
Abstract
Purpose
Disrupted mitochondrial functions and genetic variants of mitochondrial DNA (mtDNA) have been observed in different human neoplasms. Next-generation sequencing (NGS) can be used to detect even low heteroplasmy-level mtDNA variants. We aimed to investigate the mitochondrial genome in pituitary adenomas by NGS.
Methods
We analysed 11 growth hormone producing and 33 non-functioning [22 gonadotroph and 11 hormone immunonegative] pituitary adenomas using VariantPro™ Mitochondrion Panel on Illumina MiSeq instrument. Revised Cambridge Reference Sequence (rCRS) of the mtDNA was used as reference. Heteroplasmy was determined using a 3% cutoff.
Results
496 variants were identified in pituitary adenomas with overall low level of heteroplasmy (7.22%). On average, 35 variants were detected per sample. Samples harbouring the highest number of variants had the highest Ki-67 indices independently of histological subtypes. We identified eight variants (A11251G, T4216C, T16126C, C15452A, T14798C, A188G, G185A, and T16093C) with different prevalences among different histological groups. T16189C was found in 40% of non-recurrent adenomas, while it was not present in the recurrent ones. T14798C and T4216C were confirmed by Sanger sequencing in all 44 samples. 100% concordance was found between NGS and Sanger method.
Conclusions
NGS is a reliable method for investigating mitochondrial genome and heteroplasmy in pituitary adenomas. Out of the 496 detected variants, 414 have not been previously reported in pituitary adenoma. The high number of mtDNA variants may contribute to adenoma genesis, and some variants (i.e., T16189C) might associate with benign behaviour.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The mitochondrial genome consists of several copies of circular, double-stranded DNA molecules, covering 16,569 base pairs, 37 genes. Of these, 13 encode polypeptides of respiratory enzyme complexes, 22 encode transfer RNAs, and 2 encode ribosomal RNAs (12S, 16S) [1, 2]. Several copies of the mitochondrial DNA (mtDNA) can exist, causing co-existence of mutant and wild-type alleles in the same cell (or tissue), referred to as heteroplasmy [2]. The ratio of the mutant allele compared to the wild type is defined as degree of heteroplasmy.
Mitochondria have an essential role in apoptosis. Failure of apoptosis has been considered as a hallmark of oncogenesis. Since the first study about the mtDNA mutations in colorectal cancer has been published in 1998, extensive research has started to analyse the mitochondrial genome. The presence of frequently observed mitochondrial mutations in a variety of human neoplasms (e.g., breast, ovarian, colorectal, gastric, hepatocellular, pancreatic, prostate, lung, thyroid, renal cell cancers, and brain tumors [3]) along with the description of abnormal mitochondria in tumors raised the possibility that these mutations might have a casual role in tumor initiation and progression [4, 5].
Dasgupta et al. analysed the functional effect of the mitochondrially encoded cytochrome B (MT-CYB) gene mutation, and showed that overexpression of MT-CYB-induced tumor growth in vitro and in vivo in bladder cancer cells and increased the invasive phenotype, underlining the functional importance of this mutation [6]. Another study suggested that MT-CYB can be an immune target for CD4(+) T cells and, therefore, have implications for the immunosurveillance of mitochondrial aberrations in cancer patients [7]. In a trans-mitochondrial hybrid (cybrid) model (containing a common HeLa nucleus and mtDNA of interest), a pathogenic mitochondrially encoded ATP synthase subunit 6 (MT-ATP6) gene variant seemed to promote tumor growth by preventing apoptosis [8]. The increasing interest towards mitochondrial variants in cancer is probably due to the recognition that understanding these molecular mechanisms would take us closer to develop novel biomarkers and therapeutic strategies [9]. Still, the question whether these variants are cause or consequence of tumor progression remain unanswered.
Previous studies mostly used Sanger sequencing and MitoChip (microarray-based sequencing following PCR-amplified mitochondrial DNA) for analysing the whole mitochondrial genome. Next-generation sequencing (NGS) is a cost-effective, high-throughput, and sensitive method that has become a suitable tool to detect any DNA variants even if they are present at low level. Thus, this method is likely to be superior for identification of heteroplasmy [2, 10, 11].
Pituitary adenomas are common neoplasms accounting for 10–15% of intracranial tumors [12]. The majority (95%) occur sporadically, and despite extensive research, the molecular mechanisms of their pathogenesis are largely unknown [13]. Besides GNAS [guanine nucleotide-binding protein (G protein), alpha stimulating] mutations that frequently (approx. 40%) occur in somatotropinomas [14], mutations in classical tumor suppressor genes (e.g., TP53 and RB1) and oncogenes (e.g., Ras) are rarely found in benign pituitary adenomas [15,16,17,18], suggesting the contribution of alternative mechanisms. Earlier studies identified epigenetic modifications, altered expression of cell cycle regulator cyclins and cyclin-dependent kinases (CDKs), growth factors, and their receptors, and disturbances of signal transduction pathways [18]. However, none of these changes can entirely explain the complex mechanism behind adenoma genesis.
In line with these observations, whole-exome (WES) and whole-genome sequencing (WGS) data revealed lower mutation rate in pituitary adenomas compared to other tumor types. These studies also stated that most of the variants are single-nucleotide variants (SNVs) [13, 19, 20]. Newey et al. examined non-functioning adenomas (NFPAs) by WES. They could not identify somatic mutations in genes previously reported in pituitary tumorigenesis, or associated with familial pituitary syndromes or in typical oncogenes/tumor suppressor genes. The authors also showed that there were no recurrent mutations within specific genes, suggesting that there is no common driver genes responsible for pathogenesis of NFPA [13]. Similar results were shown by another group performing WGS of growth hormone (GH) secreting pituitary adenomas [20]. Lan et al. compared invasive and non-invasive pituitary adenomas and found that the DPCR1 (diffuse panbronchiolitis critical region 1), EGFL7 (EGF like domain multiple 7), PRDM family (PR/SET domain family), and LRRC50 (leucine rich repeat containing 50) can function as genetic modifiers and most likely contribute to the development of oncocytic change and invasive tumor phenotype [19].
Mitochondrial variants have only been assessed in oncocytic type of pituitary tumors so far. These studies showed high prevalence of Complex I variants which were described in association with benign behaviour [21, 22].
Here, we report for the first time data obtained using next-generation sequencing technology for analysis of the whole mitochondrial genome of pituitary tumors of different histological types, including GH-producing (GH) and clinically non-functioning [gonadotroph (GO) and hormone-immunonegative (HN)] pituitary adenomas.
Materials and methods
Patients
The study was approved by the Scientific and Research Committee of the Medical Research Council of Hungary (0618/15), and the samples were obtained after acquiring written informed consent from all patients.
Tissue samples were obtained from 44 patients diagnosed with pituitary adenoma, comprising 11 GH-secreting and 33 clinically non-functioning pituitary adenomas (NFPAs), including 22 gonadotroph (GO), and 11 hormone-immunonegative (HN) tumors (Table 1). Pituitary adenoma tissues were surgically removed at the National Institute of Clinical Neurosciences, Budapest, Hungary between 2015 and 2017. Histological diagnoses were performed at the 1st Department of Pathology and Experimental Cancer Research, Semmelweis University, Budapest. The clinical diagnosis of adenomas was based on patients’ hormone levels and routine histological diagnosis including immunostaining for anterior lobe hormones.
DNA isolation
Total DNA was isolated from pituitary adenoma tissues using QIAamp Fast DNA Tissue Kit (Qiagen). DNA purity and concentration were measured using NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific). The presence of normal pituitary tissue was determined similarly to Välimäki et al. [20]. In our cohort, 43 of 44 samples contained no healthy cells, and in the remaining one tissue, the percentage of healthy pituitary cells was less than 5%.
Next-generation sequencing for mtDNA analysis
DNA library was prepared using the VariantPro™ Mitochondrion Panel Library Preparation Kit (LC Sciences). The presence of the desired fragments and the purity of the indexed libraries were analysed on Agilent 2100 Bioanalyzer (Agilent Technologies) using High-Sensitivity DNA Analysis Kit (Agilent Technologies). The concentrations of the libraries were measured using Qubit Fluorometer (Thermo Fisher Scientific). Equimolar amounts of the 45 indexed libraries were pooled to obtain a 4 nM library mixture. After denaturing, and further diluting, the final 10 pM of library mixture was loaded into Illumina cartridge. Sequencing was performed using the Illumina MiSeq Reagent v2 kit (500 cycles) on the Illumina MiSeq instrument following the manufacturer’s instructions (Illumina).
Bioinformatical analysis of next-generation sequencing
Sequencing data processing was performed following the Genome Analysis Toolkit (GATK) best practices guideline as follows: paired end sequencing data were exported to FASTQ file format. Reads were trimmed using Trim Galore (Babraham Bioinformatics, http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and cutadapt [23] to remove adapters and bases, where the quality value was less than 20.
The trimmed reads were aligned with Burrows–Wheeler Aligner (BWA) [24]. Picard tools were used to sort reads (http://broadinstitute.github.io/picard). Local realignment around indels and Base Quality Score Recalibration (BQSR) were performed using GATK [25]. Variant discovery was performed in two steps: single sample variant calling was performed using MuTect2 in GATK tumor_only_mode to call all variants. This was followed by CombineVariants the single sample gVCFs (genome Variant Call Format) to the multi sample VCF (Variant Call Format). Variant effects were predicted using SnpEff [26]. Revised Cambridge Reference Sequence (rCRS) of the Human Mitochondrial DNA (NC_012920.1 gi:251831106) was applied as reference mitochondrial sequence [27]. Variants were annotated using MITOMAP mtDNA Coding Region and RNA Sequence Variants.
Reference alleles were coded as 0, variant alleles as 1 and heteroplasmy ratio was calculated by variant read number/total read for each variant and each sample. A 3% cutoff was used to identify heteroplasmy 3% as previously described [28].
Statistical analysis
Nonparametric Kruskal–Wallis Rank test was applied for evaluating prevalence of variants among different histological groups. Multiple comparisons of mean ranks for all groups to compute post hoc comparisons of all pairs of groups were used [29]. For three groups’ comparison (with all pairs), we used the Conover–Iman test of multiple comparisons using rank sums as post hoc test in R package. We adjusted p values for multiple comparisons with the Benjamini and Hochberg method (BH) [30].
For comparison between two groups, the nonparametric Wilcoxon rank sum test was performed. BH was used for generation of multiple testing corrected p values.
For comparisons within groups, frequency of appearance of each variant (heteroplasmic or full variant number/group sample number) was also evaluated using Kruskal–Wallis Rank test and illustrated on cluster heat maps.
Proportion test was applied with “prop.test” R function which can be used for testing the null hypothesis that the proportions (probabilities of success) in several groups are the same or that they equal certain given values. This test gave similar results to Kruskal–Wallis Rank test and nonparametric Wilcoxon rank sum test.
For hierarchical cluster analysis, we used heatmap R package to draw clustered heat maps with clustering method “average” and with Euclidean distance method.
Sanger validation of mitochondrial variants
The validation of the single-nucleotide variants (SNVs) was performed by Sanger sequencing. We sequenced the same 44 tissue samples used in NGS. To avoid co-amplification of nuclear DNA, we used a well-established method for the exclusive amplification of mitochondrial fragments as described by Ramos et al. [31, 32]. The specific mitochondrial DNA fragments were PCR-amplified and cleaned using the Clean Sweep PCR Purification Kit (A29895, Thermo Fisher Scientific). Sanger sequencing run was performed on Applied Biosystems 3130 Genetic Analyzer (Thermo Fisher Scientific) using the BigDye™ Direct Cycle Sequencing Kit (4458687, Thermo Fisher Scientific).
Results
Whole mitochondrial genome sequencing by NGS
Sequencing run generated an average 52,399 reads per sample (min 24,019; max 242,870) and 95 ± 1% of the reads were aligned to the mitochondrial genome. Total coverage was avg. 10,439,722 read per sample and coverage depth was 630 ± 370 (avg ± SE) reads per base.
We detected 496 single-nucleotide variants (SNVs) in all tumors. Of these, 269 variants were protein-coding of which 135 were non-synonymous and 132 were synonymous (Online Resource 1).
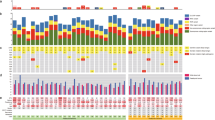
Heteroplasmy occurred in 482 of the detected 496 variants at least in one sample and overall on low level (indicated by colours on cluster heatmap, Fig. 1a). Hormone-immunonegative (HN) adenomas showed a slightly higher heteroplasmy prevalence compared to GO and GH adenomas 8.27% vs. 6.96 and 6.72%, respectively (Table 2). This remained the same after dissection of protein-coding and non-coding variants (Table 2). In addition, we did not find difference in Ki-67 proliferation index or tumor size between sample groups harbouring low (< 50%) and high (> 50%) heteroplasmy.

Possible associations with clinicopathological features. a Profile of detected variants showed overall low level of heteroplasmy and hierarchical cluster analysis could not discriminate pituitary adenoma samples based on either histological type, Ki-67 index, or recurrent/non-recurrent status. The colour scale indicates the ratio of heteroplasmy obtained by NGS, where 0 (blue) shows 100% reference allele and 1 (green) shows 100% variant allele. b Number of the found variants in pituitary adenoma samples, grouped by histological type and ranked from fewer to more variants. Analysing the samples harbouring the highest number of variants obtained that they have the highest Ki-67 indices independently of histological type. c Regarding the number of the variants, we identified 143, 58, and 52 unique variants appeared only in GO, HN, and GH-secreting adenomas, respectively. Legends: rec: recurrent, non-rec: non-recurrent, Ki-67 group 1: Ki-67 proliferation index is between 1 and 4%, Ki-67 group 2: Ki-67 proliferation index is between 5 and 10%, Hist histological subtype, GH growth hormone producing, GO gonadotroph, HN hormone immunonegative
On average, 35 variants were detected per sample. Similar results were obtained in different histological groups: we found 33, 34 and 40 variants per sample in GH-producing, gonadotroph, and hormone-immunonegative adenomas, respectively. Furthermore, samples harbouring the highest number of variants (Fig. 1b) had the highest Ki-67 indices [Ki-67: 8% (GH), 7–10% (GO), and 5% (HN)] independently of histological type.
When analysing distinct histological groups, we identified 143, 58, and 52 unique variants that appeared only in GO, HN, and GH-secreting adenomas, respectively (Fig. 1c, Online Resource 2).
Using hierarchical cluster analysis on all variants, no clear separation of the samples was identified based on histological type, Ki-67 index, or recurrent/non-recurrent status (Fig. 1a).
Nonparametric ANOVA identified eight significant variants among different histological groups. The variants: A11251G, T4216C, T16126C, C15452A, T14798C, A188G, and T16093C differentiated GH producing from HN adenomas. The prevalence of variants: T14798C, G185A, A188G, and T16093C differed between GO and HN adenomas (Table 3).
T16189C variant was found in 40% (6/15) of non-recurrent adenomas compared to recurrent ones, where it was not detectable (0/11) (p = 0.02090).
Individual variant prevalence did not show associations with Ki-67 proliferation index or tumor size.
Technical validation
Four of the eight variants that showed differences among histological groups (G185A, A188G, T16093C, and T16126C) were localized in the D-loop region (displacement loop region, “non-coding” part of mtDNA). A11251G coded a synonymous variant, while T14798C and C15452A represented non-synonymous polymorphisms referred as rs28357681 and rs527236209, respectively, in the gene of mitochondrially encoded cytochrome B (MT-CYB). T4216C also represented a non-synonymous polymorphism as rs1599988 in mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 1 (MT-ND1). We selected rs28357681 and rs1599988 for technical validation, since these variants were protein-coding, non-synonymous variants, and these alterations showed no heteroplasmy; hence, Sanger sequencing was a reasonable option for validation. We validated the two variants on all the 44 samples and we found 100% concordance between NGS and Sanger sequencing results.
Discussion
Mitochondrial dysfunction and mutations in mtDNA have been implicated in several human neoplasms. In this study, we analysed the whole mitochondrial genome in pituitary adenomas by NGS. The VariantPro™ Mitochondria Panel kit we used in this study has only been applied in two studies so far [33, 34]. In one of these the whole mitochondrial genome was sequenced, similarly to our study [34], while in the other, specific primers for the targeted regions were used [33]. In agreement with these studies, we observed an excellent alignment rate to mitochondrial genome and satisfactory coverage depth. This fact together with the results of validation by Sanger sequencing (100% concordance) confirmed the excellent applicability of NGS-based methods for mitochondrial genome sequencing.
Overall, we identified 496 variants in pituitary adenoma tissues compared to the human mitochondrial reference sequence. We also identified variants specific to distinct adenoma histological types. Most of the variants showed low level of heteroplasmy. This is in line with previous publications which claimed that low level of heteroplasmy was more frequent in benign tumors and occurrence of heteroplasmy increased with metastatic potential [28]. Thus, these data support our result, showing that low-level heteroplasmy is one characteristic hallmark of pituitary adenomas. In addition, samples with high and low levels of heteroplasmy did not show difference in Ki-67 proliferation index or tumor size.
Whole mitochondrial variant pattern did not show clear association with clinicopathological features. This finding is in agreement with others showing no significant associations with clinicopathological parameters including sex, age, tumor size, and duration of clinical course in tumors of the central nervous system [35]. However, when analysing the number of individual homoplasmic mitochondrial variants we found that samples harbouring the highest number of variants had the highest Ki-67 indices independent of histological type. Interestingly, increased mtDNA copy number was previously found in gliomas and the relevance of this finding needs further evaluations in pituitary adenomas [36].
We also identified eight individual variants that showed different prevalence in the different histological groups.
Four (G185A, A188G, T16093C, and T16126C) were localized in the D-loop region. Displacement loop (D-loop) region (16,024–16,569 nucleotides) is located in the mitochondrial control region (16024-576 nucleotides), often called the “non-coding” part of the mitochondrial DNA (mtDNA). The function of the D-loop is not entirely clear yet, which makes more difficult to understand the relevance of the present variants. A growing number of studies have been published about correlations between D-loop variants and clinical features [37,38,39]. It has been shown that D-loop mutations may play role in the pathogenesis of breast cancer [39], gastric cancer [40], bladder cancer, lung cancer, and hepatocellular carcinoma [41], while Lin et al. found that somatic mutations in the D-loop associated with a better survival in oral squamous cell carcinoma patients [42]. However, the exact functional contribution of D-loop alterations in tumorigenesis has to be further investigated [41].
Another four variants of eight that showed different prevalence in the different histological groups encoded MT-CYB, MT-ND1, and MT-ND4 (mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 4). MT-CYB is the only subunit of the Respiratory Complex III (Cytochrome bc1 complex) that is encoded in the mitochondrial genome, and it is essential for the assembly of the complex [43, 44]. Moreover, it was shown that disruption of Complex III assembly dramatically reduced the level of Complex I as well [45, 46]. T14798C has been implicated in non-muscle invasive bladder cancer [47]. The MT-ND1 gene has been examined in association with several diseases including recurrent pregnancy loss [48], colorectal cancer [49], bladder cancer [47], Parkinson’s disease [50, 51], and contradicting results have been published regarding it’s possible effect on multiple sclerosis [52] and Leber’s optic atrophy [53,54,55]. MT-ND4 is a subunit of the Respiratory Complex I and is needed to the assembly of the complex. Variations in ND4 sequence have been associated with macular degeneration [56], Leber’s hereditary optic neuropathy [57], mesial temporal lobe epilepsy [58], and cystic fibrosis [59]. In acute myeloid leukaemia, an association between ND4 variant and favourable prognosis has been shown [60]. In contrast, findings from other groups suggested that variations in this gene can elevate pancreatic cancer risk, or be involved in causing cisplatin resistance [61].
We found that the T16189C variant was frequent in non-recurrent adenomas, while it was absent in recurrent ones. Unfortunately, we could not analyse the primary tumor of our recurrent pituitary samples. However, a previous publication compared primary and recurrent oral squamous cell carcinomas and described that although most of the patients showed similar distribution of mutations in the index and the recurrent tumor, there were differences in the level of heteroplasmy [28].
Currently, there is no other publication about whole mtDNA analysis of different types of pituitary adenomas. A single study focused on genes encoding proteins of Respiratory Complex I in pituitary oncocytomas and another one analysed it in head and neck tumors [21, 22]. In total, 20 variants of mtDNA were identified in pituitary oncocytomas [22], and 12 others in other head and neck tumors [21]. Interestingly, none of these variants were found in more than one sample. Notably, these variants were not present in our samples emphasizing the difference between pituitary oncocytomas and pituitary adenomas of anterior lobe origin. These data are in agreement with the conclusion made by authors that unique mtDNA variants are specific for oncocytomas. Our results extend these finding by showing that unique mtDNA variants can be found in pituitary adenoma subtypes. With respect to data on pituitary tumors, the HmtDB database (http://www.hmtdb.uniba.it/) contains 19 mitochondrial variants in 19 GH ± PRL (growth hormone ± prolactin) producing adenomas. Variants described in these samples contained overlaps with our results at 82 positions including the T16189C variant (Online Resource 1). The other 414 variants identified in the current study have never been reported. However, it has to be noted that the low number of samples used in our study was a limitation of our work, and the variants identified should be further investigated on higher number of sample set.
Taken together, in this study, we reported for the first time the whole mitochondrial genome pattern of different human pituitary adenomas. We identified variants characteristic of gonadotroph, GH-producing, and hormone-immunonegative pituitary adenomas. We also observed that high number of variants may have a role in higher proliferation rate and that the T16189C variant can potentially associate with benign behaviour. Although the biological relevance of these results needs further validation, considering our data together with the low level of heteroplasmy suggest that mitochondrial genome alterations may not play a major role in the pathogenesis of pituitary adenomas. We also showed that NGS is a reliable method for the analysis of mtDNA variants with no false-positive results, and can be used for accurate quantification of even a low level of heteroplasmy.
References
Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG (1981) Sequence and organization of the human mitochondrial genome. Nature 290:457–465
Lightowlers RN, Chinnery PF, Turnbull DM, Howell N (1997) Mammalian mitochondrial genetics: heredity, heteroplasmy and disease. Trends Genet 13:450–455. https://doi.org/10.1016/S0168-9525(97)01266-3
Chatterjee A, Mambo E, Sidransky D (2006) Mitochondrial DNA mutations in human cancer. Oncogene 25:4663–4674. https://doi.org/10.1038/sj.onc.1209604
Carew JS, Huang P (2002) Mitochondrial defects in cancer. Mol Cancer 1:9
Máximo V, Lima J, Soares P, Sobrinho-Simões M (2009) Mitochondria and cancer. Virchows Arch 454:481–495. https://doi.org/10.1007/s00428-009-0766-2
Dasgupta S, Hoque MO, Upadhyay S, Sidransky D (2008) Mitochondrial cytochrome B gene mutation promotes tumor growth in bladder cancer. Cancer Res 68:700–706. https://doi.org/10.1158/0008-5472.CAN-07-5532
Voo KS, Zeng G, Mu J-B, Zhou J, Su X-Z, Wang R-F (2006) CD4 + T-cell response to mitochondrial cytochrome b in human melanoma. Cancer Res 66:5919–5926. https://doi.org/10.1158/0008-5472.CAN-05-4574
Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, Manfredi G, Oda H, Ohta S (2005) Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res 65:1655–1663. https://doi.org/10.1158/0008-5472.CAN-04-2012
Larman TC, DePalma SR, Hadjipanayis AG, Protopopov A, Zhang J, Gabriel SB, Chin L, Seidman CE, Kucherlapati R, Seidman JG (2012) Spectrum of somatic mitochondrial mutations in five cancers. Proc Natl Acad Sci USA 109:14087–14091. https://doi.org/10.1073/pnas.1211502109
Huang T (2011) Next generation sequencing to characterize mitochondrial genomic DNA heteroplasmy. Curr Protoc Hum Genet Chapter 19(Unit19):8. https://doi.org/10.1002/0471142905.hg1908s71
Tang S, Huang T (2010) Characterization of mitochondrial DNA heteroplasmy using a parallel sequencing system. BioTechniques 48:287–296. https://doi.org/10.2144/000113389
Ezzat S, Asa SL, Couldwell WT, Barr CE, Dodge WE, Vance ML, McCutcheon IE (2004) The prevalence of pituitary adenomas. Cancer 101:613–619. https://doi.org/10.1002/cncr.20412
Newey PJ, Nesbit MA, Rimmer AJ, Head RA, Gorvin CM, Attar M, Gregory L, Wass JAH, Buck D, Karavitaki N, Grossman AB, McVean G, Ansorge O, Thakker RV (2013) Whole-exome sequencing studies of nonfunctioning pituitary adenomas. J Clin Endocrinol Metab 98:E796–E800. https://doi.org/10.1210/jc.2012-4028
Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L (1989) GTPase inhibiting mutations activate the α chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 340:692–696
Karga HJ, Alexander JM, Hedley-Whyte ET, Klibanski A, Jameson JL (1992) Ras mutations in human pituitary tumors. J Clin Endocrinol Metab 74:914–919. https://doi.org/10.1210/jcem.74.4.1312542
Levy A, Hall L, Yeudall WA, Lightman SL (1994) p53 gene mutations in pituitary adenomas: rare events. Clin Endocrinol (Oxf) 41:809–814
Poncin J, Stevenaert A, Beckers A (1999) Somatic MEN1 gene mutation does not contribute significantly to sporadic pituitary tumorigenesis. Eur J Endocrinol 140:573–576. https://doi.org/10.1530/eje.0.1400573
Vandeva S, Vasilev V, Vroonen L, Naves L, Jaffrain-Rea M-L, Daly AF, Zacharieva S, Beckers A (2010) Familial pituitary adenomas. Ann Endocrinol (Paris) 71:479–485. https://doi.org/10.1016/j.ando.2010.08.005
Lan X, Gao H, Wang F, Feng J, Bai J, Zhao P, Cao L, Gui S, Gong L, Zhang Y (2016) Whole-exome sequencing identifies variants in invasive pituitary adenomas. Oncol Lett 12:2319–2328. https://doi.org/10.3892/ol.2016.5029
Välimäki N, Demir H, Pitkänen E, Kaasinen E, Karppinen A, Kivipelto L, Schalin-Jäntti C, Aaltonen LA, Karhu A (2015) Whole-genome sequencing of growth hormone (GH)-secreting pituitary adenomas. J Clin Endocrinol Metab 100:3918–3927. https://doi.org/10.1210/jc.2015-3129
Kurelac I, MacKay A, Lambros MBK, Di Cesare E, Cenacchi G, Ceccarelli C, Morra I, Melcarne A, Morandi L, Calabrese FM, Attimonelli M, Tallini G, Reis-Filho JS, Gasparre G (2013) Somatic complex I disruptive mitochondrial DNA mutations are modifiers of tumorigenesis that correlate with low genomic instability in pituitary adenomas. Hum Mol Genet 22:226–238. https://doi.org/10.1093/hmg/dds422
Porcelli AM, Ghelli A, Ceccarelli C, Lang M, Cenacchi G, Capristo M, Pennisi LF, Morra I, Ciccarelli E, Melcarne A, Bartoletti-Stella A, Salfi N, Tallini G, Martinuzzi A, Carelli V, Attimonelli M, Rugolo M, Romeo G, Gasparre G (2010) The genetic and metabolic signature of oncocytic transformation implicates HIF1alpha destabilization. Hum Mol Genet 19:1019–1032. https://doi.org/10.1093/hmg/ddp566
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498. https://doi.org/10.1038/ng.806
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80–92. https://doi.org/10.4161/fly.19695
Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 23:147. https://doi.org/10.1038/13779
Kloss-Brandstätter A, Weissensteiner H, Erhart G, Schäfer G, Forer L, Schönherr S, Pacher D, Seifarth C, Stöckl A, Fendt L, Sottsas I, Klocker H, Huck CW, Rasse M, Kronenberg F, Kloss FR (2015) Validation of next-generation sequencing of entire mitochondrial genomes and the diversity of mitochondrial dna mutations in oral squamous cell carcinoma. PLoS One 10:e0135643. https://doi.org/10.1371/journal.pone.0135643
Siegel S, Castellan NJ Jr (1988) Nonparametric statistics for the behavioral sciences, 2nd edn. Mcgraw-Hill Book Company, New York
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol) 57:289–300
Ramos A, Santos C, Alvarez L, Nogués R, Aluja MP (2009) Human mitochondrial DNA complete amplification and sequencing: a new validated primer set that prevents nuclear DNA sequences of mitochondrial origin co-amplification. Electrophoresis 30:1587–1593. https://doi.org/10.1002/elps.200800601
Ramos A, Santos C, Barbena E, Mateiu L, Alvarez L, Nogués R, Aluja MP (2011) Validated primer set that prevents nuclear DNA sequences of mitochondrial origin co-amplification: a revision based on the New Human Genome Reference Sequence (GRCh37). Electrophoresis 32:782–783. https://doi.org/10.1002/elps.201000583
Dai Y, Wang C, Nie Z, Han J, Chen T, Zhao X, Ai C, Ji Y, Gao T, Jiang P (2018) Mutation analysis of Leber’s hereditary optic neuropathy using a multi-gene panel. Biomed Rep 8:51–58
Li X, Liu L, Xi Q, Zhao X, Fang M, Ma J, Zhu Z, Wang X, Shi C, Wang J, Zhu H, Zhang J, Zhang C, Hu S, Ni M, Gu M (2016) Short-term serum deprivation causes no significant mitochondrial DNA mutation in vascular smooth muscle cells revealed by a new next generation sequencing technology. Acta Biochim Biophys Sin (Shanghai) 48:862–864. https://doi.org/10.1093/abbs/gmw059
Vega A, Salas A, Gamborino E, Sobrido MJ, Macaulay V, Carracedo A (2004) mtDNA mutations in tumors of the central nervous system reflect the neutral evolution of mtDNA in populations. Oncogene 23:1314–1320. https://doi.org/10.1038/sj.onc.1207214
Liang BC, Hays L (1996) Mitochondrial DNA copy number changes in human gliomas. Cancer Lett 105:167–173
Miyazono F, Schneider PM, Metzger R, Warnecke-Eberz U, Baldus SE, Dienes HP, Aikou T, Hoelscher AH (2002) Mutations in the mitochondrial DNA D-Loop region occur frequently in adenocarcinoma in Barrett’s esophagus. Oncogene 21:3780–3783. https://doi.org/10.1038/sj.onc.1205532
Sharma H, Singh A, Sharma C, Jain SK, Singh N (2005) Mutations in the mitochondrial DNA D-loop region are frequent in cervical cancer. Cancer Cell Int 5:34. https://doi.org/10.1186/1475-2867-5-34
Ye C, Shu XO, Pierce L, Wen W, Courtney R, Gao Y-T, Zheng W, Cai Q (2010) Mutations in the mitochondrial DNA D-loop region and breast cancer risk. Breast Cancer Res Treat 119:431–436. https://doi.org/10.1007/s10549-009-0397-y
Zhao Y-B, Yang H-Y, Zhang X-W, Chen G-Y (2005) Mutation in D-loop region of mitochondrial DNA in gastric cancer and its significance. World J Gastroenterol 11:3304–3306. https://doi.org/10.3748/wjg.v11.i21.3304
Chatterjee A, Dasgupta S, Sidransky D (2011) Mitochondrial subversion in cancer. Cancer Prev Res (Phila) 4:638–654. https://doi.org/10.1158/1940-6207.CAPR-10-0326
Lin J-C, Wang C-C, Jiang R-S, Wang W-Y, Liu S-A (2015) Impact of somatic mutations in the D-Loop of mitochondrial DNA on the survival of oral squamous cell carcinoma patients. PLOS One 10:e0124322. https://doi.org/10.1371/journal.pone.0124322
di Rago JP, Macadre C, Lazowska J, Slonimski PP (1993) The C-terminal domain of yeast cytochrome b is essential for a correct assembly of the mitochondrial cytochrome bc1 complex. FEBS Lett 328:153–158
Rana M, de Coo I, Diaz F, Smeets H, Moraes CT (2000) An out-of-frame cytochrome b gene deletion from a patient with parkinsonism is associated with impaired complex III assembly and an increase in free radical production. Ann Neurol 48:774–781
Bruno C, Santorelli FM, Assereto S, Tonoli E, Tessa A, Traverso M, Scapolan S, Bado M, Tedeschi S, Minetti C (2003) Progressive exercise intolerance associated with a new muscle-restricted nonsense mutation (G142X) in the mitochondrial cytochrome b gene. Muscle Nerve 28:508–511. https://doi.org/10.1002/mus.10429
Budde SM, van den Heuvel LP, Janssen AJ, Smeets RJ, Buskens CA, DeMeirleir L, Van Coster R, Baethmann M, Voit T, Trijbels JM, Smeitink JA (2000) Combined enzymatic complex I and III deficiency associated with mutations in the nuclear encoded NDUFS4 gene. Biochem Biophys Res Commun 275:63–68. https://doi.org/10.1006/bbrc.2000.3257
Guney AI, Ergec DS, Tavukcu HH, Koc G, Kirac D, Ulucan K, Javadova D, Turkeri L (2012) Detection of mitochondrial DNA mutations in nonmuscle invasive bladder cancer. Genet Test Mol Biomarkers 16:672–678. https://doi.org/10.1089/gtmb.2011.0227
Colagar AH, Mosaieby E, Seyedhassani SM, Mohajerani M, Arasteh A, Kamalidehghan B, Houshmand M (2013) T4216C mutation in NADH dehydrogenase I gene is associated with recurrent pregnancy loss. Mitochondrial DNA 24:610–612. https://doi.org/10.3109/19401736.2013.772150
Akouchekian M, Houshmand M, Akbari MHH, Kamalidehghan B, Dehghan M (2011) Analysis of mitochondrial ND1 gene in human colorectal cancer. J Res Med Sci 16:50–55
Kirchner SC, Hallagan SE, Farin FM, Dilley J, Costa-Mallen P, Smith-Weller T, Franklin GM, Swanson PD, Checkoway H (2000) Mitochondrial ND1 sequence analysis and association of the T4216C mutation with Parkinson’s disease. Neurotoxicology 21:441–445
Swerdlow RH, Weaver B, Grawey A, Wenger C, Freed E, Worrall BB (2006) Complex I polymorphisms, bigenomic heterogeneity, and family history in Virginians with Parkinson’s disease. J Neurol Sci 247:224–230. https://doi.org/10.1016/j.jns.2006.05.053
Andalib S, Emamhadi M, Yousefzadeh-Chabok S, Salari A, Sigaroudi AE, Vafaee MS (2016) MtDNA T4216C variation in multiple sclerosis: a systematic review and meta-analysis. Acta Neurol Belg 116:439–443. https://doi.org/10.1007/s13760-016-0675-5
Brown MD, Voljavec AS, Lott MT, Torroni A, Yang CC, Wallace DC (1992) Mitochondrial DNA complex I and III mutations associated with Leber’s hereditary optic neuropathy. Genetics 130:163–173
Howell N, Kubacka I, Halvorson S, Howell B, McCullough DA, Mackey D (1995) Phylogenetic analysis of the mitochondrial genomes from Leber hereditary optic neuropathy pedigrees. Genetics 140:285–302
Johns DR, Berman J (1991) Alternative, simultaneous complex I mitochondrial DNA mutations in Leber’s hereditary optic neuropathy. Biochem Biophys Res Commun 174:1324–1330
Restrepo NA, Mitchell SL, Goodloe RJ, Murdock DG, Haines JL, Crawford DC (2015) Mitochondrial variation and the risk of age-related macular degeneration across diverse populations. Pac Symp Biocomput 20:243–254
Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, Nikoskelainen EK (1988) Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 242:1427–1430
Gurses C, Azakli H, Alptekin A, Cakiris A, Abaci N, Arikan M, Kursun O, Gokyigit A, Ustek D (2014) Mitochondrial DNA profiling via genomic analysis in mesial temporal lobe epilepsy patients with hippocampal sclerosis. Gene 538:323–327. https://doi.org/10.1016/j.gene.2014.01.030
Valdivieso AG, Marcucci F, Taminelli G, Guerrico AG, Alvarez S, Teiber ML, Dankert MA, Santa-Coloma TA (2007) The expression of the mitochondrial gene MT-ND4 is downregulated in cystic fibrosis. Biochem Biophys Res Commun 356:805–809. https://doi.org/10.1016/j.bbrc.2007.03.057
Damm F, Bunke T, Thol F, Markus B, Wagner K, Göhring G, Schlegelberger B, Heil G, Reuter CWM, Püllmann K, Schlenk RF, Döhner K, Heuser M, Krauter J, Döhner H, Ganser A, Morgan MA (2012) Prognostic implications and molecular associations of NADH dehydrogenase subunit 4 (ND4) mutations in acute myeloid leukemia. Leukemia 26:289–295. https://doi.org/10.1038/leu.2011.200
van Gisbergen MW, Voets AM, Starmans MHW, de Coo IFM, Yadak R, Hoffmann RF, Boutros PC, Smeets HJM, Dubois L, Lambin P (2015) How do changes in the mtDNA and mitochondrial dysfunction influence cancer and cancer therapy? Challenges, opportunities and models. Mutat Res Rev Mutat Res 764:16–30. https://doi.org/10.1016/j.mrrev.2015.01.001
Acknowledgements
Open access funding provided by Semmelweis University (SE).
Funding
This work has been funded by Hungarian Scientific Research Grant (OTKA PD116093) and by Semmelweis Research-Innovation Fund (STIA-KF-17) to Henriett Butz, and EFOP-3.6.3-VEKOP-16-2017-00009 by Semmelweis University to Kinga Németh. Henriett Butz is a recipient of Bolyai Research Fellowship of Hungarian Academy of Sciences. Kinga Németh received NTP-EFÖ-P-15 Grant from The Ministry of Human Capacities.
Author information
Authors and Affiliations
Contributions
HB and AP conceived the study. KN, HB, and AP designed the research. NSz and PI recruited, diagnosed, and treated the patients. SC performed the neurosurgeries and sample acquisition. LR performed immunostaining and histological analysis. KN performed sample preparation and next-generation sequencing. OD and IL performed the bioinformatic analysis. KN, BSz, KPA, and KL performed Sanger sequencing. KN, AP, and HB analysed data, wrote, and edited the manuscript. All authors gave the final approval for publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This article does not contain any studies with animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
40618_2019_1005_MOESM1_ESM.docx
Online Resource 1 Variants identified in pituitary adenomas. Common variants between current study and pituitary adenoma samples available in HmtDB are indicated. Current study included 44 [11 GH-producing and 33 non-functioning (gonadotroph and hormone-immunonegative)] adenomas. HmtDB samples represent only GH positive adenomas (n = 19). Out of 496 variants 414 were firstly identified by the current study (DOCX 56 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Németh, K., Darvasi, O., Likó, I. et al. Next-generation sequencing identifies novel mitochondrial variants in pituitary adenomas. J Endocrinol Invest 42, 931–940 (2019). https://doi.org/10.1007/s40618-019-1005-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-019-1005-6