Burris TP, et al. Identification of SR2211: a potent synthetic RORgamma-selective modulator. ACS Chem Biol. 2012;7:672–7.
Xiao S, Yosef N, Yang J, Wang Y, Zhou L, Zhu C, et al. Small-molecule RORgammat antagonists inhibit T helper 17 cell transcriptional network by divergent mechanisms. Immunity. 2014;40:477–89. Several of small molecules are identified to inhibit Th17 differentiation via interference of ROR-γt with binding to target genes. These small molecules are effective to treat animal models of autoimmune inflammatory diseases and are potentially to be developed as drugs.
Skepner J, Ramesh R, Trocha M, Schmidt D, Baloglu E, Lobera M, et al. Pharmacologic inhibition of RORgammat regulates Th17 signature gene expression and suppresses cutaneous inflammation in vivo. J Immunol. 2014;192:2564–75.
Freeley M, Long A. Advances in siRNA delivery to T-cells: potential clinical applications for inflammatory disease, cancer and infection. Biochem J. 2013;455:133–47.
Rangachari M, Zhu C, Sakuishi K, Xiao S, Karman J, Chen A, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3-mediated cell death and exhaustion. Nat Med. 2012;18:1394–400.
Jin HS, Liao L, Park Y, Liu YC. Neddylation pathway regulates T-cell function by targeting an adaptor protein Shc and a protein kinase Erk signaling. Proc Natl Acad Sci U S A. 2013;110:624–9.
Zhou J, Neff CP, Swiderski P, Li H, Smith DD, Aboellail T, et al. Functional in vivo delivery of multiplexed anti-HIV-1 siRNAs via a chemically synthesized aptamer with a sticky bridge. Mol Ther J Am Soc Gene Ther. 2013;21:192–200.
Ni X, Castanares M, Mukherjee A, Lupold SE. Nucleic acid aptamers: clinical applications and promising new horizons. Curr Med Chem. 2011;18:4206–14.
Pisitkun P, Claudio E, Ren N, Wang H, Siebenlist U. The adaptor protein CIKS/ACT1 is necessary for collagen-induced arthritis, and it contributes to the production of collagen-specific antibody. Arthritis Rheum. 2010;62:3334–44.
Ha HL, Wang H, Pisitkun P, Kim JC, Tassi I, Tang W, et al. IL-17 drives psoriatic inflammation via distinct, target cell-specific mechanisms. Proc Natl Acad Sci U S A. 2014;111:E3422–31.
Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–56.
Kang Z, Altuntas CZ, Gulen MF, Liu C, Giltiay N, Qin H, et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity. 2010;32:414–25.
Pisitkun P, Ha HL, Wang H, Claudio E, Tivy CC, Zhou H, et al. Interleukin-17 cytokines are critical in development of fatal lupus glomerulonephritis. Immunity. 2012;37:1104–15.
Claudio E, Sonder SU, Saret S, Carvalho G, Ramalingam TR, Wynn TA, et al. The adaptor protein CIKS/Act1 is essential for IL-25-mediated allergic airway inflammation. J Immunol. 2009;182:1617–30.
Acknowledgments
Dr. Chu’s work is supported by grants from NIH (AR055254), Rheumatology Research Foundation, and National Psoriasis Foundation.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Ling Zhao declares that he has no conflict of interest. Daniela Ghetie declares that she has no conflict of interest. Zhenyu Jiang declares that he has no conflict of interest.
Cong-Qiu Chu is a consultant of Sobi and Regeneron.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Spondyloarthritis and Psoriatic Arthritis
Rights and permissions
About this article

Cite this article
Zhao, L., Ghetie, D., Jiang, Z. et al. How Can We Manipulate the IL-23/IL-17 Axis?. Curr Treat Options in Rheum 1, 182–196 (2015). https://doi.org/10.1007/s40674-015-0017-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40674-015-0017-4