
Abstract
Examination of the fetus with congenital anomalies is essential to identify the nature of the defects and determine if they constitute a part of a recognized abnormality of development and morphogenesis. A structured approach, including a comprehensive history and details of the anomalies present, assists in making a definitive diagnosis. Dysmorphic diagnosis in the fetus is a challenge as the complete phenotype may not be manifested, coupled with the possibility of subtle dysmorphism that is difficult to recognize. Correlation of the antenatal scan findings with those at autopsy, combined with the experienced eye of a clinical geneticist, is helpful in making a syndrome diagnosis. Specialized databases, reference textbooks, and previously published malformation syndromes are important aids for the geneticist. An accurate diagnosis is important to counsel families of the prognosis, available treatment options, and recurrence risks in future conceptions. New technologies for genetic testing are available to confirm a syndrome diagnosis and consequently, it is vital that appropriate fetal samples are stored in all cases. A team approach with expert input from a fetal medicine specialist, perinatal pathologist, geneticist, radiologist, and the genetics laboratory are important components of fetal dysmorphic syndrome identification.
Similar content being viewed by others



Avoid common mistakes on your manuscript.
Introduction
While the modern era of dysmorphology was founded in the 1960s, many recent advances have increased the relevance and output in this field. Dr David W Smith, the “father of dysmorphology” started to study abnormalities of structure as an offshoot of his endocrinology practice and addressed dysmorphology in his seminal paper in 1966 [1]. The field has expanded exponentially, especially over the last decade, with application of advanced molecular techniques, which in addition to defining new malformation syndromes, has also made it possible to understand the underlying cause of birth defects and syndromes.
The relevance of dysmorphology in fetal medicine relates to the identification of fetal anomalies on an antenatal scan. Most anomalies arise early in embryonic or fetal life and are present in 30 % of all conceptions. However, only 3 % of all births finally have malformations, and this is due to natural losses and termination after antenatal detection [2]. With improved instruments and expertise, it is now possible to identify anomalies as early as the first trimester of gestation. This improves pregnancy management with the option of early termination for lethal anomalies and high-risk monitoring of others for progression, identification of associated anomalies, and appropriate care [3].
Importance of an Accurate Diagnosis
Families with an ongoing pregnancy are unprepared for the news of an anomalous ultrasound. Appropriate counseling and comprehensive care are important integral components of the management in such situations. A survey of 30 women undergoing termination of pregnancy for fetal abnormality identified fragmented care related to poor communication and disorganized medical systems, delays in subspecialty appointments, and absence of availability of genetic counseling services. This was compounded with limited ability of the obstetrician to provide genetic counseling, including making a definite diagnosis and guiding the family through decision-making [4]. Therefore, the primary care provider, often the obstetrician, plays a vital role in the management of pregnancies with an identified fetal structural anomaly. The knowledge of a systematic approach, a search for likely associated malformations, and syndrome identification, are vital to counsel families so that they can make an informed decision to continue or interrupt the pregnancy [5].
The fallacies of concluding outcomes on antenatally detected malformations without knowledge of dysmorphology and syndromology were recognized more than a decade ago [6]. The obstetrician needs to be familiar with fetal anomalies and their associations and also be informed of the value of fetal autopsy to make a final diagnosis for pregnancies that are discontinued [7]. Options of fetal radiographs with external examination in skeletal anomalies [8], fetal photographs [9], and newer techniques of minimally-invasive autopsy [10] should also be considered in specific circumstances.
Dysmorphology Diagnosis
Brian Hall described the art of dysmorphology as a detailed historical and clinical evaluation and an inexpensive visual aid to arrive at a diagnosis in a child with dysmorphism [11]. This concept extends to the evaluation of a dysmorphic fetus, as these defects originate early in embryonic and fetal life. The ultimate aim of fetal dysmorphology, antenatal or postnatal, is to arrive at a differential of diagnostic probabilities and apply definitive tests to establish the diagnosis. An advancing gestation in pregnancy is the greatest constraint and prompt and efficient evaluation to confirm a diagnosis and facilitate appropriate counseling is important.
Common causes of congenital malformations include a genetic etiology due to chromosomal or single gene defects, environmental etiology comprising intrauterine infections, drugs and medications, maternal metabolic disorders, ionizing radiation, and the multifactorial group. For some birth defects and malformation, syndromes etiologies are still not defined. Evaluation of birth defects involves recognition of the abnormality and its etiopathogenesis.
The Process of Syndrome Diagnosis
The process of syndromic diagnosis includes a detailed history and an examination of the fetus for major and minor anomalies, ancillary investigations, and an integration of all the information as depicted in Fig. 1.

The process of syndrome diagnosis
History (Includes a Detailed Family History As Well As the Current Pregnancy Details)
Family History
Consanguineous marriage or that within the same caste increases the risk for autosomal recessive disorders. A three-generation family history with phenotypic details of other affected siblings or relatives, if present, should be obtained. If a defined diagnosis exists in another affected family member, it must be correlated with any anomaly or dysmorphism observed in the fetus. Consider a patient who gave birth to a child with multiple malformations, growth restriction, and trisomy 18, as confirmed by fluorescent in situ hybridization. The second pregnancy of this couple was similarly affected. A more detailed family history revealed that the patient’s sister also interrupted a pregnancy with antenatally identified similar fetal malformations. Chromosomal analysis confirmed both sisters to be carriers of a balanced translocation involving chromosome 4 and 18 (Fig. 2). A simple family history could have altered the pregnancy outcomes in this family.

Carrier of a balanced translocation—46, XX, t(4;18) (p14; q11.2)
It is often useful to ask for photographs of the proband and other affected and unaffected relatives to make a diagnosis and exclude nonsyndromic familial traits. This is especially relevant when direct examination of the affected individuals is not possible or in situations where the phenotype of the malformation syndrome evolves with time (e.g., Prader–Willi syndrome, Noonan syndrome, tuberous sclerosis). Parental ages at conception have relevance for increased risk of chromosomal aneuploidies in advanced maternal age and the increased risk of autosomal dominant disorders due to de novo mutations with advance paternal age.
Method of Conception
Some studies report an almost threefold increased risk of congenital malformations in pregnancies conceived by artificial techniques compared to natural conception [12, 13], whereas others fail to identify this association [14]. All categories of birth defects are observed except those of head, face, neck, and eye. A systematic review and meta-analysis reported an increased incidence of imprinting disorders (e.g., Beckwith–Wiedemann syndrome) in children conceived by assisted reproductive techniques [15].
Maternal Drugs/Alcohol Intake
A history of drug intake especially for known teratogens must be elicited. Common teratogenic drugs include anticonvulsants such as sodium valproate, carbimazole, warfarin, retinoic acid, angiotensin converting enzyme inhibitors, and angiotensin receptor antagonists as well as alcohol. A systematic analysis in Netherlands identified that as much as 5 % of all pregnant women received a potentially teratogenic drug during pregnancy and 0.66 % received a drug contraindicated in pregnancy due to evidence of fetal malformations associated with the drug intake in pregnancy [16]. Therefore, increased awareness among health care professionals is indicated.
Maternal Infections
Common infections with an increased risk of malformation include rubella, cytomegalovirus, syphilis, and toxoplasmosis.
Maternal Disorders
Pre-existing and gestational diabetes is associated with a 2–3 fold increased risk of congenital malformations [17]. Other maternal disorders associated with adverse fetal outcomes include pregnancy induced hypertension, systemic lupus erythematosus, and metabolic disorders like inadequately treated phenylketonuria. Table 1 lists some of the frequent malformations observed with common teratogenic drugs, infections, and maternal disorders.
Serial Antenatal Scans
All scans need to be reviewed to assess fetal biometry, identify fetal growth restriction, shortening of tubular bones for a skeletal dysplasia as well as major and minor malformations. In the presence of a CNS malformation, fetal MRI may clarify anatomy that is not well delineated by antenatal ultrasound examination. Common malformations for which MRI is particularly valuable include ventriculomegaly, posterior fossa anomalies, and corpus callosum agenesis [18].
Abnormal Screening Results
Review the markers in the first and second trimester screening for association with structural anomalies and adverse pregnancy outcomes.
Results of Any Invasive Procedures
Check outcomes from fetal karyotype, microarray, or fluorescent in situ hybridization (FISH) analysis for microdeletions and duplication syndromes, DNA-based tests (e.g., mutational analysis for achondroplasia) or other ancillary tests.
Review Records of Previous Affected Children
Prenatal ultrasonogram records may provide information about abnormal growth pattern, if available. Events at birth including fetal distress, oligohydramnios or polyhydramnios, birth weight, length and head circumference, and neonatal behavior and feeding history should be sought. Details of development history and behavior with formal assessment are very important. Examination of old records, noted findings, and treatment as well as anthropometry details with growth charted over time can assist to reach a conclusive diagnosis in some cases.
Examination of the Fetus for Malformations and Dysmorphism
A thorough head-to-toe examination of the fetus is essential, whether performed antenatally by ultrasound or after discontinuation of the pregnancy. Evaluation of morphology with a keen and systematic eye by a good dysmorphologist is the key to making a diagnosis. Knowledge of the nature and pattern of anomalies, as illustrated below, is important and the first step to making a diagnosis. Recognition of the pattern of anomalies and diagnosis of a malformation syndrome is essential to counsel families. However at times, despite all efforts, a diagnosis cannot be made. In these cases, recognition of the nature and pattern of the anomalies, referred to below, may help determine the underlying cause, prognosis, and recurrence risks for appropriate counseling.
Single Primary Defect or Multiple Birth Defects
When an anomaly is identified, it is important to differentiate a single primary anomaly from those associated with multiple defects [19]. This simplified categorization of the pattern of anomalies according to the type of developmental error that produced the defect is useful to determine the genetic basis of the birth defect and in counseling for recurrence risks.
A malformation is an anomaly of structure that occurs due to an error in morphogenesis. A malformation can be major or minor. A major malformation is one that is severe and interferes with function, e.g., duodenal atresia, neural tube defects, cleft lip and palate, or heart defects like rhabdomyoma, atrioventricular canal defects. Some major malformations, like the following, suggest a specific disorder:
-
Left-sided heart defects (e.g., coarctation of aorta, bicuspid aortic stenosis) in Turner syndrome
-
Rhabdomyoma in tuberous sclerosis (Fig. 3)
Fig. 3 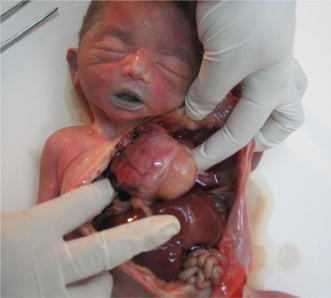
Fetus with rhabdomyoma. In this case the parents should be evaluated for tuberous sclerosis, an autosomal dominant genetic disorder
-
Atrioventricular canal defects in Down syndrome
-
Duodenal atresia in Down syndrome

Minor malformations are mainly of cosmetic significance and may be difficult to pick up on an antenatal scan. However, they should be looked for postnatally, as multiple minor malformations are important phenotypic features of some syndromes (Fig. 4). The presence of two or more minor anomalies also increases the likelihood of a major malformation that must be looked for, if not overt.

Fetus at 36 weeks gestation, delivered after intrauterine demise. Examination showed multiple minor malformations—a Depressed bridge of nose, upslant of left eye, small, low-set ears and simian crease with clinodactyly. b The diagnosis of trisomy 21 was confirmed by FISH
A malformation should be differentiated from other single primary defects in development like deformation, disruption and dysplasia. The type of the anomaly is a consequence of the stage of embryogenesis at which the teratogenic or genetic factor occurred and this information can be useful to guide prognosis. However, it is important to note that these distinctions may not be absolute. The clinical relevance of differentiating birth defects as above is to help formulate a differential diagnosis to serve as a guide for further investigations.
Deformations are abnormalities of shape and form of a normally differentiated structure. They usually occur late in fetal life compared to malformations that are of embryonic origin. The common causes include extrinsic abnormal mechanical forces (e.g., fetal crowding due to uterine abnormalities, multiple gestation, oligohydramnios, breech presentation) and intrinsic defects such as fetal neuromuscular disorders. A common example is talipes equinovarus, which can occur secondary to oligohydramnios or as a result of fetal akinesia due to a primary hereditary neuromuscular etiology. Though the common deformations that occur due to extrinsic forces have a 2–5 % recurrence risk, provided the initiating event does not recur, it is important to remember that those due to intrinsic defects will have a recurrence based on the underlying cause.
Disruptions are anomalies that occur due to abnormal extrinsic factors in a previously normal tissue. The common disruptive forces include floating amniotic bands and interruption of blood supply. An example is the amniotic band syndrome presenting with asymmetric limb amputations and associated bizarre malformations as shown in the Fig. 5. Most disruptions are sporadic events with a negligible recurrence risk, but some vascular disruptions can be related to genetic defects that affect vessel integrity and/or thrombosis [20, 21].

Amniotic band syndrome with omphalocele, syndactyly, oligodactyly, and asymmetrical transverse limb defect. The left ear was also absent. A fibrous band is seen attached to the fingers of the right hand
The presence of multiple anomalies in a fetus requires recognition of these as a malformation syndrome, a malformation sequence or an association. This helps to evaluate for a defined etiology, counsel the families of the prognosis, management, and recurrence risk in subsequent pregnancies.
After one anomaly is identified, a detailed fetal examination should be done to evaluate for associated anomalies and a multiple malformation syndrome. This refers to a recognizable pattern of a combination of major and minor malformations that occurs due an underlying etiology. In the fetus depicted in Fig. 6a, b, c, the pregnancy was discontinued after counseling in view of multiple anomalies identified by ultrasound study. These included bilateral polycystic kidneys and occipital encephalocele. Examination of the fetus identified associated polydactyly. The collective findings are consistent with a ciliopathy disorder, most likely Meckel–Gruber syndrome or Joubert syndrome. Mutation testing in fetal DNA identified a homozygous mutation in CEP290 gene (c.1419_1423delAATAA) confirming Meckel–Gruber syndrome. Meckel–Gruber syndrome is autosomal recessive with a 25 % recurrence risk and confirmation of the diagnosis in the fetus allows for definite prenatal testing in subsequent pregnancies.

Meckel–Gruber syndrome. a Cystic kidneys. b Encephalocele. c Polydactyly
A fetus with scoliosis, hemivertebrae and hypoplastic first metacarpal, cardiac malformation, and tracheoesophageal fistula has anomalies consistent with VACTERL (vertebral, anal, cardiac, tracheoesophageal, renal, radial, and limb). VACTERL is an example of an association that includes a group of unrelated anomalies, which come together more often than expected by chance alone. It is important to recognize them, as their risk of recurrence is low. With recent advances in technology and diagnostic abilities, the molecular basis for some previously described associations are now understood. An example is CDH7 mutation in CHARGE (C—coloboma of iris or retina, H—heart defects A—atresia of the choanae, R—retardation of growth and development, G—genital anomalies, mostly in the male, and E—ear abnormalities.) syndrome [22].
In addition to the above, multiple defects in the fetus might represent a sequence, which is a specific pattern of abnormality that occurs due to a cascade of events initiated by a primary malformation or disruption. For example, in Potter sequence, renal agenesis is the primary malformation that causes oligohydramnios that, in turn, results in clubfeet, flat facies, and hypoplastic lungs. Pierre Robin sequence due to micrognathia and consequent glossoptosis and U-shaped cleft palate is another example.
In the process of examination of a fetus with anomalies, specific features or “pearls of dysmorphology” should bring to mind a specific diagnosis or at least narrow the search. Examples that may be identified in the fetus include webbed neck in Turner and Noonan syndromes, “mitten hands” in Apert syndrome, or broad thumb and great toe in Rubinstein–Taybi syndrome. Familiarity with these key findings requires experience and preparation.
Making a Clinical Diagnosis
Making a dysmorphology diagnosis in a fetus can be a greater challenge compared to that in a child. The latter allows for an easier appreciation of a “facial gestalt”, variations of facial expressions, behavior patterns, and evolution of a phenotype over time. Nonetheless, a general impression of some common syndromes can be suspected in a fetus and then confirmed by appropriate tests. Down syndrome (Fig. 4a, b), Cornelia de Lange syndrome and Rubinstein–Taybi syndrome (Fig. 7a, b) are some examples of a gestalt diagnosis in a fetus. Astute clinical observation sharpened over time with experience will narrow down the diagnostic possibilities, for which further laboratory tests and correlation with similar reports and photographs in published are paramount to making a malformation syndrome diagnosis.

Gestalt diagnosis—facial impression of Rubinstein–Taybi syndrome
Syndrome diagnosis in a fetus in the antenatal period or at autopsy begins with identification of a/multiple major malformation/malformations and then looking for additional features of common syndromes associated with the malformation. It can be difficult to appreciate facial dysmorphism in the fetus and thereby careful and detailed examination for subtle signs facilitates diagnosis. For example, ventriculomegaly would entail looking for a chromosomal etiology, X-linked family history and adducted thumbs for L1 syndrome, congenital cytomegalovirus or toxoplasma infections, hydrolethalus syndrome, eye anomalies for Walker–Warburg syndrome and muscle–eye–brain disease.
A combination of pivotal signs and significant investigations can be correlated using search engines, dysmorphology books, and databases to arrive at a differential diagnosis and, when possible, a definite diagnosis for a case. Some excellent references are Winter–Baraitser London Dysmorphology Database [23], Online Mendelian Inheritance in Man [24], Smith’s Recognizable Patterns of Human Malformations [25], Orphanet [26], POSSUMweb [27], and GeneReviews [28]. The London Medical Database is a compendium of over 4700 dysmorphic and multiple congenital anomaly syndromes. These resources are “systems for experts” and not experts to reach a diagnosis. They cannot replace careful history taking and detailed clinical examination with accurate recognition and description of phenotype by an experienced dysmorphologist.
Postnatal examination of the fetus terminated for an ultrasound diagnosis of a lethal skeletal dysplasia deserves special mention. Skeletal radiographs and fetal examination are the two most important components of an autopsy to accurately diagnose the type of skeletal dysplasia, as this is usually not possible on an antenatal scan [29].
Investigations
Investigations performed help to refine the differential diagnosis and further confirm the clinical suspicion. The most suitable investigation(s) is selected on a case specific basis. For ease of understanding and application, the investigations can be grouped under various headings:
-
(a)
Radiographs Anteroposterior and lateral radiographs of the fetus are recommended to look for skeletal anomalies like absent or hypoplastic clavicle in cleidocranial dysostosis and Yunis–Varon syndrome, vertebral and abnormalities of spine in spondylocostal dysostosis and VACTERL.
-
(b)
CT Scan and MRI CT Scan in skeletal dysplasias may help to define the abnormality better. MRI is useful for posterior fossa anomalies that may not be well defined on the antenatal scan. Minimally invasive autopsy is a poorly chosen euphemism for application of radiographic techniques in place of traditional autopsy. Cost and expertise remain considerations for such procedures, which do not provide potentially invaluable histopathological information. Although postmortem MRI can confirm ventriculomegaly, additional histological data (cerebral cortical dysplasia, gonadoblastoid dysplasia of the testes) may lead to a specific diagnosis—Walker–Warburg syndrome.
-
(c)
Infections TORCH and parvovirus serology in maternal or blood and DNA or immunohistochemical studies of fetal tissues.
-
(d)
Chromosomal Analysis Common indications include
-
1.
Presence of a typically defined chromosomal syndrome like Down syndrome, trisomy 18.
-
2.
Malformations associated with a high incidence of chromosomal defect e.g., omphalocele, holoprosencephaly, or cystic hygroma.
-
3.
Multiple malformations in the fetus not conforming to a defined syndrome, or in a setting of a history of recurrent spontaneous abortions/family history of intellectual disability or dysmorphism.
-
(e)
FISH This is indicated in suspected microdeletion syndromes. In fetuses, microdeletion should be suspected due to the presence of a specific anomaly(ies), e.g., 22q11.1 deletion (velocardiofacial/DiGeorge syndrome) in fetuses with cardiac malformations or cleft palate, 7q11.2 deletion (Williams–Beuren syndrome) when supravalvular aortic stenosis or peripheral pulmonary stenosis is identified.
-
(f)
Chromosomal Microarray Analysis (CMA) Chromosomal microarray tests for copy number variations at a higher resolution than a routine karyotype. It is beneficial in the presence of isolated fetal malformations such as diaphragmatic hernia, holoprosencephaly, in cases of intrauterine fetal demise, and a combination of malformations not consistent with a specific malformation syndrome. Abnormalities on microarray beyond those identified on karyotype are reported in 6–10 % fetuses with anomalies [30, 31]. Actively dividing cells are not required for microarray analysis. Unlike a karyotype, microarray will not detect balanced chromosomal rearrangements.
-
(g)
Molecular Diagnosis to identify the mutation in syndromes where the gene responsible for the phenotype is identified. This not only confirms the clinical diagnosis but also allows for counseling of genetic risks and available reproductive options. It is important to store fetal DNA before termination of an affected pregnancy or at the time of fetal autopsy. Appropriate antenatal samples include chorionic villi, and amniocytes or fetal blood. Postnatal samples should be collected carefully with special care to prevent maternal contamination. Appropriate samples include fetal blood at the time of delivery, tissue from the fetal surface of the placenta, umbilical cord, and fetal spleen. For DNA-based studies, samples should be collected as soon a possible after delivery and either frozen or stored in normal saline or appropriate transport medium.
-
(h)
Metabolic Tests Some syndromes like peroxisomal disorders, Smith–Lemli–Opitz syndrome, and hypophosphatasia are diagnosed by measuring metabolites like very long chain fatty acids and plasmalogens, and serum alkaline phosphatase, respectively, in fetal blood. Prompt and appropriate collection and handling of blood samples is important to insure valid results.
Counseling
When a fetal anomaly is identified on an antenatal scan, the family wants to know the possible outcomes, long-term prognosis, and the burden of disease. If a definite diagnosis is made, the counseling is specific and families can be counseled appropriately, based on available information for the disorder. For example, a fetus with duodenal atresia due to trisomy 21 will have a different outcome compared to that of a fetus with an isolated duodenal atresia, which would have a better quality of life after surgery. In situations where definitive answers cannot be arrived at, the counseling is challenging. In both situations, it is essential to recommend examination of the neonate or fetus after delivery. Arriving at an accurate diagnosis is important to identify families at risk and determine the recurrence risk in subsequent pregnancies and the options for early identification or prevention of the recurrence.
Conclusion
Fetal dysmorphology and syndrome delineation follow the same stepwise approach as in a child but with some distinctions specific to the fetal period. The eye does not see what the mind does not know and therefore, an understanding to making a diagnosis is important in situations such as fetal malformations, fetal growth restriction, and nonimmune hydrops fetalis. Fetal samples for diagnostic studies must be saved appropriately. A multidisciplinary approach involving a fetal medicine specialist, pathologist, dysmorphologist/clinical geneticist and radiologist works best to arrive at a diagnosis in an anomalous fetus [32]. Where these resources are not available, a goal should be to document phenotypic data as thoroughly as possible, and collect and store tissue samples appropriately so that interpretation and ancillary testing can be done later, if needed.
References
Smith DW. Dysmorphology (teratology). J Pediatr. 1966;69:1150–69.
Davies DP, Evans DJR. Clinical dysmorphology: understanding congenital abnormalities. Paediatr Child Health. 2003;13:288–97.
Jones D, Fiozzo F, Waters B, McKnight D, Brown S. First-trimester diagnosis of Meckel–Gruber syndrome by fetal ultrasound with molecular identification of CC2D2A mutations by next-generation sequencing. Ultrasound Obstet Gynecol. 2014;44:719–21.
Gawron LM, Cameron KA, Phisuthikul A, Simon MA. An exploration of women’s reasons for termination timing in the setting of fetal abnormalities. Contraception. 2013;88:109–15.
Chitayat D, Babul-Hirji R. Genetic counselling in prenatally diagnosed non-chromosomal fetal abnormalities. Curr Opin Obstet Gynecol. 2000;12:77–80.
Donnenfeld AE. Fetal dysmorphology. Ultrasound Obstet Gynecol. 1997;9:73–5.
Puri RD, Verma IC. The role of radiographs in fetal autopsy. J Fetal Med. 2014;1:7–9.
Deka D, Naha M, Dadhwal V, Kabra M, Gupta N. At least an infantogram if not perinatal autopsy. J Fetal Med. 2014;1:33–9.
Movva S, Kotecha U, Sharma D, Puri RD, Verma IC. Prenatal diagnosis and elucidation of a novel molecular basis in Carpenter syndrome. J Fetal Medicine. 2014;1:89–93.
Sebire NJ. Towards the minimally invasive autopsy? Ultrasound Obstet Gynecol. 2006;28:865–7.
Hall BD. The state of the art of dysmorphology. Am J Dis Child. 1993;147:1184–9.
Gutarra-Vilchez R, Santamariña-Rubio E, Salvador J, Borrell A. Birth defects in medically assisted reproduction pregnancies in the city of Barcelona. Prenat Diagn. 2014;34:327–34.
Mayor S. Risk of congenital malformations in children born after assisted reproduction is higher than previously thought. BMJ. 2010;340:c3191.
Uyar A, Seli E. The impact of assisted reproductive technologies on genomic imprinting and imprinting disorders. Curr Opin Obstet Gynecol. 2014;26:210–21.
Lazaraviciute G, Kauser M, Bhattacharya S, Haggarty P, Bhattacharya S. A systematic review and meta-analysis of DNA methylation levels and imprinting disorders in children conceived by IVF/ICSI compared with children conceived spontaneously. Hum Reprod Update. 2014;20:840–52.
Zomerdijk I, Ruiter R, Houweling L, Herings R, Straus S, Stricker B. Dispensing of potentially teratogenic drugs before conception and during pregnancy: a population-based study. BJOG. 2015;122:1119–29.
Allen VM, Armson BA, Wilson RD, Allen VM, Blight C, Gagnon A, et al. Teratogenicity associated with pre-existing and gestational diabetes. J Obstet Gynaecol Can. 2007;29:927–44.
Weisstanner C, Kasprian G, Gruber GM, Brugger PC, Prayer D. MRI of the fetal brain. Clin Neuroradiol. 2015;25(Suppl 2):189–96.
Homfray T, Farndon PA. Fetal anomalies—the geneticist’s approach. In: Coady AM, Bower S, editors. Twinning’s textbook of fetal abnormalities. 3rd ed. China: Churchill Livingstone Elsevier; 2015. p. 139–60.
Torfs CP, Christianson RE, Iovannisci DM, Shaw GM, Lammer EJ. Selected gene polymorphisms and their interaction with maternal smoking, as risk factors for gastroschisis. Birth Defects Res A Clin Mol Teratol. 2006;76:723–30.
Schram A, Kroes HY, Sollie K, Timmer B, Barth P, van Essen T. Hereditary fetal brain degeneration resembling fetal brain disruption sequence in two sibships. Am J Med Genet A. 2004;127A:172–82.
Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–7.
London Medical Databases. Winter–Baraitser dysmorphology database. www.lmdatabases.com. Accessed 30 June 2015.
Jones KL, Jones MC, del Campo M. Smith’s recognizable patterns of human malformation. 7th ed. Philadelphia: Elsevier Saunders; 2013.
Johns Hopkins University and National Center for Biotechnology Information. Online Mendelian inheritance in man. http://www.ncbi.nlm.nih.gov/omim. Accessed 30 June 2015.
Orphanet. www.orpha.net (accessed 30 June 2015).
POSSUMweb. http://www.possum.net.au. Accessed 30 June 2015.
GeneTests. www.geneclinics.org. Accessed 30 June 2015.
Puri RD, Thakur S, Verma IC. Spectrum of severe skeletal dysplasias in North India. Indian J Pediatr. 2007;74:995–1002.
Wapner RJ, Martin CL, Levy B, Ballif BC, Eng CM, Zachary JM, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. 2012;367:2175–84.
Hillman SC, McMullan DJ, Hall G, Togneri FS, James N, Maher EJ, et al. Use of prenatal chromosomal microarray: prospective cohort study and systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2013;4:610–20.
Laury A, Sanchez-Lara PA, Pepkowitz S, Graham JM Jr. A study of 534 fetal pathology cases from prenatal diagnosis referrals analyzed from 1989 through 2000. Am J Med Genet A. 2007;143A:3107–20.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Rights and permissions
About this article

Cite this article
Puri, R.D. Fetal Dysmorphology. J. Fetal Med. 2, 151–159 (2015). https://doi.org/10.1007/s40556-015-0057-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40556-015-0057-8