
Abstract
Sustained elevation of eosinophils above 5 × 109 /l in peripheral blood (PB) should prompt further investigation. Clonal eosinophilia accounts for the much smaller proportion of eosinophilias (< 10%), but exclusion of such a neoplasia is prognostically and therapeutically relevant. Molecular genetic analysis from PB, cytogenetics from bone marrow, and bone marrow histology are primarily used to exclude clonal eosinophilia. Far more common is reactive eosinophilia, the cause of which may be drugs, allergies, solid tumors, lymphomas, worm infections, autoimmune diseases, or idiopathic hypereosinophilic syndrome (HES). Because of the diverse organ infiltration patterns in eosinophilia, a specific search for possible organ involvement (including heart, lung, gastrointestinal tract, kidney, skin, etc.) should be performed, depending on the patient’s symptoms. The diagnosis of HES is made when organ infiltration with consecutive dysfunction is diagnosed in persistent eosinophilia after exclusion of other causes. Therapeutically, oral corticosteroids (OSC) are used in HES. This can also be helpful in the differential diagnosis, as patients with clonal eosinophilia are usually not expected to achieve remission with OCS. When OCS requirements are high, other immunosuppressants (e.g., methotrexate [MTX], cyclophosphamide) and the interleukin (IL)-5 antagonist mepolizumab are used. In clonal eosinophilia, tyrosine kinase inhibitors are the first-line therapy, depending on the underlying genetic alteration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Classification of eosinophilia-associated diseases
Eosinophilia exists when peripheral blood (PB) eosinophil granulocytes are elevated above 0.5 × 109 /l. If eosinophilia persists over a longer period, further clarification should be performed with regard to the distinction between a reactive (nonclonal) and a clonal eosinophilia, which is essential from a prognostic and therapeutic point of view. The proportionally smaller part (< 10%) of eosinophilia is of clonal origin (neoplasia); here, diseases of the hematopoietic stem cell compartment are present, which are characterized by genetic aberrations, frequently elevated serum tryptase, and, less frequently, by proliferation of blasts. Clinically, these diseases may present as chronic myeloid neoplasms, in case of advanced disease also as acute leukemias (myeloid or lymphoid), lymphoproliferative neoplasms (e.g., T‑lymphoblastic lymphoma), or myelosarcoma. Organ involvement (e.g., splenomegaly) is frequently present, sometimes associated with organ dysfunction, especially of the heart.
More than 90% of eosinophilia-associated diseases are reactive in nature and arise as an accompanying phenomenon in allergies, atopias, infections, autoimmune diseases, and malignancies, or in response to drugs. Eosinophilia is often caused by overproduction of eosinophilopoietic cytokines such as interleukin (IL) 3, IL‑5, or granulocyte–macrophage colony-stimulating factor (GM-CSF), which stimulate eosinophilic proliferation [1, 2]. In some solid tumors, eosinophils can be detected as part of an antitumorigenic response, have prognostic relevance, or are found without direct cause in terms of so-called “uninvolved third parties” [3, 4].
Idiopathic hypereosinophilic syndrome (HES) belongs to the reactive eosinophilias as a distinct entity and is a systemic disease. It is defined as the combination of a persistently elevated peripheral blood (PB) eosinophilia ≥ 1.5 × 109 /l (confirmed in two measurements at least 4 weeks apart) and evidence of eosinophil organ infiltration with consecutive organ dysfunction.
Clinical appearance
Clinical presentation in HES depends on the number of involved organs and the extent of organ involvement. Frequently affected organs include skin, sinuses, lungs, heart, gastrointestinal tract, or nervous system; in principle, any organ may be involved [5]. While in some patients eosinophilia is diagnosed by chance during a routine check-up of the PB in asymptomatic or only mildly symptomatic patients (e.g., itching, urticaria), in other cases severe, sometimes life-threatening courses occur due to organ infiltration and consecutive dysfunction. Cardiac involvement is particularly problematic, with endomyocardial fibrosis and intracardiac thrombi (Löffler’s endocarditis), thromboembolic complications, peri/myocarditis, pericardial effusion, and heart failure. Pulmonary involvement is most commonly manifested by bronchial asthma, pulmonary infiltrates, pleural effusion, or pulmonary fibrosis. Gastrointestinal involvement includes esophagitis, gastroenteritis, serositis, ascites, liver dysfunction, and splenomegaly [5]. Clinically, the picture may resemble that of autoimmune diseases, especially when sinusitis and bronchial asthma are present with or without cardiac involvement. In this case, differential diagnosis from anti-neutrophil cytoplasmic antibody (ANCA)-negative eosinophilic granulomatosis with polyangiitis (EGPA) may be very difficult or even impossible. [6, 7]. The extent and number of organs affected allows conclusions to be drawn about the genesis of the eosinophilia. In clonal eosinophilia, there is often only isolated splenomegaly or involvement of rather few other organs, for example heart or skin. A high number of affected organs and involvement of the upper/lower respiratory tract and/or the gastrointestinal tract are more indicative of the presence of HES.
Diagnostics and differential diagnostics
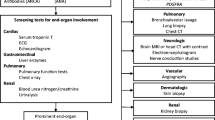
Causes of reactive eosinophilia can be clarified in many cases by anamnesis (drugs, allergies, solid tumors, lymphomas). While the level of eosinophils in the differential blood count often contributes little from a differential diagnostic point of view, monocytosis or even an increase in blasts are signs of a clonal genesis. Laboratory parameters such as lactate dehydrogenase (LDH), troponin I (TNI), pro-BNP (brain natriuretic peptide), C-reactive protein (CRP), immunoglobulin E (IgE), and autoantibodies (e.g., antinuclear antibodies [ANA], ANCA, single- and double-stranded antibodies, rheumatoid factor, cyclic citrullinated peptide [CCP], C3, C4, anti-C1q, and others), tryptase, and vitamin B12 play an important role in the differential diagnosis (Table 1).
Parasitic disease (mostly worm infection) is a rather rare cause in our geographical latitudes. In cooperation with a tropical institute, it should primarily be diagnosed or ruled out as well as possible by means of serological antibody diagnostics (screening) and subsequent stool examination (confirmation, specification).
The detection/exclusion of organ involvement should be performed actively, i.e., even in asymptomatic patients. Because of the organs most frequently involved and the potentially life-threatening complications, the lungs and heart are initially the most important organs to be checked for infiltration. With regard to clinically inapparent myocardial involvement, the determination of proBNP and TNI as well as echocardiography is recommended. However, cardiac magnetic resonance imaging (cardiac-MRI) is far more sensitive; the crucial difference to echocardiography is the detection of late gadolinium enhancement (LGE), which is indicative of either active infiltration or fibrosis of the myocardium. Pulmonary involvement should be clarified by pulmonary function testing, high-resolution computed tomography (HR-CT), and bronchoscopy with bronchoalveolar lavage (BAL), as well as biopsy; if pleural effusion is present, puncture and cytologic/immunocytologic (especially to detect malignant cells) examination of the same. Sinusitis is confirmed by imaging, and an ear, nose, and throat (ENT) consultation with endoscopy and biopsy may be considered, if necessary. Depending on the clinical presentation (e.g., nausea, vomiting, diarrhea, malabsorption, ascites), esophagogastroduodenoscopy (EGD) and colonoscopy with biopsies should be performed as the macroscopy is usually unremarkable. Rarely, serositis can be detected on abdominal contrast-enhanced imaging if the clinical picture is appropriate. If ascites is present, puncture with cytologic/immunocytologic examination is performed, and laparoscopy with biopsy may be performed.
The most important differential diagnosis to HES is clonal eosinophilia. Although much rarer, it must always be excluded because of the prognostic and therapeutic differences. Pathogenetically, clonal eosinophilias are usually based on recurrent fusion genes with consecutive tyrosine kinase activation or, more rarely, somatic mutations. The most common fusion genes involve the tyrosine kinase genes PDGFRA, PDGFRB, FGFR1, or JAK2, which are listed as a separate entity in the World Health Organization (WHO) classification: “myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRA, PDGFRB, FGFR1, or PCM1::JAK2 fusion gene.” In total, over 70 different eosinophilia-associated tyrosine kinase fusion genes have been identified to date.
If clonal eosinophilia is suspected, for example due to splenomegaly, monocytosis, or the presence of peripheral blasts, testing by polymerase chain reaction (PCR) or fluorescence in situ hybridization (FISH) analysis for the FIP1L1::PDGFRA fusion gene associated with the highest incidence should be performed first. For reasons as yet unknown, it is found almost exclusively in males [1, 5]. The second most common genetic aberration in the workup of eosinophilia is the KIT D816V mutation associated with systemic mastocytosis, which is also detectable by PCR (higher sensitivity) or next-generation sequencing (NGS) in peripheral blood. Bone marrow aspiration is important for cytology and cytogenetics and histology/immunohistology should be performed from the bone marrow trephine. Besides eosinophilia, morphology should assess cellularity, proliferation, and dysplasia of all cell series as well as monocytes, blasts, mast cells, and fibrosis in order to morphologically diagnose an underlying myeloid neoplasia or systemic mastocytosis. Bone marrow morphology is often only indicative in combination with the clinical appearance and molecular genetic findings and depends on the expertise of the examining pathologist [8]. While the FIP1L1::PDGFRA fusion gene is not detectable by conventional cytogenetics, the majority of other eosinophilia-associated tyrosine kinase fusion genes are detectable via reciprocal translocations, for example t(5:12)(q31–33;p12) as a correlate of an ETV6::PDGFRB fusion gene. In case of further suspicion of clonal eosinophilia, screening for the most common somatic point mutations by NGS should also be performed. These include KIT D816V for systemic mastocytosis and JAK2 V617F for eosinophilia-associated myeloproliferative neoplasms [9].
Laboratory findings that may indicate clonal eosinophilia include additional blood count changes, such as left-shifted leukocytosis, blasts, monocytosis, or thrombocytosis, and elevations of vitamin B12 (nonspecific but frequently elevated in myeloid neoplasms) and serum tryptase (elevated in systemic mastocytosis, fusion genes, and sometimes in other myeloid diseases). Table 1 describes the diagnostic procedure for eosinophilia.
In the rare case of persistent eosinophilia without evidence of an underlying cause (i.e., if genetics, autoantibodies, tumor search, etc. are negative) and without evidence of organ manifestation despite extensive diagnostics (active exclusion of organ manifestation), the preliminary working diagnosis of hypereosinophilia of unclear significance (HEUS) can be made. In this case, a close-meshed control with, if necessary, repetition of the performed diagnostics at a later time should be carried out. Table 2 lists the differential diagnoses of HES.
Therapy
Basic therapy in HES consists of administration of oral corticosteroids (OCS; 0.5–1 mg/kg prednisolone equivalent), with dose adjustment (tapering) according to clinical presentation. There is usually a rapid decrease in blood eosinophilia. Persistence of eosinophils may be indicative of a clonal genesis. If a dose above the Cushing’s threshold of 7.5 mg prednisolone equivalent/day is required to control symptoms, pathologic findings, and eosinophilia, OCS-sparing therapy must be considered early. Considering the severity, suitable drugs for this include methotrexate (MTX) and azathioprine, and in more severe cases, mycophenolate mofetil and cyclophosphamide; all of the above drugs are used in an off-label-use setting. In cases of cardiac involvement, life-threatening involvement of another organ, or severe multiorgan involvement, therapy should follow the treatment recommendations in EGPA; for example, primary induction therapy with intravenous cyclophosphamide and OCS [6].
Hydroxycarbamide and interferon alpha (INF-α) are frequently used in clonal eosinophilia [5]. Again, their use is off label.
In December 2021, the IL‑5 antagonist mepolizumab was also approved for treatment of HES ([10]; Table 3). By inhibiting IL‑5, the production of eosinophils is reduced. Mepolizumab is approved for HES and EGPA. Other antibodies against IL‑5 (depemokimab) or the IL‑5 receptor (benralizumab) are under investigation in clinical trials [11]. The aim is to reduce “flares,” i.e., worsening of clinical symptoms (e.g., pruritus, dyspnea, urticaria) and increase in eosinophils with increased need of OCS or other immunosuppressants. Experience has shown that OCS can rarely be completely discontinued in HES, and most patients require low-dose OCS baseline therapy to control disease activity despite the addition of immunosuppressants and/or biologicals.
Specific tyrosine kinase inhibitors (TKIs) are available for the treatment of clonal eosinophilia, depending on the underlying fusion gene. While patients with a FIP1L1::PDGFRA fusion gene or different fusion genes involving PDGFRB show excellent long-term survival under monotherapy with imatinib, patients with other fusion genes (e.g., involving JAK2 or FGFR1) often show only a transient response under appropriate TKI therapy, so that allogeneic stem cell transplantation should be considered depending on age, comorbidities, and donor availability. In case of clonal eosinophilia, a durable response to OCS is not expected, so that initiation and response or non-response occasionally has diagnostic value.
Forecast
The prognosis of HES also depends on the type and extent of organ involvement. With definitive exclusion of the diseases associated with clonal eosinophilia, especially the patients with cardiac involvement have potentially lethal courses due to endomyocarditis, thromboembolic complications, and heart failure. The prognosis of pulmonary involvement is associated with the severity of bronchial asthma and recurrent pneumonia. However, due to the not uncommon permanent steroid dependence, steroid-associated side effects, such as the development of osteopenia/osteoporosis or diabetes mellitus, are common.
Conclusion
Due to potentially vital threatening organ involvement and the potentially good treatability, a persistent eosinophilia above 0.5 × 109 /l should always be subjected to further evaluation.
HES is a diagnosis of exclusion and should be made only after reactive and clonal eosinophilia have been excluded. If a clonal genesis is suspected (e.g., splenomegaly, monocytosis, blasts), appropriate diagnostic steps must be taken (bone marrow aspiration, cytogenetics, molecular genetics) to enable these patients to receive targeted or at least cytoreductive therapy (hydroxyurea, in rare cases pegylated interferon). Depending on the molecular genetics, even allogeneic stem cell transplantation may have to be included in the therapeutic considerations.
For adequate treatment of HES, careful clarification of the qualitative and quantitative organ involvement pattern is essential. If therapy is required (symptoms, organ involvement), OCS-sparing immunosuppression is administered in addition to initial OCS administration (analogous to other autoimmune diseases), if necessary. The frequently described drugs cyclosporine, hydroxyurea, and interferon should rather not be used in HES. Recently, the IL‑5 antibody mepolizumab was approved for treatment of HES.
Case study
A 42-year-old female patient presented to the central emergency department due to increasing left arm swelling. She had a history of mild allergic bronchial asthma and had been taking an occasional asthma inhaler for about 1.5 years. Sonographically, thrombosis could be excluded. However, there were clearly enlarged left axillary and paraclavicular lymph nodes (LN) and mild splenomegaly (13 cm longitudinally). Laboratory chemistry revealed leukocytosis of 13 × 109 /l, eosinophilia of 5 × 109 /l, and elevated CRP of 80 mg/l (norm < 5). Other abnormal laboratory parameters included elevated IgE of 1500 kU/l, elevated rheumatoid factor of 50 IU/ml (norm < 15), and elevated IgG4 of 3.6 g/l (norm < 1.4). Cardiac enzymes, autoantibodies, serology/stool analysis for helminths, various viral serologies, and urine diagnostics were negative. Chest radiography showed a widened mediastinum, which appeared as mediastinal lymphadenopathy on CT thorax. A left axillary LN extirpation was performed. Histological workup showed infiltration by eosinophilic granulocytes and no evidence of malignancy. Bronchoscopy with BAL and transbronchial biopsy showed evidence of eosinophils without granuloma formation. Bone marrow aspiration with cytology, histology, and cytogenetics was not informative except for eosinophilia, and molecular genetics (FIP1L1::PDGFRA, KIT D816V, JAK2 V617F) yielded no relevant findings. Cranial magnetic resonance imaging (cMRI) showed evidence of sinusitis, and cardiac-MRI showed evidence of marked late gadolinium enhancement as a surrogate parameter of cardiac involvement (Fig. 1). Myocardial biopsy showed fibrosis without eosinophilia. A diagnosis of HES with sinusitis with histologically, pulmonary, and MRI-suspected cardiac involvement was made. In the interdisciplinary autoimmune board, intensive treatment with OCS and cyclophosphamide was proposed, analogous to the treatment recommendations for EGPA with cardiac involvement. However, the patient agreed to therapy with OCS and MTX (20 mg/week) only. She also received local endonasal steroid therapy and bronchodilator therapy. Under treatment, there was rapid remission of lymphadenopathy, but the OCS dose could not be reduced below 7.5 mg/day to achieve symptom freedom. Therefore, after 6 months, the patient was switched to azathioprine (2 mg/kg per day); However, the OCS dose could not be reduced below 7.5 mg/day. After a further 12 months, therapy could be extended to include mepolizumab 300 mg s.c. every 4 weeks, as part of a compassionate use program. Under this combination therapy, azathioprine could be discontinued in due course and the OCS dose was reduced to 2.5 mg/day.

Organ involvement in hypereosinophilic syndrome. a Axillary and mediastinal lymphadenopathy (arrows). b Sinusitis. c Late gadolinium enhancement in cardiac involvement (courtesy of the Institute of Clinical Radiology, University Medical Center Mannheim, Germany)
The patient has been completely asymptomatic for 5 years.
Abbreviations
- ANA:
-
Antinuclear antibodies
- ANCA:
-
Anti-neutrophil cytoplasmic antibodies
- BAL:
-
Bronchoscopy with bronchoalveolar lavage
- BNP:
-
Brain natriuretic peptide
- Cardiac-MRI:
-
Cardiac magnetic resonance imaging
- CMRI:
-
Cranial magnetic resonance imaging
- CRP:
-
C‑reactive protein
- EGD:
-
Esophagogastroduodenoscopy
- EGPA:
-
Eosinophilic granulomatosis with polyangiitis
- ENT:
-
Ear, nose, and throat
- FISH:
-
Fluorescence in situ hybridization
- GM-CSF:
-
Granulocyte–macrophage colony-stimulating factor
- HES:
-
Hypereosinophilic syndrome
- HEUS :
-
Hypereosinophilia of unknown significance
- HR-CT:
-
High-resolution computed tomography
- IgE:
-
Immunoglobulin E
- IL:
-
Interleukin
- INF‑α:
-
Interferon alpha
- LDH:
-
Lactate dehydrogenase
- LGE:
-
Late gadolinium enhancement
- LN:
-
Lymph node
- MTX:
-
Methotrexate
- NGS:
-
Next-generation sequencing
- PB:
-
Peripheral blood
- PCR:
-
Polymerase chain reaction
- OSC:
-
Oral corticosteroids
- TKI:
-
Tyrosine kinase inhibitors
- TNI:
-
Troponin I
- WHO:
-
World Health Organization
References
Valent P, Klion AD, Horny HP, Roufosse F, Gotlib J, Weller PF, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130:607–612.e9.
Valent P, Gleich GJ, Reiter A, Roufosse F, Weller PF, Hellmann A, et al. Pathogenesis and classification of eosinophil disorders: a review of recent developments in the field. Expert Rev Hematol. 2012;5:157–76.
Varricchi G, Galdiero MR, Loffredo S, Lucarini V, Marone G, Mattei F, et al. Eosinophils: the unsung heroes in cancer? OncoImmunology. 2018;7:e1393134.
El-Osta H, El-Haddad P, Nabbout N. Lung carcinoma associated with excessive eosinophilia. J Clin Oncol. 2008;26:3456–7.
Shomali W, Gotlib J. World Health Organization-defined eosinophilic disorders: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. 2022;97:129–48.
Chung SA, Langford CA, Maz M, Abril A, Gorelik M, Guyatt G, et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the management of antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheumatol. 2021;73:1366–83.
Hazebroek MR, Kemna MJ, Schalla S, Sanders-van Wijk S, Gerretsen SC, Dennert R, et al. Prevalence and prognostic relevance of cardiac involvement in ANCA-associated vasculitis: eosinophilic granulomatosis with polyangiitis and granulomatosis with polyangiitis. Int J Cardiol. 2015;199:170–9.
Schwaab J, Jawhar M, Naumann N, Schmitt-Graeff A, Fabarius A, Horny HP, et al. Diagnostic challenges in the work up of hypereosinophilia: pitfalls in bone marrow core biopsy interpretation. Ann Hematol. 2016;95:557–62.
Schwaab J, Umbach R, Metzgeroth G, Naumann N, Jawhar M, Sotlar K, et al. KIT D816V and JAK2 V617F mutations are seen recurrently in hypereosinophilia of unknown significance. Am J Hematol. 2015;90(9):774–7. https://doi.org/10.1002/ajh.24075.
Roufosse F, Kahn JE, Rothenberg ME, Wardlaw AJ, Klion AD, Kirby SY, et al. Efficacy and safety of mepolizumab in hypereosinophilic syndrome: a phase III, randomized, placebo-controlled trial. J Allergy Clin Immunol. 2020;146:1397–405.
Kuang FL, Legrand F, Makiya M, Ware J, Wetzler L, Brown T, et al. Benralizumab for PDGFRA-negative hypereosinophilic syndrome. N Engl J Med. 2019;380:1336–46.
Funding
German José Carreras Leukemia Foundation (grant no. DJCLS 08 R/2020).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
J. Schwaab discloses membership of the Advisory Board of Astra Zeneca and reports receiving honoraria and study support from GSK. A. Reiter states that he has received fees from GSK and study support from GSK and Astra Zeneca. G. Metzgeroth indicates membership of the Advisory Board of GSK. J. Lübke declares that he has no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schwaab, J., Lübke, J., Reiter, A. et al. Idiopathic hypereosinophilic syndrome—diagnosis and treatment. Allergo J Int 31, 251–256 (2022). https://doi.org/10.1007/s40629-022-00221-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40629-022-00221-w