
Abstract
Mendelian randomization (MR) is an innovative epidemiological approach that uses genetic variants as proxies for environmental exposures to provide unbiased estimates of the causal effect of a risk factor on disease. The explosion in availability of high-throughput biological data has resulted in increasing numbers of MR studies, novel extensions to the traditional single-SNP MR approach, and the potential to incorporate new-generation biological “omics” data (such as genome-wide genotype data, epigenetics, and metabolomic data). In this review, we discuss these new developments, ranging from the application of multiple genetic markers and the use of summary statistic data to MR approaches in the “omics” age. Progress in “omics” technologies has been touted as a means to revolutionize epidemiology, and the incorporation of “omics” data into MR to infer causality of potentially large numbers of novel biological markers represents one avenue in how this may be realized.
Similar content being viewed by others


Introduction
Mendelian randomization (MR) is an epidemiological method that uses genetic variants to estimate the causal effect of risk factors on disease-related outcomes from observational data [1–7]. It is aimed at overcoming problems with confounding that typically limit observational association studies. The tendency for these studies to incorrectly identify causal risk factors has been highlighted in a number of cases. For example, apparently robust observational associations of vitamin E with cardiovascular disease, beta-carotene with cancer, and estrogen with Alzheimer’s disease reported by such studies were not supported by subsequent randomized controlled trials (RCTs) [1, 8]. Incomplete control for confounders in observational studies is likely to be a key reason for this discrepancy in findings [9], and this has led to strong epidemiological interest in pursuing approaches such as MR that can strengthen causal inference [10].
In MR, a genetic variant acts as an instrumental variable (IV) or proxy for an exposure of interest that is postulated to influence a disease-related outcome. The causal effect of an exposure on a disease outcome is estimated from the association of the genetic instrument with the outcome, taking into account the genetic variant’s association with the exposure (see Fig. 1). Based on Mendel’s laws of inheritance, (i) alleles segregate at conception independent of environment, (ii) genetic variants affecting different traits can sort independently, and associations between genetic instruments and outcomes are not generally confounded by behavioral or environmental exposures at the population level [1, 6]. These provide a framework that mimics an RCT to infer causality between an exposure and a disease outcome.

Mendelian Randomization. A genetic instrumental variable (G) acts as a proxy measure for an environmental exposure (E) that is postulated to influence disease (D), where G is independent of measured and unmeasured confounders (U). G influences D only if a causal association exists between E and D. For example, the extent to which body mass index (BMI) causally affects blood pressure (BP) can be quantified using a genetic variant associated with BMI, such as FTO. This variant, which should be unrelated to the typical confounders, will be associated with BP if there is a causal relationship of BMI on BP.
There are, however, a number of known limitations [1, 11–14]. In particular, MR requires that a number of strong assumptions are met. Beyond the genetic variant’s association with the exposure trait, there must be no unmeasured common causes of the genetic variant and the outcome. In addition, the outcome must not be associated with the genetic variant except through its association with the exposure variable. Population stratification (i.e., when population subgroups differ both in disease rates and allele frequencies for variants of interest) is the most likely unmeasured common cause of both genetic variation and outcome. In addition, since a single genetic variant generally explains only a small amount of variation in a trait of interest, low statistical power is frequently an issue for an MR study, as its power is positively correlated with the amount of phenotypic variance explained by the genetic variant(s) [1, 15]. Finally, there is also the possibility of confounding being reintroduced through linkage disequilibrium (LD, correlation among SNPs) or pleiotropy (where a genetic variant has multiple functional consequences). This topic has been discussed in more detail elsewhere [16].
Despite these limitations, MR studies have already yielded many clinically relevant findings, providing evidence regarding causality for a range of risk factors on disease outcomes (see Table 1). Indeed, MR has been hailed as an approach that stands to make major contributions to understanding etiological pathways in complex disease [2]. With the recent methodological advances and the explosion in biological data being generated, particularly from high-throughput technologies and the successes of GWAS, the range of biomedical questions that can be tested using MR promises to broaden even further.
In this review, we firstly discuss the recent developments in MR, covering new approaches that incorporate a range of information and hypotheses that could potentially address a number of its existing limitations. Secondly, we explore how the original MR approach can be extended to incorporate the increasingly available “omics” data, such as genome-wide genotype data, epigenetics, and metabolomic data, to increase our understanding of new biological pathways and their potential roles in disease etiology.
Developments in MR
MR Using Multiple Instruments
Recent reductions in genotyping costs and the increased number of successful GWAS have enabled the discovery of a large number of genetic variants associated with phenotypic traits [17], which has resulted in the opportunity to extend the single-variant approach typically used in MR and to consider multiple genetic instruments. These instruments can be used either individually, taking independent genetic variants that each work through different biological pathways, or combined into a single allele score [18–20]. An allele score is the weighted or unweighted sum of the number of “risk” alleles associated with a given trait across several genotypes, where the weights are generally based on each genotype’s effect on the trait of interest (e.g., the SNP effects observed from GWAS).
A key advantage of using multiple instruments is the capacity to increase the statistical power of MR analyses. When each instrument independently explains variability in the exposure trait of interest, the use of multiple instruments can increase the precision of IV estimates [18]. This is important, since MR analyses generally require very large sample sizes due to the small amount of variation in a trait typically explained by a single genetic variant.
In addition, the multiple-instrument approach provides opportunities to test IV assumptions in a way that is not possible in single-instrument analyses [18]. The validity of the IV assumptions underlying MR analyses can be affected by LD and pleiotropy, and a multiple-instrument approach can be used to assess their likely presence. Specifically, the IV estimates from multiple instruments can be compared. If each independent instrument predicts the same causal effect of the environmentally modifiable risk factor for which the genetic variant acts as proxy, it then becomes much less plausible that confounding by pleiotropy or LD explains the associations, because the confounding would have to be acting in the same way for each independent instrument [21]. However, if there is missing data on genetic variants, a multiple-variant approach may result in a diminished sample size because only individuals with complete data on all genotypes used in an allelic score can be included, thereby reducing the power of the study [18]. Furthermore, there is also potential for increased bias in the IV estimator when weak instruments are used. This problem can be alleviated by combining the instruments into an allele score, albeit with some reduction in power [20]. Multiple-variant approaches have been applied in numerous MR studies, including recent applications using allele scores for BMI [22, 23] as well as multiple independent genetic instruments for iron [24] and LDL cholesterol [25].
MR Using Summary Statistics
The existence of published information from GWAS – which typically report regression coefficients summarizing the associations of many genetic variants with various traits – has been suggested as a potentially powerful source of data for MR studies [26]. Where a single genetic variant is used as an IV, the estimate of the causal effect using summarized data can be calculated in a straightforward manner with ratios based on summary statistic regression coefficients [3, 27, 28]. This has been successfully implemented, for example, using meta-analysis data from iron SNPs to demonstrate evidence of a potential protective effect of serum iron on risk of Parkinson’s disease [24]. Summary statistic estimates based on the genotype association alone can also be used to infer causality, as illustrated by MR studies demonstrating a relationship between alcohol intake and increased blood pressure [29] and esophageal cancer [30]. Methods using summarized data in the context of multiple variants have also recently been proposed [26]. In addition, the integration of genetic association studies using meta-analysis for genotype-exposure and genotype-outcome associations has also been explored in relation to MR [31], and this holds particular relevance for increasing power in the context of summary statistic data.
The use of summary statistics is somewhat limited by the fact that, as has been pointed out by others [26], the assumptions required for MR to be valid cannot be assessed as comprehensively as when individual-level data is used. Furthermore, for multiple-variant analyses using summary statistics, inflated precision has been observed when variants are in linkage disequilibrium [26]. Nonetheless, while individual-level data ideally should be implemented in MR studies wherever possible, when such data are unavailable, the existence of accessible summarized data can facilitate valid IV analyses for single- or multiple-instrument MR.
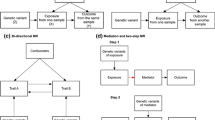
Two-sample MR
In traditional MR, estimates are produced from a single data set consisting of participants with information on the genetic variant, exposure, and outcome. However, situations may arise where information on the exposure and the outcome are available in two different data sets. In this case, it is possible to implement two-sample IV approaches with respect to MR, using ratio-based estimates [32, 33]. This approach will be particularly relevant for exposure biomarkers that are expensive to obtain or biospecimens that are either not widely available or are difficult to measure.
A subsetting approach may also be feasible when exposure data is only available in a subset of individuals in a given sample. The use of “sub-sample IV estimators” applied to MR has been shown to be effective when the IV is relatively strong [32]. In addition, a “split-sample” approach might be considered when no known genetic instruments exist for the intermediate phenotype of interest or no relevant GWAS has been undertaken with respect to appropriate genetic instruments. In this case, one could potentially undertake both the GWAS to identify SNP instruments and the MR analysis in a single sample that has been divided into two or more subsamples [23]. This ensures that the GWAS and the MR analyses are still carried out in different data subsets within the sample in order to minimize potential bias (see “Genome-wide MR” section in relation to overlapping-samples bias).
Bidirectional Approach
A bidirectional MR approach was recently proposed and implemented with the aim of facilitating a particularly clear assessment of the causal direction of an observed association [14, 34, 35]. This approach exploits the availability of two independent instruments that yield unconfounded estimates of causal effect. The direction of causality for an association between two variables is assessed through the use of one genetic proxy for one variable and a separate genetic proxy for the second variable. Two MR analyses are undertaken, one for each of the genetic proxy variables.
For example, to assess the direction of causality for an observed association between BMI and CRP, two MR analyses are performed. The first MR analysis associates BMI genetic variants with CRP, while the second MR analysis relates CRP genetic variants with BMI. If an association is observed in the first analysis but not the second, it suggests that BMI causally affects CRP levels, but not the other way around. This bidirectional approach has been successfully implemented in several studies to interrogate the direction of causation for the associations between BMI and CRP [34, 35], vitamin D [36], and uric acid [37].
Additional Extensions to MR
Factorial MR
The concept of carrying out “factorial MR” [1, 14] refers to the suggestion that the MR framework could be applied to investigate combinations of risk factors that are potentially acting together to affect disease risk. For example, obesity and alcohol intake have been found to synergistically increase the risk of liver disease, with multiplicative interactions observed [38, 39]. MR analyses could therefore be applied to generate effect estimates for co-occurring risk factors using combinations of genetic variants in which each acts as a proxy for the relevant risk factor.
Multi-phenotype MR
In addition to using MR for assessing risk factors with synergistic or combined effects, a related extension involves separating the independent effects of risk factors when multiple phenotypes are correlated with a particular SNP of interest or a set of SNPs. This multi-phenotype correlation poses problems for the MR framework, since disentangling the SNP’s effects on one particular phenotype will be challenging. Thus, attempts to address this issue were recently implemented in what has been termed multi-phenotype MR [14]. To illustrate, LDL cholesterol, HDL cholesterol, and triglycerides are strongly correlated with one another, and genetic variants identified for triglycerides tend also to be related to LDL or HDL [40]. Estimating an independent causal effect of triglycerides, therefore, is limited when such variants are used in a traditional MR design. As such, recent MR studies have attempted to isolate the effects of the correlated lipids on risk of heart disease using approaches such as (i) allele score restrictions (excluding SNPs correlated with more than one lipid); (ii) sequential statistical adjustments of the IV analysis for non-target lipids; and (iii) SNP-based regressions that correlate the target lipid SNP effect with the corresponding SNP effect for coronary artery disease, controlled for the SNP’s effects on the non-target lipids using residuals [41, 42••]. Broadly speaking, regression-based approaches, in principle, will still be subject to the typical epidemiological concerns over attempting to attribute causality using statistical control for correlated variables [43, 44]. However, the multi-phenotype framework remains a promising area, and development is ongoing [14].
Hypothesis-free Approach
The increasing availability of high-throughput biological information is resulting in extremely large data sets of genotypic and phenotypic data. Such data sets lend themselves to the appealing possibility of a powerful hypothesis-free approach [21]. In this way, one could effectively test the causality of extremely large numbers of phenotypic associations in a single study. There is promising evidence from genome-wide data that at least screening for causal associations in this way could be successfully implemented [45]. Hypothesis-free approaches will be increasingly relevant as “omics” databases, with extraordinarily large numbers of new biomarkers, continue to proliferate.
Potential MR Applications Using “OMICS” Data
There is much anticipation surrounding the prospects of “omics” technologies, with high expectations that they may revolutionize the practice of epidemiology by advancing the tools for exposure and outcome measurements [46]. Indeed, the recent explosion in availability of such “omics” data, generating hundreds of thousands of genetic markers and hundreds or thousands of biological markers, presents exciting new opportunities for increasing causal biomedical knowledge through the implementation of MR. Here, we discuss three of these “omics” data sources (genome-wide genotype data, epigenetics, and metabolomics) with respect to their potential future application in MR studies.
Genome-wide Genotypes
Within a short period of time, genome-wide association studies (GWAS) have successfully identified thousands of genetic variants robustly associated with complex traits/diseases [17]. GWAS test individual single-nucleotide polymorphisms (SNPs) covering the whole genome for their association with traits/diseases. This individual SNP association approach can identify specific loci affecting complex traits, but it doesn’t exploit the true potential of the correlated structure of the data. For example, with respect to human height, it has been shown that analyzing genome-wide SNP data simultaneously quadrupled the amount of variation explained [47] compared with that of the 180 height loci that reached genome-wide significant levels [48].
The capacity of genome-wide SNPs to explain vastly more variation in traits begs the question as to whether issues with statistical power that frequently limit MR studies might be addressed by utilizing more of the genome, for example, through the use of allele scores from genome-wide SNPs. Such allele scores are proxy measures for the trait of interest and therefore, with adequate heritability, can have strong relationships with the phenotypic trait. Indeed, genome-wide allele scores have been proposed for capturing larger amounts of phenotypic variation in traits in genetic association studies [49].
It was recently demonstrated that the application of genome-wide allele scores can be used to index biological intermediates of disease outcomes, with promising applications for large-scale screening of causal associations between potentially vast numbers of biological factors and disease outcomes [45]. However, systematic assessment of the validity of implementing genome-wide allele scores in MR studies has yet to be carried out. In particular, it should be noted that such genome-wide scores comprise variants that are data-derived, where overfitting can occur, as opposed to the use of robustly associated variants that are generally applied in traditional single-SNP MR. Furthermore, there is some evidence that bias may occur with allelic scores incorporating genetic markers at less stringent p value thresholds for associations [19].
The extent of any potential pleiotropy will also require careful assessment. Empirical studies suggest that common genetic variants are not typically related to the behavioral and socioeconomic factors considered to be important confounders in conventional observational studies [50] (although this does not necessarily hold true for detailed biological profiling, such as for lipoprotein subclasses [51]). In addition, there is some evidence that using certain allelic scores based on all SNPs across the genome may result in pleiotropic associations being observed [45]. Analogous to the suggestion that one could assess pleiotropy by using independent genetic instruments that are uncorrelated but proxy for the same phenotypic trait of interest [16], independent uncorrelated allele scores (such as scores constructed by chromosome number) could similarly be explored to assess pleiotropic effects in genome-wide analyses.
To maximize power, identifying allele scores that capture the most variation in the intermediate phenotype will be advantageous. The nature of this will likely differ among traits and will be dependent on factors such as the discovery GWAS sample size. For example, BMI allele scores with liberal thresholds for inclusion (i.e., including all SNPs, even those with very low p values for association) explain more variation than allele scores comprising only SNPs meeting the stringent p value threshold for GWAS-level significance [45]. In contrast, more stringent SNP p value thresholds are optimal for capturing most variation in LDL and CRP. In addition, weighted allele scores have been shown to be preferable to unweighted scores, at least in terms of statistical power [18, 20].
Finally, given the continually expanding GWAS consortia, it is increasingly possible that a given sample in which genome-wide allele scores are being constructed for analyses may also have been part of the GWAS study that generated the relevant SNP estimates used, for example, to weight the score. This will be problematic, as biased estimates in the variance explained in a given trait arise when allele scores that are used are constructed with SNP coefficients from a GWAS containing any of the same individuals being analyzed [49].
In sum, important methodological issues will need to be addressed before genome-wide scores can be implemented in MR analyses, in particular the potential introduction of pleiotropy and the consideration of biases such as overlapping samples. However, the availability of accessible genome-wide data in increasingly large samples and the substantial increase in variation explained in phenotypic traits by genome-wide allele scores represent exciting opportunities for developing more powerful MR studies.
Epigenetics
Enthusiasm for epidemiological studies based on epigenetic data is gaining considerable momentum in light of its potential to yield new insights into disease etiology and to provide a mechanism for gene–environment interactions [52, 53]. The epigenome comprises environmentally induced biological modifications of DNA that have the ability to regulate gene expression. Intriguing associations have been observed between these epigenetic modifications and environmental factors such as diet, alcohol, smoking, and inflammation [53], as well as associations with disease outcomes such as those in heart disease, stroke and mortality [54], schizophrenia, and bipolar disorder [55].
However, while the primary hypothesis is that environmental factors influence the epigenome, which subsequently alters the regulation of gene expression and thus modulates disease risk, the supporting evidence at this time is preliminary. Determining the causality of any observed epigenetic associations will be particularly challenging because epigenetic biomarkers are vulnerable to the typical confounders (age, sex, socioeconomic positions, diet, smoking, etc.) that also afflict many other molecular biomarkers. This is because, while genotypes are fixed, the epigenome is modified by environmental factors and is thus susceptible to the same problems of confounding and reverse causation as the environmental factors themselves [56].
A “two-step MR” approach, designed to interrogate causal mediation, has been proposed to investigate the role of epigenetic modifications in mediating risk factor associations with disease outcomes [57•]. While this two-step approach was initially described in the context of epigenetic data, in principle, it can be extended to any association involving a mediating variable. In a two-step MR, an initial IV analysis assesses the causal effect of an exposure on a mediator (e.g., an epigenetic marker), and a second IV analysis assesses the causal effect of the mediator on the disease outcome. For epigenetic markers, it has been shown that a genetic IV acting as a proxy for an exposure variable could be used to generate an unbiased estimate of the effect of the exposure on a measure of DNA methylation (an epigenetic marker), and in a second analysis, a genetic IV (specifically, a cis-variant) could be used as a proxy for the same DNA methylation measure and provide an estimate of the effect of methylation on the disease outcome [57•].
Epigenetic MR, however, is still in its infancy and, as discussed elsewhere [57•], is currently limited by several factors. Firstly, reported associations between environmental factors and both global and gene-specific DNA methylation are often modest in size. Secondly, while DNA sequence is fixed, epigenetic patterns vary across different tissue types. As such, assessing tissue-specific epigenetic patterns will be important, since the association of an epigenetic marker with a phenotype or with a genetic variant will likely vary across tissue types. While some tissue types are easily accessible (such as blood), others will be more challenging to obtain in samples sufficiently large to implement this approach. Nonetheless, the implementation of epigenetic MR appears promising, with MR already beginning to be applied to resolve issues of confounding and reverse causation with epigenetic measures. This can be seen, for example, with respect to assessing the effect of DNA methylation on postnatal growth [58] and body mass index [59•, 60].
Metabolomics
There is much interest in dissecting the role of the metabolome in health and disease. The availability of high-throughput data, advances in data handling and processing, as well as statistical tools, may provide unprecedented insights into our understanding of complex diseases [61••, 62]. The metabolome represents multiple metabolic pathways in systemic metabolism and includes, for example, lipoproteins, vitamin and cofactor levels, lipids, amino acids, and other small molecules involved in glycolysis, the citric acid cycle, and the urea cycle. Metabolites are produced endogenously as a result of chemical processes as well as from exogenous sources such diet and drugs. Circulating metabolites have been implicated in disorders of the metabolic and cardiovascular systems and have also been proven useful in the prediction of cardiometabolic disease [63]. However, the current understanding of the metabolome in disease pathogenesis is incomplete.
Recent technological developments in the analytic platforms – proton nuclear magnetic resonance (NMR) and mass spectroscopy – have enabled the measurement of hundreds to thousands of metabolites in a single procedure. Given the relationship of metabolites with environmental exposures, there is considerable potential for powerful epidemiological studies [62]. Furthermore, since many metabolites have substantial heritability and robust genetic variant associations that have already been identified [61••, 64], MR represents an ideal framework for investigating the potential causal pathways involving metabolites and disease outcomes. In particular, metabolites may act as mediators between an exposure and a disease outcome.
Given the potentially serious biases that can occur in mediation analysis using traditional epidemiological approaches as a result of unmeasured confounders and measurement error [65–67], the two-step MR approach for mediation as outlined above will be a particularly useful framework to consider causal mediating pathways involving metabolites, since it does not rely on statistical adjustments to estimate the mediation effect as is inherent in previous approaches [14]. In applying two-step MR to metabolomic mediators, two IV analyses would be applied to assess the effect of an exposure on a particular metabolite, followed by an assessment of the impact of the respective metabolite on a disease outcome of interest.
Difficulties with data harmonization are an existing concern with metabolomic data originating across different studies, with either different analytic platforms being utilized or varying methods to identify and quantify metabolites [62]. Obtaining reliable approaches for synthesizing metabolic data among studies is likely to be particularly relevant for applying MR to metabolomic data, given that MR generally requires very large samples to attain sufficient statistical power.
Conclusions
There is now a range of emerging developments in MR that are extending the established single-SNP approach to incorporate larger amounts of biological information and to test different hypotheses. These have the potential to both address some of the existing limitations of conventional single-SNP MR as well as to answer new and exciting questions involving the increasingly available “omics” data (such as genome-wide genotype data, epigenetics, and metabolomics). Several key issues will need to be considered, ranging from biases in the construction of genome-wide allele scores, to the availability of tissue-specific epigenetic patterns and limitations, to data harmonization with metabolomic measures. However, with these novel and emerging applications of the MR framework, there is the potential to explore powerful studies and to address an unprecedented range of biomedical questions in the “omics” era.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance • Of importance
Davey Smith G, Ebrahim S. “Mendelian randomization”: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22.
Sheehan NA, Didelez V, Burton PR, Tobin MD. Mendelian randomisation and causal inference in observational epidemiology. PLoS Med. 2008;5:1205–10.
Thomas D, Conti DV. Commentary: the concept of “Mendelian randomization.”. Int J Epidemiol. 2004;33(21):21–5.
Thanassoulis G, O’Donnell CJ. Mendelian randomization: nature’s randomized trial in the post-genome era. JAMA. 2009. p. 2386–8.
Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133–63.
Davey Smith G, Ebrahim S. What can mendelian randomisation tell us about modifiable behavioural and environmental exposures? BMJ. 2005;330:1076–9.
Davey Smith G, Ebrahim S. Mendelian randomization: prospects, potentials and limitations. Int J Epidemiol. 2004;33:30–42.
Tatsioni A, Bonitsis NG, Ioannidis JPA. Persistence of contradicted claims in the literature. JAMA. 2007;298:2517–26.
Lawlor DA, Davey Smith G, Kundu D, Bruckdorfer KR, Ebrahim S. Those confounded vitamins: What can we learn from the differences between observational versus randomised trial evidence? Lancet. 2004. p. 1724–7.
Davey Smith G. Assessing intrauterine influences on offspring health outcomes: Can epidemiological studies yield robust findings? Basic Clin Pharmacol Toxicol. 2008. p. 245–56.
Bochud M, Chiolero A, Elston R, Paccaud F. A cautionary note on the use of Mendelian randomization to infer causation in observational epidemiology. Int J Epidemiol. 2008;37:414–7.
Nitsch D, Molokhia M, Smeeth L, DeStavola BL, Whittaker JC, Leon DA. Limits to causal inference based on mendelian randomization: a comparison with randomized controlled trials. Am J Epidemiol. 2006;163:397–403.
Vanderweele TJ, Tchetgen Tchetgen EJ, Cornelis M, Kraft P. Methodological challenges in mendelian randomization. Epidemiology. 2014;25:427–35.
Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;(http://hmg.oxfordjournals.org/content/early/2014/07/04/hmg.ddu328.full.pdf+html).
Brion MJ, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies. Int J Epidemiol. 2013;42(5):1497–501.
Davey Smith G, Timpson N, Ebrahim S. Strengthening causal inference in cardiovascular epidemiology through Mendelian randomization. Ann Med. 2008;40:524–41.
Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, et al. The NHGRI GWAS catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42(Database issue):D1001–6.
Palmer TM, Lawlor DA, Harbord RM, Sheehan NA, Tobias JH, Timpson NJ, et al. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat Methods Med Res. 2011;21:223–42.
Burgess S, Thompson SG. Use of allele scores as instrumental variables for Mendelian randomization. Int J Epidemiol. 2013;42:1134–44.
Pierce BL, Ahsan H, Vanderweele TJ. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int J Epidemiol. 2011;40(3):740–52.
Davey Smith G. Random allocation in observational data: how small but robust effects could facilitate hypothesis-free causal inference. Epidemiology. 2011;22(4):467–8.
Holmes MV, Lange LA, Palmer T, Lanktree MB, North KE, Almoguera B, et al. Causal effects of body mass index on cardiometabolic traits and events: a Mendelian randomization analysis. Am J Hum Genet. 2014;94:198–208.
Richmond RC, Davey Smith G, Ness AR, den Hoed M, McMahon G, Timpson NJ. Assessing causality in the association between child adiposity and physical activity levels: a Mendelian randomization analysis. PLoS Med. 2014;11:e1001618.
Pichler I, Del Greco MF, Gögele M, Lill CM, Bertram L, Do CB, et al. Serum iron levels and the risk of Parkinson disease: a Mendelian randomization study. PLoS Med. 2013;10(6):e1001462.
Ference BA, Yoo W, Alesh I, Mahajan N, Mirowska KK, Mewada A, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a mendelian randomization analysis. J Am Coll Cardiol. 2012;60:2631–9.
Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37:658–65.
Harbord RM, Didelez V, Palmer TM, Meng S, Sterne JAC, Sheehan NA. Severity of bias of a simple estimator of the causal odds ratio in Mendelian randomization studies. Stat Med. 2013;32:1246–58.
Didelez V, Meng S, Sheehan NA. Assumptions of IV methods for observational epidemiology. Stat Sci. 2010;25:22–40.
Chen L, Davey Smith G, Harbord RM, Lewis SJ. Alcohol intake and blood pressure: a systematic review implementing a mendelian randomization approach. PLoS Med. 2008;5(3):e52.
Lewis SJ, Davey Smith G. Alcohol, ALDH2, and esophageal cancer: a meta-analysis which illustrates the potentials and limitations of a Mendelian randomization approach. Cancer Epidemiol Biomarkers Prev. 2005;14:1967–71.
Minelli C, Thompson JR, Tobin MD, Abrams KR. An integrated approach to the meta-analysis of genetic association studies using Mendelian randomization. Am J Epidemiol. 2004;160:445–52.
Pierce BL, Burgess S. Efficient design for mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am J Epidemiol. 2013;178:1177–84.
Inoue A, Solon G. Two-sample instrumental variables estimators. Rev Econ Stat. 2010;92:557–61.
Welsh P, Polisecki E, Robertson M, Jahn S, Buckley BM, de Craen AJM, et al. Unraveling the directional link between adiposity and inflammation: a bidirectional Mendelian randomization approach. J Clin Endocrinol Metab. 2010;95:93–9.
Timpson NJ, Nordestgaard BG, Harbord RM, Zacho J, Frayling TM, Tybjærg-Hansen A, et al. C-reactive protein levels and body mass index: elucidating direction of causation through reciprocal Mendelian randomization. Int J Obes (Lond). 2011;35:300–8.
Vimaleswaran KS, Berry DJ, Lu C, Tikkanen E, Pilz S, Hiraki LT, et al. Causal relationship between obesity and vitamin D status: bi-directional Mendelian randomization analysis of multiple cohorts. PLoS Med. 2013;10(2):e1001383.
Palmer TM, Nordestgaard BG, Benn M, Tybjærg-Hansen A, Davey Smith G, Lawlor DA, et al. Association of plasma uric acid with ischaemic heart disease and blood pressure: mendelian randomisation analysis of two large cohorts. BMJ. 2013;347:f4262.
Loomba R, Yang HI, Su J, Brenner D, Barrett-Connor E, Iloeje U, et al. Synergism between obesity and alcohol in increasing the risk of hepatocellular carcinoma: a prospective cohort study. Am J Epidemiol. 2013;177:333–42.
Hart CL, Morrison DS, Batty GD, Mitchell RJ, Davey Smith G. Effect of body mass index and alcohol consumption on liver disease: analysis of data from two prospective cohort studies. BMJ. 2010. p. c1240–c1240.
Global Lipids Genetics Consortium. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–83.
Holmes M V, Asselbergs FW, Palmer TM, Drenos F, Lanktree MB, Nelson CP, et al. Mendelian randomization of blood lipids for coronary heart disease. Eur Heart J. 2014;1–27.
Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–52. A recent and robust example of Mendelian randomization (MR) applied to a clinically relevant question, the causality of the relationship between triglyceride levels and coronary artery disease, implementing a “multiphenotype” approach.
Fewell Z, Davey Smith G, Sterne JAC. The impact of residual and unmeasured confounding in epidemiologic studies: a simulation study. Am J Epidemiol. 2007;166:646–55.
Davey Smith G, Phillips A. Confounding in epidemiological studies: why “independent” effects may not be all they seem. BMJ. 1992;305(6856):757–9.
Evans DM, Brion MJA, Paternoster L, Kemp JP, McMahon G, Munafò M, et al. Mining the human phenome using allelic scores that index biological intermediates. PLoS Genet. 2013;9(10):e1003919.
Khoury M. A primer series on -omic technologies for the practice of epidemiology. Am J Epidemiol. 2014;180(2):127–8.
Yang J, Benyamin B, McEvoy B, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42(7):565–9.
Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–8.
Wray NR, Yang J, Hayes BJ, Price AL, Goddard ME, Visscher PM. Pitfalls of predicting complex traits from SNPs. Nat Rev Genet. 2013;14(7):507–15.
Davey Smith G, Lawlor DA, Harbord R, Timpson N, Day I, Ebrahim S. Clustered environments and randomized genes: a fundamental distinction between conventional and genetic epidemiology. PLoS Med. 2007;4:1985–92.
Würtz P, Kangas A, Soininen P, Lehtimäki T, Kähönen M, Viikari J, et al. Lipoprotein subclass profiling reveals pleiotropy in the genetic variants of lipid risk factors for coronary heart disease. J Am Coll Cardiol. 2013;62(20):1906–8.
Mill J, Heijmans B. From promises to practical strategies for epigenetic epidemiology. Nat Rev Genet. 2013;14:585–94.
Relton CL, Davey Smith G. Epigenetic epidemiology of common complex disease: prospects for prediction, prevention, and treatment. PLoS Med. 2010;7(10):e1000356.
Baccarelli A, Wright R, Bollati V, Litonjua A, Zanobetti A, Tarantini L, et al. Ischemic heart disease and stroke in relation to blood DNA methylation. Epidemiology. 2010;21:819–28.
Dempster EL, Pidsley R, Schalkwyk LC, Owens S, Georgiades A, Kane F, et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet. 2011;20:4786–96.
Relton CL, Davey Smith G. Is epidemiology ready for epigenetics? Int J Epidemiol. 2012;41(1):5–9.
Relton CL, Davey Smith G. Two-step epigenetic mendelian randomization: a strategy for establishing the causal role of epigenetic processes in pathways to disease. Int J Epidemiol. 2012;41:161–76. The first description of the MR approach for mediation as applied to epigenetic data.
Groom A, Potter C, Swan DC, Fatemifar G, Evans DM, Ring SM, et al. Postnatal Growth and DNA Methylation Are Associated With Differential Gene Expression of the TACSTD2 Gene and Childhood Fat Mass. Diabetes. 2012. p. 391–400.
Dick KJ, Nelson CP, Tsaprouni L, Sandling JK, Aïssi D, Wahl S, et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet. 2014;6736:1–9. A highly systematic assessment of epigenome-wide influences on body mass index, with an MR-style approach utlising methylation SNPs for determining the direction of causality.
Murphy T, Mill J. Epigenetics in health and disease: heralding the EWAS era. Lancet. 2014;383:1952–4.
Shin S-Y, Fauman EB, Petersen A-K, Krumsiek J, Santos R, Huang J, et al. An atlas of genetic influences on human blood metabolites. Nat Genet. 2014;46(6):543–50. The most comprehensive investigation to date of genetic influences on human metabolism, reporting 145 genetic loci associated with >400 metabolites. Of particular relevance for the prospective implementation of MR studies to metabolomic data.
Tzoulaki I, Ebbels T, Valdes A, Elliott P, Ioannidis J. Design and analysis of metabolomics studies in epidemiologic research: a primer on -omic technologies. Am J Epidemiol. 2014;180(2):129–39.
Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, et al. Metabolite profiles and the risk of developing diabetes. Nat Med. 2011;17:448–53.
Kettunen J, Tukiainen T, Sarin A-P, Ortega-Alonso A, Tikkanen E, Lyytikäinen L-P, et al. Genome-wide association study identifies multiple loci influencing human serum metabolite levels. Nat Genet. 2012. p. 269–76.
Cole SR, Hernán MA. Fallibility in estimating direct effects. Int J Epidemiol. 2002;31:163–5.
Le Cessie S, Debeij J, Rosendaal FR, Cannegieter SC, Vandenbroucke JP. Quantification of bias in direct effects estimates due to different types of measurement error in the mediator. Epidemiology. 2012. p. 551–60.
Vanderweele TJ. The role of measurement error and misclassification in mediation analysis: mediation and measurement error. Epidemiology. 2012;23:561–4.
Boccia S, Hashibe M, Gallì P, De Feo E, Asakage T, Hashimoto T, et al. Aldehyde dehydrogenase 2 and head and neck cancer: a meta-analysis implementing a Mendelian randomization approach. Cancer Epidemiol Biomarkers Prev. 2009;18:248–54.
Obermayer-Pietsch BM, Bonelli CM, Walter DE, Kuhn RJ, Fahrleitner-Pammer A, Berghold A, et al. Genetic predisposition for adult lactose intolerance and relation to diet, bone density, and bone fractures. J Bone Miner Res. 2004;19:42–7.
Åsvold B, Bjørngaard J, Carslake D, Gabrielsen M, Skorpen F, Davey Smith G, et al. Causal associations of tobacco smoking with cardiovascular risk factors: Mendelian randomization analysis of the HUNT Study in Norway. Int J Epidemiol. 2014. doi:10.1093/ije/dyu113.
Stender S, Nordestgaard B, Tybæjrg-Hansen A. Elevated body mass index as a causal risk factor for symptomatic gallstone disease: a Mendelian randomization study. Hepatology. 2013;58:2133–41.
Timpson NJ, Lawlor DA, Harbord RM, Gaunt TR, Day IN, Palmer LJ, et al. C-reactive protein and its role in metabolic syndrome: Mendelian randomisation study. Lancet. 2005;366:1954–9.
Wensley F, Gao P, Burgess S, Kaptoge S, Di Angelantonio E, Shah T, et al. Association between C reactive protein and coronary heart disease: mendelian randomisation analysis based on individual participant data. BMJ. 2011;342:d548.
Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium, Hingorani AD, Casas JP. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012;379:1214–24.
IL6R Genetics Consortium Emerging Risk Factors Collaboration. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet. 2012;379(9822):1205–13.
Triglyceride Coronary Disease Genetics Consortium and Emerging Risk Factors Collaboration, Sarwar N, Sandhu MS, Ricketts SL, Butterworth AS, Di Angelantonio E, et al. Triglyceride-mediated pathways and coronary disease: collaborative analysis of 101 studies. Lancet. 2010;375:1634–9.
Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–80.
Compliance with Ethics Guidelines
Conflict of Interest
MJA Brion, B Benyamin, PM Visscher, and G Davey Smith all declare no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Marie-Jo A Brion and Beben Benyamin are joint first authorship.
Rights and permissions
About this article

Cite this article
Brion, MJ.A., Benyamin, B., Visscher, P.M. et al. Beyond the Single SNP: Emerging Developments in Mendelian Randomization in the “Omics” Era. Curr Epidemiol Rep 1, 228–236 (2014). https://doi.org/10.1007/s40471-014-0024-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40471-014-0024-2