
Abstract
Real-world data (RWD) from electronic health records (EHRs) and administrative claims databases are used increasingly to generate real-world evidence (RWE). RWE is used to support clinical evidence packages for medicines that inform decision-makers. In this review of current issues in the use of RWD-derived external comparator groups to support regulatory filings, we assess a series of topics that generally apply across many disease indications. However, most of the examples and illustrations focus on the oncology clinical research setting. The topics include an overview of current uses of RWD in drug development, a discussion of regulatory filings using RWD-derived external comparators, a brief overview of guidance documents and white papers pertaining to external comparators, a summary of some limitations and methodological issues in the use of external comparator groups and finally, a look at the future of this area and recommendations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Real-world data (RWD) from electronic health records (EHRs) and administrative claims databases are used increasingly to generate real-world evidence (RWE). RWE is used to support clinical evidence packages for medicines that inform decision-makers. For instance, there is growing attention to the use of externally derived patient data to augment control groups in randomized clinical trials and as a proxy control group in single-arm clinical trials, particularly in clinical oncology where single-arm trials are common [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18]. Much of the interest in patient data derived to contextualize clinical trials stems from recent changes in the United States regulatory landscape [7, 9, 11, 13, 19,20,21,22,23,24,25,26,27,28,29,30]. These changes include interpreting single-arm trial results for new drug applications, extending benefit-risk assessments to broader populations beyond those participating in clinical trials to patients found in RWD cohorts, and product label extensions [18, 20, 29, 31,32,33,34,35]. The regulatory shift is evinced through legislation such as the 21st Century Cures Act in 2016, the evolving Prescription Drug Users Fee Act (PDUFA), the 2018 US Food and Drug Administration (FDA) guidance on the use of RWE in regulatory decision making (FDA Framework for RWE),Footnote 1 and research initiatives such as the 2017 National Cancer Institute Cancer Moonshot [31,32,33, 36, 37].The changing regulatory landscape is not unique to the USA, as health authorities in other jurisdictions, such as the European Medicines Agency (EMA), the Pharmaceuticals and Medical Devices Agency (PDMA) in Japan, and Health Canada, have all issued recent statements regarding the developing role of RWD in drug development [38,39,40].
Changes in regulatory practice are not the only reason for the increased focus on externally derived control groups. The field of medicine has benefited from the advent and availability of next generation gene sequencing (NGS) platforms that have greatly enhanced oncology drug discovery and led to increased targeting of onco-genic mutations [41]. Many targeted oncology molecules are now following accelerated pathways such as FDA’s Breakthrough Therapy Designation, with a commensurate acceleration in the drug development cycle that can advance experimental therapies from phase 1B directly into phase III trials. For example, sortorasib was recently granted accelerated approval for KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC) [42]. In addition to speeding the drug development cycle, externally derived control groups can support situations in which randomization may not be possible or ethical, including when effective treatments are not available (e.g., novel biomarker targets) or current treatment options are suboptimal. There are numerous examples in oncology drug development where the unmet medical need is so pronounced that palliative care would be the only alternative to an experimental treatment. In yet other cases, the populations have rare biomarkers that make patient enrollment challenging. Collectively, these factors have resulted in a growing number of early phase single-arm oncology trials and increasing attention to “hybrid’ designs in later phase trials, which include randomized controls augmented with external controls.
The availability, completeness, and quality of RWD, especially EHR databases, has been steadily improving. These EHR databases can augment structured data fields with unstructured information gleaned from text fields in the medical charts. Also, the data are more contemporaneous, with very little lag between the time of a medical encounter and the data becoming available for analysis. There is also better characterization of important biomarkers as testing practices increase over time. Linkage of data sources such as EHRs with administrative claims databases provides additional granularity and completeness and fills in important missing gaps in the patients’ clinical, treatment and demographic profiles. Finally, the overall quality of important endpoint and clinical outcomes like mortality and disease response has been critically assessed and validated in some EHRs [43,44,45].
The use of RWD for externally derived comparator groups raises important methodologic considerations. To address selection bias and other forms of study bias, statistical methods to control for bias and confounding have continued to evolve [46,47,48,49,50,51,52,53]. Developing methods include the use of target trial emulation principles to avoid various forms of bias, including selection bias, the use of summary confounder scores such as a propensity score (PS) and methods for assessing and controlling unmeasured confounding, as accomplished with negative controls and use of instrumental variables [46,47,48,49,50,51].
In this review of current issues in the use of RWD-derived external comparator groups to support regulatory filings, we assess a series of topics that generally apply across many disease indications. However, most of the examples and illustrations will focus specifically on the oncology clinical research setting. The topics included in the review are as follows:
-
An overview of current uses of RWD in drug development
-
Regulatory filings using RWD-derived external comparators
-
Guidance documents and white papers pertaining to external comparators
-
Limitations and methodological issues in the use of external comparator groups
-
A look at the future of this area and recommendations
Overview of Uses of RWD-derived External Comparators
Historical controls generally refer to the use of patient cohorts derived from previously conducted clinical trials that are repurposed for use in the assessment of treatment effects or adverse events observed in other clinical trials. In addition to concerns about limited availability of critical variables, the use of historical control arms as comparators for single-arm trials have been criticized because the historical data may not reflect current standard of care. The increasing availability of more contemporary RWD enables concurrent comparisons, and accordingly, the nomenclature for these types of applications has evolved from historical controls to external, synthetic, or virtual controls/comparators [3, 6, 10, 54, 55].
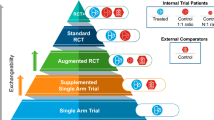
Several uses exist for combining externally derived cohorts with clinical trial data. The corresponding point in a medicine’s development lifecycle where an application is relevant is depicted in Fig. 1. These main applications are elaborated on in Table 1 and are discussed below.

Use cases using external control groups/comparators across the drug development lifecycle
Describing Disease Natural History in Early Phase Single-Arm Trial Settings
Disease burden and unmet medical need can be explored by examining early stage clinical development findings with the natural history of the disease using stand-alone externally derived cohorts [34, 56]. The overarching purpose is to characterize unmet medical need for a given disease target population, which in turn provides an understanding of the potential benefits offered by an experimental treatment in the absence of a randomized control group [34, 56]. This approach is often warranted owing to outdated literature or lack of publications focused on the specific target population of interest.
Assessing Treatment Effects in Early Phase Single-Arm Trials: Direct Comparisons Using External Comparator Arms
When appropriate RWD are available they can be used to make direct comparisons to trial experimental arms. RWD-derived external comparators can mimic the overall health, demographic, and disease characteristics of the trial arm through application of a trial’s inclusion and exclusion criteria (Fig. 2A). However, these non-randomized comparisons are subject to potential biases. For example, the prevalence of a biomarker that defines the trial population could differ in the RWD control group, introducing selection bias and threatening comparability. To reduce selection bias, the RWD-derived controls should be sampled from a similar underlying population as the treatment arm. To compensate for potential bias and confounding between the trial arm and the externally derived comparator arm, appropriate adjustment methods (e.g., propensity score matching and weighting) and sensitivity analyses should be incorporated into the analyses [6, 8].

A Diagram of external control in single-arm trial. B Diagram of hybrid randomized trial design using external control group to augment randomized controls
Despite growing interest and potential, relatively few studies have used external controls as a means of interpreting data from clinical trials. One of the first examples is a study by Gökbuget et al. comparing outcomes from a single-arm trial in relapsed/refractory acute lymphoblastic leukemia patients to an external comparator [57]. These researchers constructed an external control group using RWD to compare response and survival between the external comparator and clinical trial. The data were included in regulatory filings and played an important role in the FDA’s accelerated approval of blinatumomab [58]. The efficacy of blinatumomab versus standard of care chemotherapy was further confirmed in a phase 3 randomized controlled trial two years later [59]. The concordance of the findings provided additional support for the use of external controls.
Tan and colleagues took a different approach than the blinatumomab example by extracting data from published single-arm trials in patients treated with crizotinib for anaplastic lymphoma kinase (ALK) positive metastatic non-small cell lung cancer (mNSCLC) [60] and comparing the aggregate findings with single-arm studies in NSCLC patients treated with ceritinib. The comparison demonstrated an advantage for crizotinib in progression-free survival (PFS) and overall survival (OS) [60]. Other research using external controls have also shown early promise [12, 54, 55], including studies drawing on aggregate findings from the clinical trial literature in acute myeloid leukemia and anaplastic lymphoma kinase-targeted NSCLC; each of these efforts demonstrated the promise of external controls for interpreting findings from single-arm trials [54, 60, 61]. A recent study drew on data from 11 advanced NSCLC (aNSCLC) randomized trials and substituted data from an oncology EHR database [8] for the controls arms from the clinical trials. In most cases, the researchers were able to replicate closely the hazard ratios for overall survival from the original randomized trials [8].
Accelerating Late Phase Trials: Hybrid Designs Using External Controls
Innovative study designs for late phase oncology trials are another opportunity for using external controls. In the past, the purpose of so-called adaptive trials was to channel more patients to the RCT experimental arm that was experiencing better outcomes [35]. Hybrid trials have a couple of different meanings by regulators [35]. There are hybrid trial designs that use EHR data to collect information on patients enrolled in trials and thereby reduce costs and timelines (see Zhu et al. 2020 for a comprehensive treatment of this design [62]). Another form of hybrid trial design, which shares similar objectives to both adaptive designs and the previously mentioned hybrid trials, are trials that augment randomized controls with controls from other trials or from RWD that share similar characteristics as those in the trial [1, 26, 62]. These hybrid designs expose a smaller proportion of randomized subjects in the clinical trial to a potentially suboptimal standard of care, and the external data (historical clinical trial and/or RWD) are used to supplement the randomized control. In some cases, Bayesian approaches are used to assess commensurability between the external controls and the trial patients over time [1, 4, 62]. The assessment of the commensurability between the trial data and the external cohort takes place incrementally over the course of the trial enrollment period and may involve techniques that select controls from the external data based on the assessment of commensurability with respect to the trial outcome (e.g., the greater the commensurability, the more external patients selected to augment the trial patients). Figure 2B shows a simplified version of the hybrid trial design using a post hoc “all or none” selection of external controls approach in a fictional trial where a 3:1 randomization scheme is used. The purpose of these approaches is to accelerate trial enrollment, expose fewer patients to suboptimal treatments, and ultimately bring innovative potentially lifesaving or life-extending medicines to patients sooner.
Indirect Comparative Effectiveness
Indirect treatment comparison in the pre-approval and post-marketing settings allow for comparisons between experimental treatments or newly marketed drugs respectively, with novel or new standard of care marketed treatments. Using external RWD cohorts standardized to trial eligibility criteria, treatment effects are characterized between a trial experimental treatment and some standard of care treatment in the RWD [60, 61]. The RWD cohort is then re-standardized to a published trial that compares the same standard of care with another newer novel marketed therapy [63]. Additional adjustment can be achieved through weighting on aggregate level baseline characteristics [63]. The analysis involving the published trial data can be accomplished using software that converts digital Kaplan–Meier (KM) curves using a pixel-based conversion approach that reconstructs the published KM curve with the RWD external comparator in the same KM plot [63, 64]. The same technique may be used to extract a novel treatment from the published literature and plot a KM graph comparing the novel treatment with an experimental treatment from a trial.
Post-marketing Safety Studies and Long-term Safety Follow-up
Post-marketing safety studies containing a single exposed cohort is another setting where the use of externally derived cohorts can aid in the interpretation of study findings [19]. For example, in prospective studies examining rare populations where an internal comparator is infeasible due to enrollment challenges (e.g., idiopathic pulmonary fibrosis), an external comparator arm derived from a large administrative claims database can be beneficial for evaluating important safety endpoints. Often pregnancy safety exposure registries will capture data only on exposed patients, making interpretation of potential safety signals challenging in the absence of an unexposed control group. These studies may rely on the published data to interpret safety signals, which may be outdated and may not reflect the same patient characteristics found in the registry. Finally, long-term safety studies could be conducted using data from RWD-derived patient cohort as an extension to the trial observation period allowing for adequate follow-up of important safety endpoints [6].
Regulatory Filings Using RWD-derived External Comparators
Regulatory reviews of hematology and oncology new drug applications (NDAs) and labeling extensions submitted by drug developers using data from single-arm trials is increasing [7, 16, 24, 65,66,67] (Fig. 3). Despite the growth in single-arm study approvals, RWD have been included in very few evidence packages. This trend could change in response to the December 2016 enactment of the 21st Century Cures Act and the subsequent initiation of the NCIs Cancer Moonshot program in 2017. Bolislis et al. (2020) reviewed NDAs and labeling extensions using RWD submitted to the EMA, FDA, PMDA, and Health Canada over the past 20 years and found only 27 cases where RWD was used, primarily in the oncology disease area [16]. In addition to the blinatumomab example mentioned above, there are five other examples of favorable FDA decisions that considered RWD. These include label expansions for blinatumomab, avelumab approval for metastatic Merkel cell carcinoma, and axicabtagene ciloleucel approval for relapsed or refractory large B-cell lymphoma [16, 68]. Although not an example of an RWD external comparator analysis, palbociclib received an approval in metastatic male breast cancer based in part on evidence from RWD [69].

Oncology and hematology FDA approvals and label expansions where RWE was considered in the total evidence package.
There are also examples where drug developers have been unsuccessful using RWD-derived control arms as part of their FDA filing. Despite receiving Breakthrough Therapy Designation for relapsed or refractory multiple myeloma (RRMM), in the selinexor submission, the regulators commented on study design issues, differences in the EHR population compared to the trial patients, and methodologic issues, which led them to conclude that the RWD “is not adequate to provide context or comparison for the overall survival observed” in the clinical trial patients [70]. Additionally, the regulators expressed concerns about the RWD study not being pre-specified, as the study protocol was not submitted a priori to the FDA [70]. In 2019, the sponsor of tazemetostat, indicated in epithelioid sarcoma patients, had their RWD evidence disregarded by the FDA due to lack of pre-specification of the study protocol and concerns about the study design and methods [71]. Again in 2019, the FDA did not consider the submitted RWD-derived control arms for the filings for both erdafitinib, a fibroblast growth factor receptor positive (FGFR +)-targeted therapy for patients with advanced or metastatic urothelial cancer, and entrectinib, for ROS1 + metastatic non-small cell lung cancer. The FDA cited concerns related more to the generalizability of the EHR cohorts than to concerns related directly to their methodologies [72, 73].
In 2020, an external comparator study was submitted to the FDA (RE-MIND: NCT04150328) to support the NDA for tafasitamab indicated in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) [74]. This analysis aimed to provide context for interpreting the efficacy findings observed in the single-arm L-MIND pivotal trial for patients with DLBCL. The primary objective of the study was to isolate the contribution of tafasitamab to efficacy of a tafasitamab plus lenalidomide regimen in a cohort of RWD patients matched to trial patients from L-MIND. Data for the L-MIND patients were collected from clinical trials and data for the RE-MIND patients were collected from the medical records of patients in real-world settings. Of note, there were different study periods for the trial patients (2016 to 2018) and the RWD patients (2007 to 2019) [74]. In the FDA decision, the reviewers noted the following concerns:
The validity of the study is compromised by several limitations in the study design. Bias resulting from key differences in patient selection and unequal distribution of important measured and unmeasured prognostic indicators between treatment arms are likely to favor survival for the L-MIND patients. Most importantly, given important differences in the patient populations included in the L-MIND trial and RE-MIND study, primarily as a result of selection bias, this study does not provide sufficient evidence to isolate the contribution of tafasitamab to efficacy of tafasitamab+LEN combination therapy for DLBCL” (Page 3) [74]
This survey of reviews conducted by the FDA indicates that regarding the use of RWD-derived external comparators for contextualizing single-arm trials, these are at best early days in terms of acceptance. Concerns relate to selection bias, generalizability of RWD, and the resulting inherent differences in baseline covariates between RWD comparators and trial patients, as well as the capture and completeness of important prognostic factors in RWD.
Best Practices: Guidance Documents and White Papers
Given the growing interest in external controls, there are several recent guidance documents, best practices, and white papers in circulation devoted to the subject. FDA’s Framework for Real-World Evidence Program discusses the role of external controls in the context of single-arm trials using either historical clinical trial data or RWD:
Collection of RWD on patients currently receiving other treatments, together with statistical methods, such as propensity scoring, could improve the quality of the external control data that are used when randomization may not be feasible or ethical, provided there is adequate detail to capture relevant covariates. (page 20) [33]
The Framework suggests limitations such as a lack of standardized diagnostic criteria and study endpoints, and general concern with the comparability of RWD patient populations with trial patients [33]. Other regulatory agencies, including EMA, PMDA, and Health Canada, have commented on the role of RWD in regulatory filings [38,39,40].
Many recent commentaries discuss the use of RWD-derived external controls and their use in drug development [5, 6, 9, 10, 15, 17, 19, 22]. Common recommendations for when to use RWD include single-arm trials where randomization is infeasible or unethical, label expansions, long-term follow-up, and augmenting randomized control groups in hybrid designs. An additional rationale relates to drugs that fill high unmet medical need where filings with single-arm studies receive accelerated approval (e.g., see sortorasib example above) and are followed by randomized phase 3 study as part of a post-marketing requirement.
Methodological and Other Considerations
This section highlights analytic issues (e.g., bias and confounding) that may arise when external RWD cohorts are combined with clinical trial data as well as some general considerations (see Table 2).
FDA and other regulators have repeatedly noted concern regarding how well RWD populations reflect clinical trial patients in terms of clinical and demographic characteristics and potentially unobserved prognostic factors. Although these differences can be controlled using statistical methods, this ability depends on data, especially prognostic factors, being available in the RWD. Additionally, differences in biomarker testing practices between RWD and trials can complicate studies specific to certain genetic mutations or protein expression. In the real-world setting patients may be tested late in the disease process, whereas patients enrolled in trials will be tested at the outset of the study. This difference in timing can affect trial results especially for those trials which are conducted in patients treated in the first line.
The use of concurrent controls from the RWD can help address changes to standard of care treatments that are common in oncology. Other issues in using RWD include endpoints like treatment response and disease progression that can differ in how they are defined and collected in clinical trials versus RWD [75]. Mortality, although well captured in some inpatient EHRs, is still less complete than found in clinical trials [43, 44] and missing death data could lead to underestimation of mortality in the control arms [44]. In addition, many EHR databases only include US patients, whereas trial populations are often global and disease prognosis can vary by region.
Trials conducted in later lines of treatment can complicate selection of an appropriate index date or “time 0” for RWD analyses. A biomarker test date may occur after the initiation of treatment, introducing an “immortal” period between the time of treatment initiation and the required test result (because if the patient died, the test would not be performed, so all patients with the test result had to be alive through the date of the test) [76, 77]. Figure 4A and B illustrates this point in studies examining first- and second-line-treated patients respectively.

A Immortal time bias in initial treatment period. “Patient 1” in panel A has an immortal period following initiation of treatment and the test result. To handle this immortal time bias, patients can be excluded altogether or alternatively, patients’ time 0 can be changed from the start of treatment to the test date. B Illustration of an immortal period in second-line treatment (“Patient 3”). Also, it can be seen that “Patient 1” despite having a test date after the start of first-line treatment is still appropriately eligible for an analysis focusing only on second-line treatment
There are multiple methods available to address possible shortcomings of RWD, including negative controls, instrumental variables, propensity scores (PS), and high-dimensional PS [13, 47, 48, 50, 51, 78]. Other methods such as inverse probability of censoring and time to censoring for handling potential informative censoring bias stemming from missing outcomes like mortality can improve the integrity of the analysis [52]. Quantitative bias analysis can be used to assess the robustness of findings and assumptions [53]. Approaches such as study restriction by line of treatment, stratification or statistical adjustment by line of treatment can also be used. An assessment of important confounders not captured in RWD may point to the need for additional data abstraction of these potential confounders. Many of these issues can be addressed through thoughtful study design and analysis.
Transparency about data limitations, comparability of treatment groups and other potential sources of bias is crucial. In oncology and other diseases areas with high mortality or morbidity, regulators must make benefit/risk determinations and a common understanding of what can, and cannot, be inferred from the RWD comparisons serves regulators and sponsors, but most of all patients.
The Future of External Comparators
Changes in the regulatory landscape have led to an increased focus on the use of RWD as external comparators in the clinical evidence package used in regulatory drug submissions in oncology. To date, best practices are still being established and debated among the scientific community with many groups such as the FDA, Drug Information Association, Friends of Cancer Research, American Society for Clinical Oncology, and the International Society for Pharmacoepidemiology currently working on guidance around the use of external comparators as part of the clinical evidence package [6, 17, 25, 26, 30, 33, 78]. Adoption of these approaches will hinge on continued dialogue and scientific exchange with a view to producing credible evidence to support drug development efforts.
What does the future hold? Inevitably, there will be ever greater availability of RWD data sources in oncology with more granularity and improved quality, including growing capture of NGS and other biomarker test results. Methods to account for bias and confounding will continue to evolve. One of the major challenges will be to synthesize the empirical findings into a coherent model that captures the more salient learnings from a wide spectrum of researchers. Our ability to do this in a thoughtful and collaborative way across the various stakeholders will ultimately decide the role that RWD will play in drug development.
When viewed from the various stakeholder perspectives, whether patients, drug developers, regulatory agencies, or payers, there is a common shared interest in accelerating oncology drug development. For patients diagnosed with cancer, the prospect of novel treatments becoming available that may be able to help extend their life is obviously crucial. Biopharmaceutical companies have as their core mission the development of effective and safe treatments to combat life-threatening disease. Single-arm studies can accelerate the approval process in settings with a high unmet medical need and get medicines to patients faster. Using RWD-derived control arms to evaluate efficacy can also inform better decision making. Regulators also have as their mission to bring safe and effective medicines to patients in a timely fashion. Finally, payers would benefit from the availability of more suitable targeted treatments with enhanced prognoses that would in the long run justify the cost of newly developed treatments. With all the apparent advantages that RWE can potentially provide to the traditional drug development paradigm come myriad concerns related to poorly designed RWD/clinical trials or RWD that is used in settings where it is not appropriate or warranted. These lingering concerns makes it even more important to focus on advancing the scientific knowledge base pertaining to the use of RWD in clinical drug development.
Notes
An update to the FDA’s Guidance on the use of RWE to support regulator decision making is expected in the coming months, with a draft version released on September 29, 2021.
References
Han B, Zhan J, Zhong ZJ, Liu D, Lindborg S. Covariate-adjusted borrowing of historical control data in randomized clinical trials. Pharm Stat. 2017;16:296–308.
Hiemenz JW, Raad II, Maertens JA, et al. Efficacy of caspofungin as salvage therapy for invasive aspergillosis compared to standard therapy in a historical cohort. Eur J Clin Microbiol Infect Dis. 2010;29:1387–94.
Davi, R, Chandler M, Elashoff B, et al. Results of a randomized control arm are replicated by a synthetic control arm; a case study in NSCLC; ASCO, abstract #9108, 2019.
Yin PT, Desmond J, Day J. Sharing historical trial data to accelerate clinical development. Clin Pharmacol Ther. 2019;106:1177–9.
Gray CM, Grimson F, Layton D, Pocock S, Kim J. A framework for methodological choice and evidence assessment for studies using external comparators from real-world data. Drug Saf. 2020;43:623–33.
Seeger JD, Davis KJ, Iannacone MR, et al. Methods for external control groups for single arm trials or long-term uncontrolled extensions to randomized clinical trials. Pharmacoepidemiol Drug Saf. 2020;29:1382–92. https://doi.org/10.1002/pds.5141.
Goring S, Taylor A, Müller K, et al. Characteristics of non-randomised studies using comparisons with external controls submitted for regulatory approval in the USA and Europe: a systematic review. BMJ Open. 2019;9(2):e024895–e024895.
Carrigan G, Whipple S, Capra WB, et al. Using electronic health records to derive control arms for early phase single-arm lung cancer trials: proof-of-concept in randomized controlled trials. Clin Pharmacol Ther. 2020;107(2):369–77.
Burcu, M, Dreyer, NA, Franklin, JM, et al. Real-world evidence to support regulatory decision-making for medicines: considerations for external control arms. Pharmacoepidemiol Drug Saf. 2020;1– 8.
Mack C, Christian J, Brinkley E, Warren EJ, Hall M, Dreyer N. When context is hard to come by: external comparators and how to use them. Ther Innov Regul Sci. 2019. https://doi.org/10.1177/2168479019878672.
Jarow JP. Use of external controls in regulatory decision-making. Clin Pharmacol Ther. 2017;101:595–6.
Patel K, Ouwens M, Shire N, Khosla S. The application of electronic medical records (EMRs) as a virtual comparator arm in a lung cancer clinical trial: a case study. J Clin Oncol. 2017;35(suppl.15). Abstract e18098.
Abrahami D, Pradhan R, Yin H, Honig P, Andre EB, Azoulay L. Use of real-world data to emulate a clinical trial and support regulatory decision-making: assessing the impact of temporality, comparator choice and methods of adjustment. Clin Pharmacol Ther. 2021 Feb;109(2):452–61. https://doi.org/10.1002/cpt.2012.
Viele K, Berry S, Neuenschwander B, et al. Use of historical control data for assessing treatment effects in clinical trials. Pharm Stat. 2014;13(1):41–54.
Thorlund K, Dron L, Park JJH, Mills EJ. Synthetic and external controls in clinical trials - a primer for researchers. Clin Epidemiol. 2020;12:457–467. Published 2020 May 8.
Bolislis WR, Fay M, Kühler TC. Use of real-world data for new drug applications and line extensions. Clin Ther. 2020;42(5):926–38.
Ghadessi et al. A roadmap to using historical controls in clinical trials – by Drug Information Association Adaptive Design Scientific Working Group (DIA-ADSWG) Orphanet Journal of Rare Diseases. 2020;15:69. https://doi.org/10.1186/s13023-020-1332-x
Khozin S, Blumenthal GM, Pazdur R. Real-world data for clinical evidence generation in oncology. J Natl Cancer Inst. 2017;109:djx187.
Franklin JM, Schneeweiss S. When and how can real world data analyses substitute for randomized controlled trials? Clin Pharmacol Ther. 2017;102:924–33.
Sridhara R. External controls in cancer clinical trials –challenges and opportunities. US Food and Drug Administration. Presented at: Clinical Trials and Translational Research Advisory Committee. 2019. https://deainfo.nci.nih.gov/advisory/ctac/0719/Att%203_External%20Controls%20in%20Cancer%20Clinical%20Trials_Sridhara.pdf. Accessed 20 Dec 2020.
Schmidli H, Häring DA, Thomas M, et al. Beyond randomized clinical trials: use of external controls. Clin Pharmacol Ther. 2020;107(806–816):30.
Dreyer NA. Advancing a framework for regulatory use of real-world evidence: when real is reliable. Ther Innov Regul Sci. 2018;52:362–8.
Hatswell AJ, et al. Regulatory approval of pharmaceuticals without a randomised controlled study: analysis of EMA and FDA approvals 1999–2014. BMJ Open. 2016;6:e011666.
Baumfeld Andre E, Reynolds R, Caubel P, Azoulay L, Dreyer NA. Trial designs using real-world data: the changing landscape of the regulatory approval process. Pharmacoepidemiol Drug Saf. 2019;1–12.
Friends of Cancer Research. Exploring whether a synthetic control arm can be derived from historical clinical trials that match baseline characteristics and overall survival outcome of a randomized control arm: case study in non-small cell lung cancer. 2018. https://www.focr.org/sites/default/files/pdf/SCA%20White%20Paper.pdf. Accessed 18 Dec 2020.
Friends of Cancer Research. Characterizing the use of external controls for augmenting randomized control arms and confirming benefit. 2019.https://friendsofcancerresearch.org/wpcontent/uploads/Panel_1_Slide_Deck_AM19.pdf. Accessed 18 Dec 2020.
Jarow JP, LaVange L, Woodcock J. Multidimensional evidence generation and FDA regulatory decision making defining and using “real-world” data. JAMA. 2017;318:703–4.
Khozin S, Kim G, Pazdur R. From big data to smart data: FDA’s INFORMED initiative. Nat Rev Drug Discov. 2017;16:306.
Corrigan-Curay J, Sacks L, Woodcock J. Real-world evidence and real-world data for evaluating drug safety and effectiveness. JAMA. 2018;320:867–8.
Mahendraratnam N, Eckert J, Mercon K, et al. Using secondary data to generate real-world evidence for regulatory decision making,and demonstrating their credibility. Duke-Margolis Center for Health Policy. 2019. https://www.healthpolicy.duke.edu/sites/default/files/2020-08/Non-Interventional%20Study%20Credibility.pdf. Accessed 20 Dec 2020.
United States Congress. 21st Century Cures Act. https://www.congress.gov/bill/114th-congress/house-bill/34/ (2016).
Cancer Moonshot. https://www.cancer.gov/research/key-initiatives/moonshot-cancerinitiative. Accessed June 10, 2018.
US Food and Drug Administration. Framework for FDA’s RWE Program, 2018. https://www.fda.gov/media/120060/download (Accessed Aug 02, 2020).
US Food and Drug Administration. Guidance for industry: rare diseases: natural history studies for drug development. US Department of Health & Human Services; March 2019.
US Food and Drug Administration. Guidance for industry: adaptive designs for clinical trials of drugs and biologics. US Department of Health & Human Services. 2019:33
US Food and Drug Administration: PDUFA reauthorization performance goals and procedures fiscal years 2018 through 2022. https://www.fda.gov/media/99140/download. Accessed 18 Dec 2020.
US Congress: Prescription Drug User Fee Act. P.L. 102–571,1992.
European Medicines Agency: EMA Regulatory Science to 2025 EMA/110706/2020
Utilizing Real World Data: A PMDA Perspective https://globalforum.diaglobal.org/issue/august-2018/utilizing-real-world-data-a-pmda-perspective/ accessed 12/20/2020
Health Canada. Optimizing the use of real world evidence to inform regulatory decision making, health products and food branch notice April 16, 2019 Available at: https://www.canada.ca/en/health-canada/services/drugs-healthproducts/drugproducts/announcements/optimizing-real-world-evidenceregulatory-decisions.html
Wetterstrand KA. DNA Sequencing Costs: data from the NHGRI Genome Sequencing Program (GSP) (Available at: www.genome.gov/sequencingcostsdata. Accessed 12/18/20
US Food and Drug Administration: FDA grants accelerated approval to sotorasib for KRAS G12C mutated NSCLC: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-sotorasib-kras-g12c-mutated-nsclc: accessed 7/2/2021
Curtis MD, Griffith SD, Tucker M, et al. Development and validation of a high-quality composite real-world mortality endpoint. Health Serv Res. 2018;53:4460–76.
Carrigan G, Whipple S, Taylor MD, et al. An evaluation of the impact of missing deaths on overall survival analyses of advanced non-small cell lung cancer patients conducted in an electronic health records database. Pharmacoepidemiol Drug Saf. 2019;28:572–81.
Griffith SD, Miksad RA, Calkins G, et al. Characterizing the feasibility and performance of real-world tumor progression end points and their association with overall survival in a large advanced non–small-cell lung cancer data set. JCO Clinical Cancer Informatics. 2019;1–13
Brookhart MA, Wyss R, Layton JB, Stürmer T. Propensity score methods for confounding control in nonexperimental research.Circ Cardiovasc Qual Outcomes. 2013; 6:604–611. https://doi.org/10.1161/CIRCOUTCOMES.113.000359
Vanderweele TJ, Arah OA. Bias formulas for sensitivity analysis of unmeasured confounding for general outcomes, treatments, and confounders. Epidemiology. 2011;22(1):42–52. https://doi.org/10.1097/EDE.0b013e3181f74493.
Arnold BF, Ercumen A, Benjamin-Chung J, Colford JM Jr. Brief report: negative controls to detect selection bias and measurement bias in epidemiologic studies. Epidemiology. 2016;27(5):637–41.
Lipsitch M, TchetgenTchetgen E, Cohen T. Negative controls: a tool for detecting confounding and bias in observational studies. Epidemiology. 2010;21(3):383–8.
McGrath LJ, Spangler L, Curtis JR, et al. Using negative control outcomes to assess the comparability of treatment groups among women with osteoporosis in the United States. Pharmacoepidemiol Drug Saf. 2020;29(8):854–63.
Hernán MA, Robins JM. Instruments for causal inference: an epidemiologist’s dream? Epidemiology. 2006;17(4):360–72. https://doi.org/10.1097/01.ede.0000222409.00878.37.
Robins JM, Finkelstein DM. Correcting for noncompliance and dependent censoring in an AIDS clinical trial with inverse probability of censoring weighted (IPCW) log-rank tests. Biometrics. 2000;56(3):779–88.
Lash TL, Fox MP, Cooney D, et al. Quantitative bias analysis in regulatory settings. Am J Public Health. 2016;106(7):1227–30.
Berry DA, et al. Creating a synthetic control arm from previous clinical trials: application to establishing early end points as indicators of overall survival in acute myeloid leukemia (AML). J ClinOncol. 2017;35(suppl. 15). Abstract 7021.
Jia Z, et al. Generation of ‘“virtual”’ control groups for single arm prostate cancer adjuvant trials. PLoS ONE. 2014;9:e85010.
Spira AI, Tu H, Aggarwal S, et al. Natural history of advanced non–small-cell lung cancer in patients with KRAS p.G12C mutated or wild-type disease. Lung Cancer. 2021. https://doi.org/10.1016/j.lungcan.2021.05.026
Gokbuget N, Kelsh M, Chia V, et al. Blinatumomab vs historical standard therapy of adult relapsed/refractory acute lymphoblastic leukemia. Blood Cancer J. 2016;6:e473.
US Food and Drug Administration. Summary Review for Regulatory Action: Blincyto (Blinatumomab). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/125557Orig1s000MedR.pdf. Accessed 02 Aug 2020.
Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836–47.
Tan DS-W, et al. Comparative efficacy of ceritinib and crizotinib as initial ALK–targeted therapies in previously treated advanced NSCLC: an adjusted comparison with external controls. J Thorac Oncol. 2016;11:1550–7.
Davies J, et al. Comparative effectiveness from a single-arm trial and real-world data: alectinib versus ceritinib. J Comp Eff Res. 2018;7:855–65.
Zhu M, Sridhar S, Hollingsworth R, et al. Hybrid clinical trials to generate real-world evidence: design considerations from a sponsor’s perspective. Contemp Clin Trials. 2020;94:105856. https://doi.org/10.1016/j.cct.2019.105856.
Guyot P, Ades AE, Ouwens MJ, et al. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC Med Res Methodol. 2012;12:9.
Engauge digitizer [computer program]. Version 4.1.http://markummitchell.github.io/engauge-digitizer/. Accessed July 12, 2019
US Food and Drug Administration: New Drug Therapy Approvals 2017: https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/new-drug-therapy-approvals-2017 (accessed November 17, 2020)
US Food and Drug Administration: New Drug Therapy Approvals 2018: https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/new-drug-therapy-approvals-2018 (accessed November 17, 2020)
US Food and Drug Administration: New Drug Therapy Approvals 2019: https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/new-drug-therapy-approvals-2019 (accessed November 17, 2020)
US Food and Drug Administration. BLA multidisciplinary review and evaluation: BLA 761049.Bavencio(Avelumab). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761049Orig1s000MultidisciplineR.pdf. Accessed 02 Aug 2020.
US Food and Drug Administration. Ibrance Revised Labeling; 2019 [FDA website]. Available at: https://www.accesdata.fda.gov/drugsatfda_docs/nda/2019/207103Orig1s008. Accessed 02 Aug 2020.
Oncologic Drugs Advisory Committee Meeting, 2019 https://www.fda.gov/media/121670/download (Accessed Aug 02, 2020). (Selinexor)
Oncologic Drugs Advisory Committee Meeting December 18, 2019 NDA 211723 Tazemetostat Applicant: Epizyme. Available at: https://www.fda.gov/media/133573/download. Accessed 02 Aug 2020.
US Food and Drug Administration. Memo: review of the sponsor’s white paper on comparison of overall survival between Erdafitinib clinical study BLC2001 patients and real-world control patients from observational data: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212018Orig1s000OtherR.pdf. Accessed 02 July 2020.
US Food and Drug Administration. Comparative analysis of ROS1-positive locally advanced or metastatic non-small cell lung cancer between patients treated in entrectinib trials and crizotinib treated patients from real world data. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212725Orig1s000,%20212726Orig1s000OtherR.pdf. Accessed 02 July 2020.
An Observational Retrospective Cohort Study of Lenalidomide Monotherapy in Relapsed or Refractory Diffuse Large B-Cell Lymphoma (R/R DLBCL) to Generate a Historical Control for Clinical Trial MOR208C203; Epidemiology: Review of Tafasitamab, RWE study report, May 5, 2020, https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/761163Orig1s000OtherR.pdf. Accessed 02 July 2020.
Stewart M, Norden AD, Dreyer N, et al. An exploratory analysis of real-world endpoints for assessing outcomes among immunotherapy treated patients with advanced non-small cell lung cancer. JCO Clin Care Inform. 2019;3:1–15.
Hernán MA, Sauer BC, Hernández-Díaz S, Platt R, Shrier I. Specifying a target trial prevents immortal time bias and other self-inflicted injuries in observational analyses. J Clin Epidemiol. 2016;79:70–5.
Suissa S. Immortal time bias in pharmaco-epidemiology. Am J Epidemiol. 2008;167(4):492–9.
Lim J, Wally R, Yuan J et al. Minimizing patient burden through the use of historical subject-level data in innovative confirmatory clinical trials: review of methods and opportunities. Ther Innov Regul Sci 2018 1–14
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing Interests
Drs. Carrigan, Bradbury, and Chia are employees of Amgen with stock holdings in Amgen. Drs. Capra and Taylor are employees Genentech with stock holdings in Genentech. Dr. Sarsour is an employee of Janssen with stock holdings in Janssen. Dr. Brown has no competing interests to declare. Dr. Rothman is an employee of the Research Triangle Institute, an independent nonprofit research organization that does work for government agencies and pharmaceutical companies. Dr. Brookhart has provided past epidemiology consulting services for Amgen.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Dr. Sarsour was an employee of Genentech at the time this manuscript was drafted.
Dr. Brown was an employee of Harvard Medical School & Harvard Pilgrim Health Care Institute at the time this manuscript was drafted.
This article is part of the Topical Collection on Pharmacoepidemiology.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Carrigan, G., Bradbury, B.D., Brookhart, M.A. et al. External Comparator Groups Derived from Real-world Data Used in Support of Regulatory Decision Making: Use Cases and Challenges. Curr Epidemiol Rep 9, 326–337 (2022). https://doi.org/10.1007/s40471-022-00305-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40471-022-00305-9