
Abstract
Renin is the first and rate-limiting step of the renin-angiotensin system. The exclusive source of renin in the circulation are the juxtaglomerular cells of the kidney, which line the afferent arterioles at the entrance of the glomeruli. Normally, renin production by these cells suffices to maintain homeostasis. However, under chronic stimulation of renin release, for instance during a low-salt diet or antihypertensive therapy, cells that previously expressed renin during congenital life re-convert to a renin-producing cell phenotype, a phenomenon which is known as “recruitment”. How exactly such differentiation occurs remains to be clarified. This review critically discusses the phenotypic plasticity of renin cells, connecting them not only to the classical concept of blood pressure regulation, but also to more complex contexts such as development and growth processes, cell repair mechanisms and tissue regeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The renin-angiotensin system (RAS) plays a central role in blood pressure regulation and fluid-electrolyte homeostasis. It involves an array of enzymes, peptides and receptors with endocrine, paracrine and autocrine functions, whose actions are able to correct small extracellular volume (ECV) variations by increasing or decreasing RAS activity. According to the classical, endocrine view of the RAS, also known as the systemic RAS, renin, synthesized in the kidneys, catalyzes cleavage of a 10-amino acid peptide, angiotensin I, from the N-terminus portion of the circulating angiotensinogen, a protein produced by the liver. This decapeptide is subsequently hydrolyzed by angiotensin I-converting enzyme (ACE), a dicarboxyl-peptidase ubiquitously present on the plasma membrane of endothelial cells, yielding the octapeptide angiotensin II, the active end product of the RAS cascade, which is responsible, among others, for sodium reabsorption in the proximal convolute tubule of the nephron, and for vessel constriction, especially arterioles within the renal and systemic circulation [1]. This traditional axis was completed later, with the discovery of aldosterone. Angiotensin II stimulates the zona glomerulosa in the adrenal cortex to release aldosterone, which promotes sodium reabsorption coupled with potassium exchange in the distal convolute tubule of the nephron. The whole axis, named renin-angiotensin-aldosterone system (RAAS), is now firmly linked to ECV adjustment and blood pressure regulation [2]. However, in the last three decades, the RAS has assumed an even bigger role, in that it also participates in the pathophysiology of renal and cardiovascular diseases, through its inflammatory and pro-fibrotic actions. As a consequence, RAS blockade now is a cornerstone in the treatment of renal and cardiovascular diseases [3, 4]. The RAS also contributes to normal and abnormal growth processes and, evolutionary, the growth-promoting actions of angiotensin precede its endocrine and paracrine effects, representing one of its most highly conserved functions [5].
2 Renin
In 1898, Tigerstedt and Bergman performed a set of experiments, where they observed a rise in blood pressure after injecting rabbit kidney extracts in the jugular vein of rabbits. They characterized the substance as derived from the renal cortex, soluble in water and alcohol, thermolabile, and named it “renin”. This discovery remained dormant for more than 40 years, until a few decades ago research finally started to focus on renin [6]. Renin represents the first and the rate-limiting step of the RAS cascade. Overproduction and underproduction of renin result in severe disturbances of body fluid homeostasis. Precise regulation of renin release is therefore indispensable for proper RAS functioning [1, 7]. Renin is an aspartyl protease synthesized as an inactive zymogen, prorenin. The kidney secretes both renin and prorenin, but plasma levels of prorenin are, under normal conditions, about tenfold higher than the plasma levels of renin [8]. Renin consists of two homologous lobes, with a cleft in between, which contains the active site. Prorenin has a 43-amino acid N-terminal propeptide, which covers the enzymatic cleft between the 2 lobes, thereby preventing the access of its specific substrate, angiotensinogen. Prorenin is a glycoprotein, which can be activated in two ways: proteolytically or non-proteolytically. The former is irreversible and involves removal of the propeptide by a processing enzyme, while the latter is reversible, inducible by certain conditions such as low temperature and pH, and results from a conformational alteration, i.e., unfolding of the propeptide from the enzymatic cleft [9].
3 Juxtaglomerular Cells (JGC)
The JGC of the kidney are the only source of renin and the main source of prorenin in the circulation [1, 10]. Several extrarenal tissues, such as the adrenal gland, ovary, testis, placenta and retina, additionally produce and secrete prorenin [8]. Indeed, after a bilateral nephrectomy, prorenin, but not renin, can still be detected at low levels in blood [11]. Following its synthesis, prorenin enters the Golgi apparatus to be glycosylated, and then proceeds to one of two different pathways. The first one involves clear vesicles containing prorenin which are secreted constitutively, while the second pathway involves prorenin tagging for regulated secretion, which is accompanied by proteolytic prorenin-renin conversion, and renin storage in dense cytoplasmic granules [10], awaiting release in response to various stimuli, such as a sudden fall in blood pressure, a low salt diet, or β-adrenergic stimulation [12].
JGC are part of the juxtaglomerular apparatus, a tight structure situated on the kidney glomerular hilum, composed mainly of specialized epithelial cells (macula densa cells of the ascending limb of the nephron loop), extraglomerular mesangial cells, the terminal portion of the afferent arterioles and the proximal portion of the efferent arterioles. JGC are within these vessels and exhibit an epithelioid appearance, with a prominent endoplasmic reticulum, Golgi apparatus, and renin granules, but also containing myofibrils and adhesion plaques, typical of smooth muscle cells [13].
Efforts to characterize JGC have historically been hampered by several factors: JGC lose their secretory granules and, concomitantly, their ability to proteolytically activate prorenin when placed in culture [14]; JGC make up approximately 1/1000 of the total cell mass of the kidney, making it difficult to isolate sufficient quantities of pure cells for further characterization [15].
4 JGC Progenitors and their Embryology
JGC represent one type of the so-called repertoire of renin-producing cells (RPC), a group of cells that at some moment in their lifespan produce renin. During kidney ontogeny in mammals, renin expression changes according to a pattern (Fig. 1). It first appears in the undifferentiated metanephric blastema before vessel formation has begun. Once the vascularization begins, around the 14th embryonic day in mice and rats, RPC are distributed along the walls of the arteriolar vessels. As the process evolves, renin starts to be expressed in other cells along the distal portions of the newly formed vessels. Gradually, RPC disappear from the larger vessels, shifting toward the afferent arterioles, and, finally, assuming their position on the juxtaglomerular apparatus [12]. Gomez et al. showed that the ontogeny of renin expression by RPC depicts its own phylogenetic evolution pattern [16].

Schematic illustration of nephrogenesis. The ureteric bud (UB) interacts with the methanephric mesenchyme (MM), resulting in the subsequent formation of the cap mesenchyme (CM) and loose mesenchyme (LM). The CM harbors the transcription factors Six2 and Cited1 (in orange), and will give rise to the tubular system (proximal convolute tubule (PCT), loop of Henle (LH) and distal convolute tubule (DCT); the LM harbors the transcriptions factor FoxD1 (depicted in green), and will give rise to most of the glomeruli and vascular cells from the afferent arteriole (AA) and efferent arteriole (EA). The UB will evolve to from the collecting duct (CD) and ureter.
The expression of the renin gene is tissue-specific and developmentally regulated [17]. Renal renin concentrations are high in early life, decreasing progressively as kidney maturation evolves [18]. During this process, RPC are associated with assembling and branching of the developing kidney vasculature [19], and the ablation of these cells in mice during development results in a distinct kidney phenotype with peculiar vascular abnormalities [20]. In vivo, vascularization of the kidney is synchronized with epithelial nephrogenesis [21]. Epithelial branching morphogenesis is critical for the formation of various organs, including the vasculature and kidneys [22]. The definitive kidney in mammals originates from a complex interaction between the ureteric bud and the metanephric mesenchyme, both derived from the intermediate mesoderm. These reciprocal actions lead the ureteric bud to elongate and bifurcate toward the metanephric mesenchyme, forming on its tip, an aggregate of mesenchyme cells, the cap mesenchyme. This condensate of cells generates a vesicle, which continues to form a comma-shaped body and later, a S-shaped body that gives rise to most of glomerulus and tubular epithelia. As nephrogenesis progresses, the newly formed S-shaped body is fused to the ureteric bud, from which the collecting ducts and ureter originate [23, 24].
Cap mesenchyme expresses the transcriptional factors Six2 and Cited1 and gives rise to Bowman’s capsule, podocytes, the proximal and distal convolute tubules and the loop of Henle. There is an outer layer of loose mesenchyme cells, adjacent to the cap mesenchyme, which expresses different transcriptional factors: cKit (endothelial precursors) or forkhead box D1 (FoxD1, stromal cells). The latter factor is responsible for the generation of RPC, mesangial cells and all mural cells, including vascular smooth muscle cells (VSMC), perivascular fibroblasts and pericytes [25,26,27].
5 The Recruitment Phenomenon
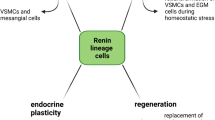
JGC lineage involves differentiation of the above metanephric mesenchymal cells. This complex process generates hemangioblasts, which will evolve to endothelial cells, and RPC precursors, the latter harboring the transcriptional factor FoxD1. During kidney ontogeny, these precursors will give rise to JGC and a subset of VSMC [25, 27]. In the adult, stress events that threaten body homeostasis, such as hypotension or extracellular fluid depletion, are normally corrected through renin release by JGC. Nevertheless, if this response does not suffice, or if renin expression is chronically stimulated, VSMC along the preglomerular arterioles undergo metaplasia to a renin cell phenotype in order to also synthesize renin, a phenomenon known as “recruitment” [28] (Fig. 2). Importantly, the upregulation of renin synthesis and release following a homeostasis threat is due to a rise in the number of RPC (hyperplasia) rather than an elevation on the renin production per cell (hypertrophy) [29, 30].

The ‘recruitment’ phenomenon, illustrated by a schematic illustration (a, c) and immunofluorescence and fluorescent in situ hybridization data from control and captopril-treated mice (b, d, modified from [64]). In adult kidneys, renin is produced by the juxtaglomerular (JG) cells located within the afferent arterioles at the entrance of the glomeruli. However, under chronic renin stimulation (e.g., following captopril treatment), renin lineage cells [like the vascular smooth muscle cells (VSMC)] along the afferent arterioles may convert into a renin-producing cell (RPC) phenotype in order to maintain the homeostasis
The recruitment phenomenon is an indispensible mechanism to maintain homeostasis. In the 1970s, Cantin et al. observed metaplasia of VSMC into JGC in the arteries and arterioles of ischemic kidneys [31]. Nowadays, it is known that this transformation involves differentiation of a non-renin-expressing cell into a phenotype that is able to synthesize renin, and occurs in the descendants of cells that previously expressed renin during development [32]. Interestingly, once the stimulation perpetuates, the recruitment intensifies, and RPC may occur all along the extension of preglomerular arterioles, larger vessels, in the extraglomerular and intraglomerular mesangium, in a pattern that resembles the development of the embryonic kidney [32]. In a recent review, Gomez emphasized that recruitment does not involve migration or replication of cells, but solely concerns a phenotype switch toward renin expression by cells whose capability to produce renin is latent, and re-acquired following appropriate stimulation [33].
Nevertheless, Dzau et al. identified liver X receptor alpha (LXRα), a nuclear receptor, as an important player in the induction of renin expression in JGC. They showed that LXRα exerts its activity as a cAMP-responsive regulator, binding to a unique upstream region of the renin promoter [34]. LXRα activation additionally upregulated a set of genes (e.g., c-myc) that are involved in cellular differentiation, proliferation and migration. Accordingly, LXRα would induce JGC hypertrophy and hyperplasia, through its coordinate interaction on the renin and c-myc promoters [29].
Moreover, mesenchymal stem cells (MSC) have also been suggested to play a pivotal role in the recruitment phenomenon. Matsushita et al. demonstrated that human and murine MSC are capable of synthesizing renin following LXRα activation. Indeed, MSC overexpressing LXRα and under continuous cAMP stimulation underwent differentiation to a RPC phenotype, which could also express α-smooth muscle actin (αSMA) [35]. Thus, on the basis of these findings, it was speculated that MSC, which are normally resident within the glomerulus, might be the origin of the RPC [35]. Wang et al. used cell lineage tracking models to further study the role of MSC in RPC recruitment. First, they isolated MSC from the adult mouse kidney and verified the expression of typical tissue stem/progenitor cell markers, including CD44, and of metanephric mesenchymal cell markers, such as FoxD1. CD44-positive MSC-like cells differentiated into RPC following exposure to an LXRα agonist, cAMP or a phosphodiesterase inhibitor [36]. Furthermore, mice submitted to a low salt diet and captopril treatment, conditions well-established to induce JGC recruitment, exhibited an expansion of RPC in the kidney, accompanied by CD44 cells, with co-localization of MSC markers and renin. These results could not be reproduced with bone marrow MSC, suggesting that only MSC resident in the adult kidney contribute to JGC recruitment [36]. The prostaglandin E2/E-prostanoid receptor 4 pathway, also known for its involvement in tubuloglomerular feedback, played a key role in the activation of renal CD44-positive MSC during conditions of JGC recruitment [37].
However, the participation of adult renal MSC in JGC recruitment has been questioned. Gomez et al. reported that MSC at most have a very minor contribution in comparison with the already existing pool of arteriolar cells undergoing a phenotype switch [33]. The latter authors ascertained that JGC also express CD44, so that it is questionable whether MSC truly represent a different cell group in the recruitment process, or are just simply descendants of the renin cell lineage, capable of switching their renin phenotype back and forth. Moreover, CD44 expression on MSC might be an in vitro artifact, since primary MSC from bone marrow lacked such expression, while it did occur after culturing the cells [38]. Further studies are needed to unravel the precise role of MSC in JGC recruitment.
6 cAMP Pathway and Recruitment
Renin is a cAMP-inducible gene. In all species tested, there is a functional cAMP response element on the renin promoter [12]. Among all the intracellular second messengers that control renin secretion, the cAMP signaling cascade appears to be the core pathway for the exocytosis of renin [10, 39, 40] (Fig. 3). Thus, prostaglandins, kinins, and β-adrenergic agonists have a stimulatory effect on renin release, in all cases because they increase cAMP generation [41].

Simplified scheme showing how cAMP and cGMP regulate renin (modified from [33]). ATP adenosine-triphosphate, cAMP cyclic adenosine-monophosphate, AC adenylate cyclase, CBP CREB-binding protein, CREB cAMP response element binding protein, cGMP cyclic guanosine-monophosphate, PDE phosphodiesterase, NO nitric oxide, pGC particulate guanylate cyclase, sGC soluble guanylate cyclase, GPRC G-protein-coupled receptor, Gsα stimulatory G protein α-subunit, Gβ inhibitory G protein β-subunit, Gγ inhibitory G protein γ-subunit, PKA protein kinase A
cAMP regulates a wide range of biological processes in cells. The binding of an extracellular signal molecule to a G-protein coupled receptor activates adenylyl cyclase (AC), an enzyme that generates cAMP from ATP, increasing its intracellular levels. This rise activates cAMP-dependent protein kinase A (PKA). PKA translocates to the nucleus to phosphorylate the gene regulatory protein CREB (cAMP responsive element binding protein). CREB recognizes a specific DNA sequence, called the cAMP response element (CRE), found in the regulatory region of many genes. Once phosphorylated, CREB recruits the coactivator CBP (CREB-binding protein), which stimulates gene transcription [42].
Interestingly, conditional deletion of G-protein subunits in RPC has a great impact on their function. Indeed, mice with protein Gsα (stimulatory subunit α) deficiency in JGC have very low plasma renin concentrations, with resulting low levels of aldosterone and arterial blood pressure. Moreover, such deletion also resulted in abnormalities in the preglomerular arterial tree [43, 44].
Elegant evidence on the regulation of renin production by cAMP was obtained by Gomez et al., who labeled cells of renin lineage with cyan fluorescent protein (CFP), and cells producing renin with yellow fluorescent protein (YFP) [45]. This yielded suitable amounts of cells which could still produce renin after several passages. CFP-labeled cells expressed VSMC markers like α-SMA and smooth muscle myosin heavy chain (SM-MHC or Myh11), but not renin. However, after stimulation with forskolin (an AC stimulator) or cAMP analogs, they began to express YFP and renin, and decreased α-SMA and Myh11 expression. This response was even bigger with more intense or longer stimuli, suggesting that the recruitment response is graded [45].
7 Role of Calcium and cGMP
Calcium plays an important role in the biology of secretory cells. In general, a rise in cytosolic calcium leads to the release of their content. However, the opposite occurs in parathyroid cells and JGC, where calcium inhibits renin exocytosis. This is known as the “calcium paradox”, and it remained as a mystery for decades [41, 46].
JGC harbor 2 isoforms of AC (types V and VI), which are inhibited by cytosolic calcium. Thus, particularly in JGC, a decrease in cytosolic calcium would stimulate AC, resulting in cAMP synthesis and, consequently, renin release [47, 48]. Initially, it had been reported that calcium inhibits renin gene transcription and destabilizes renin mRNA [49]. More recently, it became clear that calcium stimulates, via the calcium sensing receptor, a calcium calmodulin-activated phosphodiesterase 1C (PDE1), an enzyme that degradates cAMP, thereby providing an additional explanation of the calcium paradox [50].
The contribution of cGMP to renin release is more complex, with both stimulatory and inhibitory effects [46]. The cGMP and cAMP pathways are cross-linked. Nitric oxide (NO) activates soluble guanylate cyclase (GC) to generate cGMP, which in turn inhibits phosphodiesterase 3, a cAMP-degrading enzyme. Consequently, cAMP levels will go up, and renin release will rise [46, 51]. However, ligands that increase cGMP via activation of particulate GC (like atrial natriuretic peptide) inhibit renin exocytosis through activation of cGMP-dependent protein kinase type II [10, 46]. Interestingly, Neubauer et al. recently demonstrated that RPC recruitment is dependent on NO availability and the NO-GC signaling pathway [52].
8 Epigenetic Mechanisms and microRNA (miRNA)
Acetylation and deacetylation of histones are important epigenetics mechanisms involved in gene transcription regulation. Acetylation is mediated by histone acetyltransferase (HAT), which leads to the transfer of an acetyl functional group to histone molecules, promoting nucleosomal relaxation and transcriptional activation. Deacetylation is mediated by histone deacetylase (HDAC), and results in chromatin condensation and transcriptional repression [53].
Using the double-fluorescent reporter mouse model described above, Gomez et al. observed that chromatin remodeling contributes to the recruitment process, at the cAMP level, through histone H4 acetylation [45]. The underlying mechanism involved the CREB-recruited cofactors CBP and p300, which exhibit HAT activity and, in the CRE region, promote nucleosomal relaxation and, consequently, transcriptional activation. In support of this concept, forskolin increased histone H4 acetylation in the CRE region [45]. Studies carried out in mice with conditional deletion of CBP and p300 in RPC revealed that individual deletion of one of these cofactors did not affect renin expression, while simultaneous deletion reduced renin in adult life, and resulted in renal interstitial fibrosis [54]. CBP/p300 were also indispensable for re-expression of renin in the arteriolar VSMC of mice exposed to low sodium + captopril [55]. Taken together, RPC have a poised chromatin landscape suitable to respond properly to threats, allowing these cells to switch the renin phenotype on and off [33].
In addition to the epigenetic control mechanisms, microRNAs (miRNAs) also control JGC activity. miRNAs are endogenous small non-coding RNA molecules, containing around 18–22 nucleotides. They exert their function by targeting mRNA, inducing decay or translational repression [56]. miRNA genes are phylogenetically conserved, and are involved in many cell processes such as developmental timing, death and proliferation [57]. miRNA transcription, usually by RNA polymerase II, results in a long transcript, whose structure will be cleaved by the RNase III endonuclease Drosha and the cofactor DGCR8, yielding a miRNA precursor (pre-miRNA). This precursor is processed by the RNase III enzyme Dicer, and the product is incorporated into the RNA-induced silence complex, where gene silencing proceeds [58].
Gomez et al. established the importance of Dicer for the JGC and, even further, for the morphologic and physiologic integrity of the kidney. They discovered that conditional deletion of Dicer in cells of the renin lineage in mice reduced the number of JGC and decreased renin gene expression, thus leading to reduced plasma renin and blood pressure levels. The animals also presented kidneys with vascular abnormalities and striped fibrosis [59]. Noteworthy, this vascular pattern was quite similar to that found in mice with ablation of the RPC, through the expression of diphtheria toxin A chain driven by the renin promoter [20]. Surprisingly, kidney vessels exhibited normal wall thickness and lumen size, in contrast to the concentric hypertrophy and thick vessel walls seen when RAS genes are deleted, including renin. Apparently, RPC are responsible for arterial wall thickening, by producing angiogenic and trophic factors [20, 21].
Furthermore, Medrano et al. identified two miRNA, miR-330 and miR-125b-5p, whose opposite actions are crucial for the recruitment phenomenon. miR-125b-5p is normally expressed in the VSMCs of the afferent arteriole, and responsible for sustaining a contractile phenotype. Yet, it is also expressed in JGC, in order to preserve their contractile function. However, under a homeostasis threat, miR-125b-5p expression diminishes in VSMC, allowing them to undergo a metaplastic transformation into a renin phenotype, and remains unaltered in JGC. miR-330 is expressed in JGC only, and inhibits contractile features, thus favoring renin production [60].
9 JGC Signature: The Myo-Endocrine Profile
Although several factors have been identified that regulate renin production and secretion, RPC-specific markers were limited to renin itself, and the gene Zis transcript [14]. To overcome this problem, Brunskill et al. targeted YFP to mouse JGC and used fluorescence-activated cell sorting (FACS) to enrich tagged cells for transcriptome analysis. This approach yielded a set of 369 core genes, responsible for the JGC gene regulatory network [61]. Furthermore, this distinct array of genes that governs JGC identity is unique when compared to other cell types in the kidney. Mainly, it encompasses genes highly expressed in both smooth muscle and endocrine cells [61].
Among the genes identified in the transcriptome analysis, renin was the highest transcribed gene. The second highest one was aldo-keto reductase family 1, member B7 (Akr1b7), responsible for detoxification of steroidogenesis products. Akr1b7 is a member of aldo-keto reductase superfamily, which reduces harmful aldehydes and ketones produced from the breakdown of lipid peroxides to their respective alcohols [62]. Remarkably, despite its high expression on JGC, mice with Akr1b7-deficient kidneys had no abnormalities in renin production and secretion, while renin deletion also did not affect Akr1b7 expression [61, 63]. Therefore, Akr1b7 gene might function as a novel JGC marker, independently of renin. Other highly expressed transcripts involved genes related to the smooth muscle phenotype, like α-SMA, Myh11 and calponin (Cnn1) [61].
Interestingly, the aforementioned genetic regulatory network allows RPC to possess either an endocrine or a contractile phenotype. Hence, this bivalent profile sustains the ability of RPC to switch phenotype, depending on the situation. Therefore, during a homeostasis challenge, renin lineage cells have the gene program to differentiate into an endocrine cell, synthesizing and releasing renin, in order to reestablish the homeostasis. Moreover, due to their position, at the glomerular hilum, JGC should also retain contractile properties, allowing them to contract or relax and, subsequently, to adjust renal blood flow and glomerular filtration rate [33, 61].
Martini et al. performed RNA transcriptome analysis on human reninomas, as an approach to further understand JGC biology [64]. Reninomas are rare, renin-producing tumors, arising from a proliferation of JGC in the kidney cortex, and are often detected because of the appearance of fulminant hypertension in young patients [65, 66]. Deep sequencing of 4 human reninomas with subsequent transcript mapping in the kidney of mice under different conditions yielded a list of genes (36 of which had never been described before) specifically expressed in RPC. When evaluating 10 of these genes on (pro)renin producing As4.1 cells, it was observed that platelet-derived growth factor beta (PDGFB) suppressed (pro)renin release and renin gene expression. Moreover, PDGFB-exposed cells displayed a phenotypic shift from myo-endocrine to inflammatory, evidenced by αSMA downregulation and interleukin-6 upregulation, and a more elongated shape (Fig. 4). Here it should be mentioned that neither conditional deletion of the PDGF receptor in RPC, nor deletion of endothelial PDGFB production affected the normal development of renal RPC [67]. This may not be too surprising, given the fact that PDGFB actually is a negative regulator of renin expression, possibly coming into play only under pathological conditions. In summary, the reninoma data provide a novel role for PDGFB as a regulator of RPC.

a Renin levels in medium obtained from renin-producing As4.1 cells incubated for 48 h with medium derived from HEK293 cells transfected with platelet-derived growth factor beta (PDGFB). Renin, α-smooth muscle actin (αSMA) and interleukin-6 (IL-6) gene expression are also provided. Data (mean ± SEM of n = 5–11) are modified from [64], and have been expressed as a percentage of the levels in cells exposed to medium of control-transfected cells, i.e., cells treated with the transfection mix, but without cDNA. *P < 0.05. b, c As4.1 cells under control conditions and after exposure to PDGFB (×10 magnification)
10 Notch Signaling Pathway
The Notch signaling pathway is a highly conserved system, present in all animal species, which plays a pivotal role in local cell-cell communication, determining cell fate decisions and controlling patterns formation during ontogenesis. Its dysfunction is linked to severe developmental defects and pathologies [68]. The Notch signaling cascade starts with the binding of specific ligands to the Notch transmembrane receptor, leading to cleavage of the intracellular Notch receptor domain (NIC). NIC translocates to the nucleus, where it binds to RBPJ (recombination signal binding protein for Ig-κJ region), a transcription factor that normally represses Notch target genes by recruiting a co-repressor complex. However, once it is coupled to NIC, RBPJ recruits cofactors that activate the transcriptional machinery [69].
Interestingly, among the 369 core genes that confer the JGC identity according to the approach performed by Brunskill et al., there are members of the Notch pathway, including RBPJ [61]. In fact, RBPJ had already been related to RPC plasticity by Castellanos Rivera et al. [70]. They generated a conditional knockout (cKO) of RBPJ in renin lineage cells. This resulted in a striking reduction in the JGC number and renin expression, with subsequently low plasma renin levels and a low blood pressure. Furthermore, under conditions that trigger the recruitment phenomenon, such as sodium depletion and captopril treatment, RBPJ-cKO mice were unable to exhibit renin expression along the afferent arterioles, i.e., the ability of their VSMCs to regain the renin phenotype was impaired.
Additionally, mice with RBPJ-cKO in renal cells of FoxD1 lineage displayed significant renal abnormalities, including a decline in the arteries and arterioles number, with a thinner smooth muscle cell layer and renin depletion, further upholding the concept that RBPJ is a major determinant of the transformation of FoxD1 cells progenitors into a healthy kidney vasculature [71]. Experiments in transgenic renin reporter zebrafish fully support the essential role of the Notch signaling pathway for developmental renin expression and its association with proper angiogenesis [72].
RBPJ also has a pivotal role in the maintenance of the JGC myo-endocrine gene program. Using a bacterial artificial chromosome reporter, it was observed that RBPJ activates the renin promoter directly [15]. A double-fluorescent mice reporter model subsequently revealed that RBPJ deletion does not affect RPC endowment. RPC were at their proper location, although unable to express renin and Akr1b7, and, surprisingly, also did not express the VSMC markers α-SMA, Myh11 and Cnn1. Yet, they displayed an upregulation of genes related to the immunological system, such as lipocalin-2, lysozyme-2, chemokine ligand-9 and interleukin-6 [15]. Furthermore, Belyea et al. identified renin progenitors in mouse bone marrow, and found that in the presence of RPBJ-cKO in the renin cell lineage, those progenitors gave rise to a very aggressive lymphoblastic leukemia [73].
JGC are surrounded by multiple cell types, including pericytes, endothelial cells, epithelial cells, VSMCs, mesangial cells, and macula densa cells. Direct cell-to-cell interaction will undoubtedly contribute to normal RPC functioning. It is therefore not surprising that, among the highly expressed transcripts identified by Brunskill et al. in JGC, is connexin 40 [61]. Connexins are transmembrane proteins, six of which assemble to mold a hemichannel. When 2 hemichannels of adjacent cells align, a channel is formed which allows the cytoplasma of both cells to connect. A set of these channels in parallel builds a gap junction, a structure that permits the cells to share small molecules and, therefore, to respond to extracellular signals in a coordinated way [16]. Unexpectedly, in mice lacking connexin 40, RPC were found in the periglomerular interstitium, and not at their normal juxtaglomerular location. Besides, the recruitment phenomenon (following severe sodium depletion) did not occur at its usual location, i.e., in the wall of the upstream preglomerular arterioles [74]. Apparently therefore, cell-to-cell communication via gap junctions is essential for the correct juxtaglomerular positioning and recruitment of RPC.
11 Plasticity and Regeneration
More than 100 years have passed since renin’s discovery, and the RAS now turns out to have multiple effects beyond its role in blood pressure regulation. As outlined in this review, renin cells function as pluripotent progenitors for other kidney cells types [32, 75]. Normally, a low perfusion pressure leads to an expansion of RPC. Kurt et al. investigated to what degree this relates to hypoxia per se, by deleting the von Hippel–Lindau protein in JGC, i.e., the negative regulator of the hypoxia-inducible transcriptional factor-2 (HIF-2α). Remarkably, following the upregulation of HIF-2α by this procedure, RPC started to express erythropoietin instead of renin, and the normal renin upregulation after low salt was no longer seen. In fact, JGC were reprogrammed into fibroblast-like cells, expressing collagen I and PDGF receptor β (PDGFR-β) [76, 77]. Clearly therefore, hypoxia-inducible genes are essential for normal development and physiologic adaptation of RPC. The upregulated PDGFR-β expression in these cells is in full agreement with the results from the human reninoma studies showing that the PDGFB-PDGFR-β pathway downregulates renin expression [64].
Pippin et al. proposed that cells of renin lineage present alongside the preglomerular arteries participate in the glomerular regeneration after podocyte injury [78]. In support of this concept, renin cells from the juxtaglomerular apparatus were shown to migrate to the glomerular tuft, in order to replace podocytes [79]. This is quite striking, since podocytes are normally derived from a different embryonic structure, the cap mesenchyme cells.
Easier to understand is the participation of renin progenitors in glomerular mesangium regeneration, since mesangium cells, considered as specialized pericytes, are derived from FoxD1 stromal cells present at the loose mesenchyme. Indeed, in a mesangiolysis murine model, renin lineage cells repopulated the intraglomerular mesangium [80]. Interestingly, these cells stopped producing renin and expressed PDGFR-β, i.e., the receptor linked to renin downregulation [64]. Furthermore, Stefanska et al. established that pericytes are RPC in the human kidney [81]. Traditionally, pericytes are perivascular cells that wrap around blood capillaries. They are highly plastic cells contributing to multiple processes like angiogenesis, tumorigenesis and vasculopathy progression [82]. The concept that RPC are or derive from pericytes is very interesting, since pericytes possess regenerative potential and are additionally known for the fact that they express the PDGFR-β [81]. In fact, in zebrafish, Rider et al. observed that renin cells express this marker [83].
12 Concluding Remarks
How exactly renin cells are able to differentiate into many different phenotypes remains to be clarified. The signals allowing RPC to shift phenotype, or to migrate to an injury site are now only beginning to be unraveled. Many questions still need to be answered, most importantly, whether regeneration would eventually allow a restoration of normal function. This is of particular relevance in chronic glomerulopathies, where glomerular cells are destroyed, with subsequent overproduction of extracellular matrix, fibrosis and architectural disruption, all leading to physiological impairment. Apparently, under such severe pathological conditions, tissue regeneration by RPCs either does not occur of is insufficient. One possibility is that RPCs under pathological conditions transform to fibroblast-like cells, thus contributing to renal fibrosis themselves. Currently, RAS blockade is the cornerstone of renal and cardiovascular diseases. Simultaneously, it is well known that RAS blockers, like most anti-hypertensive drugs, induce RPCs recruitment. The long-term effect of this chronic activation is still unknown, nor do we know to what degree it contributes to (further) impairment of renal function. Detailed knowledge of this process would lead to better treatment options and more favorable outcomes. Clearly, this whole area not only illustrates the complexity of the renin cell, but also represents an exciting new field that needs to be explored in the coming years.
References
Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM. Classical renin-angiotensin system in kidney physiology. Compr Physiol. 2014;4:1201–28.
Williams JS, Williams GH. 50th anniversary of aldosterone. J Clin Endocrinol Metab. 2003;88:2364–72.
Brewster UC, Perazella MA. The renin-angiotensin-aldosterone system and the kidney: effects on kidney disease. Am J Med. 2004;116:263–72.
Unger T, Li J. The role of the renin-angiotensin-aldosterone system in heart failure. J Renin Angiotensin Aldosterone Syst. 2004;5(Suppl 1):S7–10.
Gomez RA, Norwood VF. Developmental consequences of the renin-angiotensin system. Am J Kidney Dis. 1995;26:409–31.
Phillips MI, Schmidt-Ott KM. The discovery of renin 100 years ago. News Physiol Sci. 1999;14:271–4.
Hackenthal E, Paul M, Ganten D, Taugner R. Morphology, physiology, and molecular biology of renin secretion. Physiol Rev. 1990;70:1067–116.
Campbell DJ. Critical review of prorenin and (pro)renin receptor research. Hypertension. 2008;51:1259–64.
Danser AH, Deinum J. Renin, prorenin and the putative (pro)renin receptor. J Renin Angiotensin Aldosterone Syst. 2005;6:163–5.
Schweda F, Friis U, Wagner C, Skott O, Kurtz A. Renin release. Physiology (Bethesda). 2007;22:310–9.
Krop M, de Bruyn JH, Derkx FH, Danser AH. Renin and prorenin disappearance in humans post-nephrectomy: evidence for binding? Front Biosci. 2008;13:3931–9.
Castrop H, Hocherl K, Kurtz A, Schweda F, Todorov V, Wagner C. Physiology of kidney renin. Physiol Rev. 2010;90:607–73.
Jennette JC, Heptinstall RH. Heptinstall’s pathology of the kidney. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2007.
Karginova EA, Pentz ES, Kazakova IG, Norwood VF, Carey RM, Gomez RA. Zis: a developmentally regulated gene expressed in juxtaglomerular cells. Am J Physiol. 1997;273:F731–8.
Castellanos-Rivera RM, Pentz ES, Lin E, Gross KW, Medrano S, Yu J, Sequeira-Lopez ML, Gomez RA. Recombination signal binding protein for Ig-kappaJ region regulates juxtaglomerular cell phenotype by activating the myo-endocrine program and suppressing ectopic gene expression. J Am Soc Nephrol. 2015;26:67–80.
Sequeira Lopez ML, Gomez RA. Novel mechanisms for the control of renin synthesis and release. Curr Hypertens Rep. 2010;12:26–32.
Pan L, Gross KW. Transcriptional regulation of renin: an update. Hypertension. 2005;45:3–8.
Gomez RA, Pupilli C, Everett AD. Molecular and cellular aspects of renin during kidney ontogeny. Pediatr Nephrol. 1991;5:80–7.
Reddi V, Zaglul A, Pentz ES, Gomez RA. Renin-expressing cells are associated with branching of the developing kidney vasculature. J Am Soc Nephrol. 1998;9:63–71.
Pentz ES, Moyano MA, Thornhill BA, Sequeira Lopez ML, Gomez RA. Ablation of renin-expressing juxtaglomerular cells results in a distinct kidney phenotype. Am J Physiol Regul Integr Comp Physiol. 2004;286:R474–83.
Sequeira Lopez ML, Gomez RA. Development of the renal arterioles. J Am Soc Nephrol. 2011;22:2156–65.
Michos O. Kidney development: from ureteric bud formation to branching morphogenesis. Curr Opin Genet Dev. 2009;19:484–90.
Gilbert SF. Developmental biology. 6th ed. Sunderland: Sinauer Associates; 2000.
Hohenstein P, Pritchard-Jones K, Charlton J. The yin and yang of kidney development and Wilms’ tumors. Genes Dev. 2015;29:467–82.
Sequeira Lopez ML, Pentz ES, Robert B, Abrahamson DR, Gomez RA. Embryonic origin and lineage of juxtaglomerular cells. Am J Physiol Renal Physiol. 2001;281:F345–56.
Gomez RA, Belyea B, Medrano S, Pentz ES, Sequeira-Lopez ML. Fate and plasticity of renin precursors in development and disease. Pediatr Nephrol. 2014;29:721–6.
Sequeira-Lopez ML, Lin EE, Li M, Hu Y, Sigmund CD, Gomez RA. The earliest metanephric arteriolar progenitors and their role in kidney vascular development. Am J Physiol Regul Integr Comp Physiol. 2015;308:R138–49.
Gomez RA, Chevalier RL, Everett AD, Elwood JP, Peach MJ, Lynch KR, Carey RM. Recruitment of renin gene-expressing cells in adult rat kidneys. Am J Physiol. 1990;259:F660–5.
Matsushita K, Zhang Z, Pratt RE, Dzau VJ. Molecular mechanism of juxtaglomerular cell hyperplasia: a unifying hypothesis. J Am Soc Hypertens. 2007;1:164–8.
Everett AD, Carey RM, Chevalier RL, Peach MJ, Gomez RA. Renin release and gene expression in intact rat kidney microvessels and single cells. J Clin Invest. 1990;86:169–75.
Cantin M, Araujo-Nascimento MD, Benchimol S, Desormeaux Y. Metaplasia of smooth muscle cells into juxtaglomerular cells in the juxtaglomerular apparatus, arteries, and arterioles of the ischemic (endocrine) kidney. An ultrastructural-cytochemical and autoradiographic study. Am J Pathol. 1977;87:581–602.
Sequeira Lopez ML, Pentz ES, Nomasa T, Smithies O, Gomez RA. Renin cells are precursors for multiple cell types that switch to the renin phenotype when homeostasis is threatened. Dev Cell. 2004;6:719–28.
Gomez RA, Sequeira-Lopez ML. Novel functions of renin precursors in homeostasis and disease. Physiology (Bethesda). 2016;31:25–33.
Tamura K, Chen YE, Horiuchi M, Chen Q, Daviet L, Yang Z, Lopez-Ilasaca M, Mu H, Pratt RE, Dzau VJ. LXRalpha functions as a cAMP-responsive transcriptional regulator of gene expression. Proc Natl Acad Sci USA. 2000;97:8513–8.
Matsushita K, Morello F, Wu Y, Zhang L, Iwanaga S, Pratt RE, Dzau VJ. Mesenchymal stem cells differentiate into renin-producing juxtaglomerular (JG)-like cells under the control of liver X receptor-alpha. J Biol Chem. 2010;285:11974–82.
Wang H, Gomez JA, Klein S, Zhang Z, Seidler B, Yang Y, Schmeckpeper J, Zhang L, Muramoto GG, Chute J, Pratt RE, Saur D, Mirotsou M, Dzau VJ. Adult renal mesenchymal stem cell-like cells contribute to juxtaglomerular cell recruitment. J Am Soc Nephrol. 2013;24:1263–73.
Yang Y, Gomez JA, Herrera M, Perez-Marco R, Repenning P, Zhang Z, Payne A, Pratt RE, Koller B, Beierwaltes WH, Coffman T, Mirotsou M, Dzau VJ. Salt restriction leads to activation of adult renal mesenchymal stromal cell-like cells via prostaglandin E2 and E-prostanoid receptor 4. Hypertension. 2015;65:1047–54.
Qian H, Le Blanc K, Sigvardsson M. Primary mesenchymal stem and progenitor cells from bone marrow lack expression of CD44 protein. J Biol Chem. 2012;287:25795–807.
Della Bruna R, Kurtz A, Schricker K. Regulation of renin synthesis in the juxtaglomerular cells. Curr Opin Nephrol Hypertens. 1996;5:16–9.
Klar J, Sandner P, Muller MW, Kurtz A. Cyclic AMP stimulates renin gene transcription in juxtaglomerular cells. Pflugers Arch. 2002;444:335–44.
Beierwaltes WH. The role of calcium in the regulation of renin secretion. Am J Physiol Renal Physiol. 2010;298:F1–11.
Alberts B. Molecular biology of the cell. 5th ed. New York: Garland Science; 2008.
Chen L, Kim SM, Oppermann M, Faulhaber-Walter R, Huang Y, Mizel D, Chen M, Lopez ML, Weinstein LS, Gomez RA, Briggs JP, Schnermann J. Regulation of renin in mice with Cre recombinase-mediated deletion of G protein Gsalpha in juxtaglomerular cells. Am J Physiol Renal Physiol. 2007;292:F27–37.
Neubauer B, Machura K, Chen M, Weinstein LS, Oppermann M, Sequeira-Lopez ML, Gomez RA, Schnermann J, Castrop H, Kurtz A, Wagner C. Development of vascular renin expression in the kidney critically depends on the cyclic AMP pathway. Am J Physiol Renal Physiol. 2009;296:F1006–12.
Pentz ES, Lopez ML, Cordaillat M, Gomez RA. Identity of the renin cell is mediated by cAMP and chromatin remodeling: an in vitro model for studying cell recruitment and plasticity. Am J Physiol Heart Circ Physiol. 2008;294:H699–707.
Schweda F, Kurtz A. Cellular mechanism of renin release. Acta Physiol Scand. 2004;181:383–90.
Grunberger C, Obermayer B, Klar J, Kurtz A, Schweda F. The calcium paradoxon of renin release: calcium suppresses renin exocytosis by inhibition of calcium-dependent adenylate cyclases AC5 and AC6. Circ Res. 2006;99:1197–206.
Ortiz-Capisano MC, Ortiz PA, Harding P, Garvin JL, Beierwaltes WH. Decreased intracellular calcium stimulates renin release via calcium-inhibitable adenylyl cyclase. Hypertension. 2007;49:162–9.
Klar J, Sigl M, Obermayer B, Schweda F, Kramer BK, Kurtz A. Calcium inhibits renin gene expression by transcriptional and posttranscriptional mechanisms. Hypertension. 2005;46:1340–6.
Ortiz-Capisano MC, Liao TD, Ortiz PA, Beierwaltes WH. Calcium-dependent phosphodiesterase 1C inhibits renin release from isolated juxtaglomerular cells. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1469–76.
Friis UG, Madsen K, Stubbe J, Hansen PB, Svenningsen P, Bie P, Skott O, Jensen BL. Regulation of renin secretion by renal juxtaglomerular cells. Pflugers Arch. 2013;465:25–37.
Neubauer B, Machura K, Kettl R, Lopez ML, Friebe A, Kurtz A. Endothelium-derived nitric oxide supports renin cell recruitment through the nitric oxide-sensitive guanylate cyclase pathway. Hypertension. 2013;61:400–7.
Wang Z, Qin G, Zhao TC. HDAC4: mechanism of regulation and biological functions. Epigenomics. 2014;6:139–50.
Gomez RA, Pentz ES, Jin X, Cordaillat M, Sequeira Lopez ML. CBP and p300 are essential for renin cell identity and morphological integrity of the kidney. Am J Physiol Heart Circ Physiol. 2009;296:H1255–62.
Pentz ES, Cordaillat M, Carretero OA, Tucker AE, Sequeira Lopez ML, Gomez RA. Histone acetyl transferases CBP and p300 are necessary for maintenance of renin cell identity and transformation of smooth muscle cells to the renin phenotype. Am J Physiol Heart Circ Physiol. 2012;302:H2545–52.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–5.
Watson JD. Molecular biology of the gene. 7th ed. Boston: Pearson; 2014.
Sequeira-Lopez ML, Weatherford ET, Borges GR, Monteagudo MC, Pentz ES, Harfe BD, Carretero O, Sigmund CD, Gomez RA. The microRNA-processing enzyme dicer maintains juxtaglomerular cells. J Am Soc Nephrol. 2010;21:460–7.
Medrano S, Monteagudo MC, Sequeira-Lopez ML, Pentz ES, Gomez RA. Two microRNAs, miR-330 and miR-125b-5p, mark the juxtaglomerular cell and balance its smooth muscle phenotype. Am J Physiol Renal Physiol. 2012;302:F29–37.
Brunskill EW, Sequeira-Lopez ML, Pentz ES, Lin E, Yu J, Aronow BJ, Potter SS, Gomez RA. Genes that confer the identity of the renin cell. J Am Soc Nephrol. 2011;22:2213–25.
Liu MJ, Takahashi Y, Wada T, He J, Gao J, Tian Y, Li S, Xie W. The aldo-keto reductase Akr1b7 gene is a common transcriptional target of xenobiotic receptors pregnane X receptor and constitutive androstane receptor. Mol Pharmacol. 2009;76:604–11.
Machura K, Iankilevitch E, Neubauer B, Theuring F, Kurtz A. The aldo-keto reductase AKR1B7 coexpresses with renin without influencing renin production and secretion. Am J Physiol Renal Physiol. 2013;304:F578–84.
Martini AG, Xa LK, Lacombe M-J, Blanchet-Cohen A, Mercure C, Haibe-Kains B, Friesema ECF, van den Meiracker AH, Gross KW, Azizi M, Corvol P, Nguyen G, Reudelhuber T, Danser AHJ. Transcriptome analysis of human reninomas as an approach to understanding juxtaglomerular cell biology. Hypertension. 2017;69:1145–55.
Wong L, Hsu TH, Perlroth MG, Hofmann LV, Haynes CM, Katznelson L. Reninoma: case report and literature review. J Hypertens. 2008;26:368–73.
Gottardo F, Cesari M, Morra A, Gardiman M, Fassina A, Dal Bianco M. A kidney tumor in an adolescent with severe hypertension and hypokalemia: an uncommon case–case report and review of the literature on reninoma. Urol Int. 2010;85:121–4.
Neubauer B, Machura K, Rupp V, Tallquist MD, Betsholtz C, Sequeira-Lopez ML, Ariel Gomez R, Wagner C. Development of renal renin-expressing cells does not involve PDGF-B-PDGFR-beta signaling. Physiol Rep. 2013;1:e00132.
Lai EC. Notch signaling: control of cell communication and cell fate. Development. 2004;131:965–73.
Borggrefe T, Oswald F. The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell Mol Life Sci. 2009;66:1631–46.
Castellanos Rivera RM, Monteagudo MC, Pentz ES, Glenn ST, Gross KW, Carretero O, Sequeira-Lopez ML, Gomez RA. Transcriptional regulator RBP-J regulates the number and plasticity of renin cells. Physiol Genomics. 2011;43:1021–8.
Lin EE, Sequeira-Lopez ML, Gomez RA. RBP-J in FOXD1 + renal stromal progenitors is crucial for the proper development and assembly of the kidney vasculature and glomerular mesangial cells. Am J Physiol Renal Physiol. 2014;306:F249–58.
Rider SA, Mullins LJ, Verdon RF, MacRae CA, Mullins JJ. Renin expression in developing zebrafish is associated with angiogenesis and requires the Notch pathway and endothelium. Am J Physiol Renal Physiol. 2015;309:F531–9.
Belyea BC, Xu F, Pentz ES, Medrano S, Li M, Hu Y, Turner S, Legallo R, Jones CA, Tario JD, Liang P, Gross KW, Sequeira-Lopez ML, Gomez RA. Identification of renin progenitors in the mouse bone marrow that give rise to B-cell leukaemia. Nat Commun. 2014;5:3273.
Kurtz L, Schweda F, de Wit C, Kriz W, Witzgall R, Warth R, Sauter A, Kurtz A, Wagner C. Lack of connexin 40 causes displacement of renin-producing cells from afferent arterioles to the extraglomerular mesangium. J Am Soc Nephrol. 2007;18:1103–11.
Pippin JW, Kaverina NV, Eng DG, Krofft RD, Glenn ST, Duffield JS, Gross KW, Shankland SJ. Cells of renin lineage are adult pluripotent progenitors in experimental glomerular disease. Am J Physiol Renal Physiol. 2015;309:F341–58.
Kurt B, Paliege A, Willam C, Schwarzensteiner I, Schucht K, Neymeyer H, Sequeira-Lopez ML, Bachmann S, Gomez RA, Eckardt KU, Kurtz A. Deletion of von Hippel–Lindau protein converts renin-producing cells into erythropoietin-producing cells. J Am Soc Nephrol. 2013;24:433–44.
Kurt B, Gerl K, Karger C, Schwarzensteiner I, Kurtz A. Chronic hypoxia-inducible transcription factor-2 activation stably transforms juxtaglomerular renin cells into fibroblast-like cells in vivo. J Am Soc Nephrol. 2015;26:587–96.
Pippin JW, Sparks MA, Glenn ST, Buitrago S, Coffman TM, Duffield JS, Gross KW, Shankland SJ. Cells of renin lineage are progenitors of podocytes and parietal epithelial cells in experimental glomerular disease. Am J Pathol. 2013;183:542–57.
Kaverina NV, Kadoya H, Eng DG, Rusiniak ME, Sequeira-Lopez ML, Gomez RA, Pippin JW, Gross KW, Peti-Peterdi J, Shankland SJ. Tracking the stochastic fate of cells of the renin lineage after podocyte depletion using multicolor reporters and intravital imaging. PLoS One. 2017;12:e0173891.
Starke C, Betz H, Hickmann L, Lachmann P, Neubauer B, Kopp JB, Sequeira-Lopez ML, Gomez RA, Hohenstein B, Todorov VT, Hugo CP. Renin lineage cells repopulate the glomerular mesangium after injury. J Am Soc Nephrol. 2015;26:48–54.
Stefanska A, Kenyon C, Christian HC, Buckley C, Shaw I, Mullins JJ, Peault B. Human kidney pericytes produce renin. Kidney Int. 2016;90:1251–61.
Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005;7:452–64.
Rider SA, Christian HC, Mullins LJ, Howarth AR, MacRae CA, Mullins JJ. Zebrafish mesonephric renin cells are functionally conserved and comprise of two distinct morphological populations. Am J Physiol Renal Physiol. 2017;312:F778–90.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by a CAPES Foundation Grant (99999.006209/2015-07) to AGM.
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Martini, A.G., Danser, A.H.J. Juxtaglomerular Cell Phenotypic Plasticity. High Blood Press Cardiovasc Prev 24, 231–242 (2017). https://doi.org/10.1007/s40292-017-0212-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40292-017-0212-5