
Abstract
Background
The Gulf Centralised Committee for Drug Registration (GCC-DR), as part of the Gulf Health Council (GHC), enables the consolidated registration of pharmaceutical products throughout the member states of the Gulf Cooperation Council.
Objectives
The objectives of this study were to provide an update of the performance of the GCC-DR centralised procedure; evaluate the review times for new products submitted to the GCC Centralised Registration between January 2015 and December 2020; assess the impact of applying facilitated regulatory pathways and implementing a reliance strategy; identify the strengths and weaknesses of the centralised review process; and propose strategies that could enhance the GCC regulatory review process leading to improved access to medicines for patients.
Methods
A standardised data collection template enabled the structured documentation of information collected by the Senior Regulatory Affairs and Regulatory Affairs Specialists from the Executive Board of the Health Ministers Council for GCC States to determine the GHC structure, resources, review models and milestones and timelines. The total number of applications approved was provided together with the average yearly timelines for new active substances and generics from January 2015 to December 2020 including both scientific assessment time from the agency as well as applicant response time to questions raised. Actual approval times for each product were calculated from the date of submission to the date of approval.
Results
The fewest (58) new products were approved in 2019 and the most (200) in 2020. The average review times for new medicines were the longest (838 calendar days) in 2015 and the shortest (321 calendar days) in 2019. Important changes recently implemented include an increase in the number of GCC-DR meetings, adoption of a standardised electronic common technical document and GCC regulatory review template, removal of authorisation dependence on pricing agreements and introduction of a reliance strategy. Additional recommendations include Executive Committee mandates for dossier review, target times for dossier validation, scientific review and Expert Committee recommendation and training for quality decision making.
Conclusions
GCC procedures and decision-making processes have been positively influenced by a variety of expert reviewers, unified guidelines and the implementation of a reliance strategy. Certain barriers must still be overcome to enhance the quality of the review, and to shorten regulatory review times without compromising the scientific robustness of the review.
Similar content being viewed by others

This study describes an updated evaluation (2015–2020) of the Gulf Centralised Committee for Drug Registration (GCC-DR), which enables consolidated registration (this is defined as the process that leads to a joint assessment opinion. However, the national medical regulatory authorities have to register the product locally). |
Information collected from the Health Ministers Council revealed that recent important changes such as an increase in the number of GCC-DR meetings and the introduction of a regulatory reliance strategy have shortened review times. |
Certain barriers must still be overcome to enhance the quality of the GCC-DR review and to shorten regulatory review times without compromising the scientific robustness of the review. |
1 Background
Across the world, the review of medicines is performed by regulatory agencies according to the national regulations established by their individual countries. In recent years, however, regional groups such as the Gulf Cooperation Council (GCC), which represents the seven Gulf States, have attempted to harmonise these regulations and procedures for the purpose of a regional review.
These initiatives, including those with mature regulatory systems such as the European Union as well as those in developing pharmaceutical markets such as the Caribbean Community and Sub-Saharan Africa, have been established to maximise the use of regulatory resources and help expedite patients’ access to new quality medicines [1,2,3].
1.1 The Gulf Cooperation Council
The Gulf Health Council (GHC) is an autonomous agency that is independent of the Health Ministry administration of the GCC and that regulates medical products for human use, medical devices and diagnostics and medicinal products for veterinary use. The GHC scope of activities includes marketing authorisations, post-marketing surveillance, price regulation and site inspections. The agency consists of six staff, five of whom are pharmacists, who coordinate product licence applications.
Formed in 1981, the GCC is a regional political and economic coalition of the seven Gulf countries, United Arab Emirates, Bahrain, Saudi Arabia, Oman, Qatar, Kuwait and Yemen. The countries in the GCC encompass over 1 million square miles, with a population of over 54 million people [4]. The total gross domestic product (GDP) of the GCC nations is US$1,649,731 M or US$29,460 per capita [5]. The population of the GCC, which has grown over 30% since 2000, is projected to continue to increase, and it is estimated that by 2050, the percentage of elderly people will reach 15% of the total population [6]. This, plus an anticipated rise in the incidence and treatment of chronic diseases, may result in a rise in government healthcare expenditure to as much as 20% of the GDP [6].
1.2 Gulf Centralised Committee for Drug Registration (GCC-DR)
In 1976, the GCC Health Ministers’ Council issued a decree regarding the formation of a study group to report on the establishment of a centralised registration review system to monitor the marketing of medicines and develop common guidelines for the participating authorities. By 1999, the Gulf Centralised Committee for Drug Registration (GCC-DR) was formed [7] as part of the GHC. The GCC-DR consists of two representatives from each member state and its primary function is the registration of pharmaceutical companies as well as the registration/authorisation of safe, effective and high-quality medicinal products through a centralised procedure [8].
Data collected from the GCC central office in 2014 showed that in the first 11 years of its operation (1999–2010), the GCC-DR received 1824 medicinal product applications and approved 1165 [8]. Between 2006 and 2010, a total of 413 products were approved, increasing from 60 products in 2006 to 130 in 2009, while the median approval time increased from 107 calendar days in 2006 to 265 calendar days in 2010 [8]. At this stage, all products were subjected to a full review. However, it should be noted that the overall review times include both agency scientific assessment time and the manufacturer’s response time.
In 2015, Al-Rubaie and colleagues outlined the different steps in the GCC-DR review process and the way in which these influenced the overall timelines. The study mapped the key milestones and associated activities and evaluated the quality measures employed. Information was obtained to identify the practices that accelerate or delay marketing authorisation and procedural improvements were proposed [7]. Since 2015, the regulatory landscape within the Middle East has changed, with an increasing use of reliance as the strategy for the regulatory review [9].
1.3 Objectives
The objectives of this study were to update the information on the GCC Centralised Procedure and
-
appraise the GCC centralised regulatory review process;
-
evaluate the review times for new products, including generics and new active substances (NASs) submitted to the GCC Centralised Registration between January 2015 to December 2020;
-
assess the impact of applying facilitated regulatory pathways (FRPs) and implementing a reliance strategy;
-
identify the strengths and weaknesses of the centralised review process; and
-
propose strategies that could enhance the GCC regulatory review process leading to improved access to medicines for patients.
2 Methods
A standardised data collection template was used to enable the structured documentation of information to be collected by the Senior Regulatory Affairs and Regulatory Affairs Specialists from the Executive Board of the Health Ministers Council for GCC States in Riyadh, Saudi Arabia to determine the GHC structure, resources, review models and milestones as well as the timelines that contribute to the quality of the decision-making process and the review practices that have been adopted to improve consistency, transparency, timeliness and predictability in the regulatory review process. The data were then audited and confirmed by the director and the deputy general manager of the GHC regulatory programme. In addition, the total number of applications approved was provided together with the average yearly timelines for NASs and generics from January 2015 to December 2020. It should be noted that the average time includes both scientific assessment time from the agency as well as applicant response time to questions raised. Actual approval times for each product were calculated from the date of submission to the date of approval.
2.1 Reliance Strategy
A reliance-based regulatory model is defined by the World Health Organization (WHO) as “an act whereby a regulatory authority in one jurisdiction may take into account/ give significant weight to work performed by another regulator or other trusted institution in reaching its own decision” [10].
National regulatory authorities (NRAs) are currently considering the use of FRPs such as reliance to conserve their limited resources and avoid duplication of regulatory effort. Using FRPs can contribute toward decreased timelines for the evaluation of market authorisations, as the NRA may rely on or recognise the regulatory decisions made by a reference agency. In this way, the NRA does not need to conduct a full review of the data submitted to support the application for marketing authorisation and can realistically achieve shorter overall review timelines.
If the product is registered in two Gulf countries, then application can be made to the GHC for a centralised certificate, thereby putting reliance on the reviewing Gulf States, enabling other Gulf States to decide whether they will register the product in their jurisdiction.
3 Results
3.1 Review Models
McAuslane and colleagues previously characterised three types of scientific regulatory review. Type 1 or verification review requires the previous approval of a medicine by two or more reference agencies, with the agency verifying that the locally marketed medicine conforms with the approved product. The type 2 or abridged review requires the previous approval of a medicine by at least one reference agency and the review centres on aspects of the medicine that must be assessed in the local environment. This type of review is often limited to an assessment of the country-specific requirements for product quality and the clinical data associated with the local benefit risk assessment of the product. Type 3 or full reviews consist of a complete review of a medicine’s quality, safety (pre-clinical), and efficacy (clinical) data, with type 3A also requiring previous approval by a reference agency, which is not a requirement for type 3B [11]. The GCC-DR conducts type 3A reviews for all major applications (NASs and major line extensions). A priority review track is in place for vaccines and anti-carcinogenic therapies. However, if a product has been approved by two Gulf States or by the United States Food and Drug Administration (US FDA), European Medicines Agency (EMA) or the WHO prequalification (as reference agencies) then the GCC-DR will conduct an abridged review. If a product is registered in all member states, then the GHC will conduct a verification review, which is normally achieved within 21 days.
3.2 Data Requirements
A Certificate of Pharmaceutical Product (CPP) is required before final authorisation and no other documentation is accepted as evidence of registration. However, consideration should be given to no longer requiring a formal CPP but substituting alternative ways of verifying registration in the reference countries such as letters of authorisation or Internet reference. A full ICH electronic common technical document (eCTD) is required, including full non-clinical and clinical data (Modules 3, 4, 5).
3.3 Key Points in the GCC-DR Review Process
-
1.
Validation of marketing authorisation applications consists of a verification of legal, good manufacturing processes, patent and intellectual property status, confirmation of an acceptable format and content of the application and payment of fees.
-
2.
Queueing for the scientific assessment can take 60–180 calendar days, although priority products may be taken out of the queue to be the subject of an expedited review.
-
3.
All member states receive dossier files and have the option to review, but two member states are specifically named by the Expert Committee to review the files for quality, safety and efficacy in parallel and provide their recommendations to the GCC-DR.
-
4.
Questions to the sponsor are batched and there is a target time of 180 calendar days for the sponsor to respond to queries.
-
5.
The Expert Committee provides a peer review of the dossier and issues its recommendation, which the GHC is mandated to follow. However, authorisation does not depend on pricing agreements.
-
6.
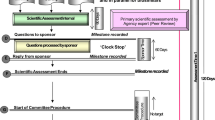
There are no target times for validation, scientific assessment, or Expert Committee evaluation, but there is an overall review target time of 365 days for a full review and 120 days for priority review and this includes both agency and manufacturer’s response time (Fig. 1).

Gulf Centralised Committee for Drug Registration procedure process map and milestones, simplified to show when an application goes to approval in one cycle
3.4 Number of Product Approvals
Over the period 2015–2020 (Fig. 2), the fewest NASs (4) were approved in 2016 (4) and the most (13) in 2019; while the fewest generics (45) were approved in 2019 and the most (189) in 2020. The substantial increase in the number of medicines approved in 2020 was due to several factors, including the introduction of a significant digital transformation for all stakeholders, which included a shared file storage system in which applicants’ dossiers were easily accessible by all member states, resulting in a faster review process for new and pending applications.

Total number of medicines approved by the Gulf Centralised Committee for Drug Registration 2015–2020. NAS new active substance
3.5 Review Times
Review times also significantly decreased with the implementation of a reliance strategy by the end of 2019. The average approval time for generics and NASs decreased from 838 calendar days in 2015 to 321 calendar days in 2019 and 411 calendar days in 2020 (Fig. 3). In addition, when products were registered in two Gulf States, an abridged review was conducted in an average review time of 61 calendar days and when products were registered in all member states, a verification review was conducted in an average of 21 calendar days. However, the product cannot be the subject of a GCC joint procurement unless it has a centralised certificate. Reasons for the reduction in review times included an increase in the number of committee meetings. In 2019, the number of drug registration committee meetings was increased to once a month by teleconferencing in addition to a face-to-face regulatory meeting every quarter, for a total of 16 meetings per year. Prior to that, there were only 5 committee meetings in 2017 and 10 meetings in 2018.

Average approval time for all new active substances and generics approved by the Gulf Centralised Committee for Drug Registration by approval route 2015–2020
Moreover, the following pharmaceutical legislation and regulatory procedures were implemented as part of the principle of reliance:
-
Products registered in the six GCC member states are directly approved (this indicates that the registration department of the GCC will be responsible for issuing the registration certificate). Products registered in two or more GCC member states are registered and the registration certificate is issued after 60 days, unless observations or comments from GCC member states are received on the reports issued. The review process involves a comparative assessment between the peripherally submitted dossiers and the centrally submitted ones, alongside a validation of submitted certificates.
-
If a product is registered in two or more GCC states, then this means that the GHC can issue a product certificate whilst the manufacturer is still undergoing the process centrally.
-
The inspection visit to the manufacturer is bypassed if a visit has been made by one of the member states to the manufacturer within the past 2 years and no issues have been raised.
-
GCC member states rely on approvals issued by the GHC for variation requests of pharmaceuticals products, where the company has the right to start implementing the approved variation in the GCC countries, and the company must provide member states with product files according to the life cycle of the registration of the product if they differ from that presented centrally (eCTD sequence).
Electronic communication with applicants and member states has also expedited the review process for new and pending application requests, also resulting in reduced approval times. Altogether, these factors led to the reduction in approval times seen in 2019 and 2020.
Prior to the implementation of reliance, approval times had been reduced when member states began to depend on their own scientific reports for the national procedure, but this practice was replaced by reliance at the end of 2019.
3.6 Good Review Practices
The importance of establishing good review practices (GRevPs) is recognised by the GHC and these have been implemented in the GCC-DR review; they include standard operating procedures (SOPs) for the guidance of scientific assessors, the use of a standardised assessment templates, shared/joint reviews within the Gulf Region, and a formal training programme for assessors as well as an electronic tracking system for monitoring the progress of applications. However, some GRevPs, such as an internal quality policy, SOPs for the guidance of advisory committees and internal or external peer review, remain to be implemented.
To bring about continuous improvement in the assessment process, GCC-DR assesses the feedback of reviewers and other stakeholders and takes necessary action, monitors the progress of applications through an electronic internal tracking system and carries out training through both internal and external courses, on-the-job learning and participation in external workshops. Training is tested through examinations and is required for professional advancement. In addition, the assessment report (AR) is shared with other regulatory authorities if requested and the sponsor also get a copy of the full AR. However, currently the ARs are not released to the public.
3.7 Decision-Making Frameworks
The GHC has a systematic structured approach to decision making, with clearly assigned roles and responsibilities. They assign values and relative importance to decision criteria, evaluate both internal and external influences/biases, re-evaluate decisions as new information becomes available and effectively communicate to the pharmaceutical company the basis of the decision.
4 Discussion
Since the 2014 study of the GCC Central Process [8], several important recommended changes have been implemented. Sample laboratory analyses, which were formerly carried out sequentially after the evaluations of quality, safety, and efficacy are now performed in parallel, which saves time. The number of GCC-DR meetings has been increased to once a month by teleconferencing, while every quarter there is a face-to-face regulatory meeting. A standardised electronic common technical document format has been adopted and a standardised template is now used by all GCC countries for their regulatory review, while product authorisations are no longer dependent on pricing agreements.
As a result of these enhancements, the average approval time for all new medicines through the Gulf Centralised Procedure decreased from the average of 838 calendar days in 2015 to 321 days in 2019 and 411 days in 2020 (Fig. 3), despite the considerable increase in the number of products reviewed (Fig. 2). However, the major reason for the improvement in 2019–2020 was the implementation of a reliance strategy in which account was taken of the reviews by reference agencies. In the GCC, when products were previously registered in two of the Gulf States, the review time was significantly reduced to 61 calendar days. The average review of new active substances also showed an improvement from 1180 calendar days in 2016 to 182 days in 2019 (Fig. 4) and again, this improvement was due to the reliance strategy that was put in place.

Average approval time for all new active substances approved by the Gulf Centralised Committee for Drug Registration by approval route 2015–2020
As part of their own national review processes, the seven GCC countries all provide a full (type 3) review; however, when the medicine submitted to the GCC is identical to that approved by two reference agencies, the GCC conducts an abridged review [12,13,14]. Verification or abridged reviews are of particular value as the number of submissions to GCC-DR has continued to increase, to a high of 200 submissions in 2020. Where a Gulf State has obtained a national approval based on reliance and secured an assessment report from the reference agency then the GHC is responsible for communicating this to other GCC member states.
Currently, the GCC centralised process does not include the identification of target times for dossier validation, scientific review, or Expert Committee recommendation of new drug applications. As the regulatory processes of the GHC evolve, documenting performance against targets would help to assess where time is spent and identify areas where improvement is needed. For this reason, the WHO Global Benchmarking Tool includes the proactive and consistent measurement of regulatory agency performance against stated target times [15]. Furthermore, although the reviewer’s questions to the sponsor regarding applications are batched at fixed points in the review process, the time companies take to respond to these questions is not recorded and this response time therefore cannot be measured and differentiated in the overall processing time.
The GHC stated that the three most important reasons to introduce quality into the review process are to increase efficiency, improve process predictability and enhance transparency. Indeed, the GHC puts a high priority on building quality into its processes and has measures in place to monitor and improve quality. However, GHC GRevPs do not yet include an internal quality policy, SOPs for advisory committees or an internal or external peer review. Furthermore, scientific review templates are not based on the templates of other agencies and there are no SOPs for how to complete the assessment reports. Assessment reports themselves are shared with other regulatory authorities as well as sponsors but are not yet published on the GHC website.
The importance of implementing quality decision-making practices is underlined in the WHO Global Benchmarking Tool. Awareness of biases inherent in decision making and the ongoing evaluation of decision-making practices can enhance efficiency and outcome and enable predictability, consistency and timeliness in evaluations [16]. However, there are no measures in place to minimise the impact of subjective influences/biases on GHC decision making and no formal assessment takes place to periodically measure the quality of decision making within the agency; there is a lack of training in the area of quality decision making.
The relationship between the national and centralised procedure is a two-way process. When a recommendation is made from the centralised process to the National Medicines Regulatory Agency (NMRA), then the NMRA will not re-evaluate the application but will review it for a pricing decision. However, when a decision is passed from the National to the centralised procedure, it requires two GCC member states for it to be recommended for marketing in all member states. In the future it is hoped that the process of reliance will be transferred to other sectors such as the registration of veterinary medical devices. Once a new medicine receives the central registration approval, then the pharmaceutical company can market the medicine once it has been granted a national certificate, which will include the price of the product. Whilst it is recognised that the website should contain practical information such as actual approval review procedures as well as new drug approvals, at the present time this is addressed through workshops and interactions with the industry.
4.1 Recommendations
To further enable the successful ongoing evolution of the GCC Central process, several actions are recommended:
-
1.
Performance measurement In order to effectively benchmark the regulatory review process and monitor performance, the GHC should consider recording the regulatory timelines for each of these milestones as well as setting targets. Measuring regulatory metrics should be carried out so that there is a differentiation in the review time between the agency and the manufacturer’s response time. In addition, practices that accelerate or delay marketing authorisation should include a focus not only on products approved but on all products assessed.
-
2.
Quality decision making the GHC should consider establishing a formal assessment of decision making as well as implementing a training programme in this important area.
-
3.
Increased transparency the GHC should consider publishing approval times and a summary basis of the approval decision for products on their website.
-
4.
Good review practices Whilst several GRevPs are implemented in the GHC review process, consideration should be given to implementing an internal quality policy, SOPs for the guidance of the advisory committee and the establishment of an internal or external peer review.
5 Conclusions
The objectives of this study were to update the information on the GCC Centralised Procedure and appraise the GCC centralised regulatory review process; evaluate the review times for new products, including generics and NASs submitted to the GCC Centralised Registration between January 2015 to December 2020; assess the impact of applying FRPs and implementing a reliance strategy; identify the strengths and weaknesses of the centralised review process; and propose strategies that could enhance the GCC regulatory review process.
Since the 2015 review by Al Rubiae and colleagues [7], key recommended changes have been implemented in the GCC Centralised Process. Sample laboratory analyses are now performed in parallel with evaluations of quality, safety and efficacy, the number of GCC-DR meetings has been increased, a standardised electronic common technical document format and standardised template are now used for regulatory review and product authorisations are no longer dependent on pricing agreements. The average approval time for all new medicines through the Gulf Centralised Procedure decreased from the average of 838 calendar days to 321 days in 2019 and 411 days in 2020. While electronic communication with applicants and member states is one factor in these reduced approval times, the major reason for the shortened approval times was the implementation of a reliance strategy. Strategies for improvement of observed process limitations include the development of performance management metrics, a formal decision-making framework and a programme of increased transparency as well as the implementation of an internal quality policy, SOPs for the guidance of the advisory committee and the establishment of an internal or external peer review.
References
Pignatti F, Boone H, Moulon I. Overview of the European regulatory approval system. J Ambul Care Manage. 2004;27:89–97. https://doi.org/10.1097/00004479-200404000-00003.
Sithole T, Mahlangu G, Salek S. Evaluating the success of ZaZiBoNa, the Southern African development community collaborative medicines registration initiative. Ther Innov Reg Sci. 2020;54:1319–29. https://doi.org/10.1007/s43441-020-00154-y.
Preston C, Chahal HS, Porrás A, Cargill L, Hinds M, Olowokure B, Cummings R, Hospedales J. Regionalization as an approach to regulatory systems strengthening: a case study in CARICOM member states. Rev Panam Salud Publica. 2016;39:262–8.
World Population Review. GCC Countries. 2021. https://worldpopulationreview.com/countries/gcc-countries/. Accessed 4 Feb 2022.
Country Economy.com. Country groupings GCC – Cooperation Council for the United States of the gulf. https://countryeconomy.com/countries/groups/cooperation-council-arab-states-of-the-gulf. Accessed 4 Feb 2022.
Stevens IL. The GCC demographic shift. Arab Health. 2017;5. https://www.arabhealthonline.com/magazine/en/latest-issue/Issue-5/The-GCC-Demographic-Shift.html
Al-Rubaie MR, Salek SS, Walker SR. An evaluation of the efficiency of the Gulf Cooperation Council’s Centralized Procedure by the Gulf Regulatory Authorities and Pharmaceutical Companies: recommendations for an improved model. Ther Innov Reg Sci. 2015;49:560–8. https://doi.org/10.1177/2168479015570336.
Al-Rubaie MR, Walker SR, Salek SS. An evaluation of the Gulf Cooperation Council centralized procedure: the way forward. Ther Innov Reg Sci. 2014;48:709–16.
Hashan H, Aljuffali I, Patel P, Walker S. The Saudi Arabia Food and Drug Authority: an evaluation of the registration process and good review practices in Saudi Arabia in comparison with Australia, Canada and Singapore. Pharm Med. 2016;30:37–47.
WHO (2021) WHO Expert Committee on Specifications for Pharmaceutical Preparations: Fifty-fifth report (WHO Technical Report Series, No. 1033). Annex 10—Good reliance practices in the regulation of medical products: high level principles and considerations. https://apps.who.int/iris/bitstream/handle/10665/340323/9789240020900-eng.pdf. Accessed 25 May 2021.
McAuslane N, Cone M, Collins J, Walker S. Emerging markets and emerging agencies: a comparative study of how key regulatory agencies in Asia, Latin America, the Middle East and Africa are developing regulatory processes and review models for new medicinal products. Drug Info J. 2009;43:349–59.
Al-Essa R, Salek S, Walker S. An appraisal of good regulatory review practices in the Gulf Cooperation Council States. Drug Inf J. 2012;46:57–64.
Al-Essa R, Salek S, Walker S. Regulatory review process in the Gulf Cooperation Council States: similarities and differences. Drug Inf J. 2012;46:65–72.
Chehimi I. The regulatory reliance review model: adoption in the Middle East. Glob Forum. 2019.https://globalforum.diaglobal.org/issue/july-2019/the-regulatory-reliance-review-model-adoption-in-the-middle-east/
World Health Organization. WHO Global Benchmarking Tool (GBT) for evaluation of national regulatory systems. 2019 https://www.who.int/medicines/regulation/benchmarking_tool/en/. Accessed 3 July 2019.
Bujar M, Donelan R, McAuslane N, Walker S, Salek S. Assessing the quality of decision making in the development and regulatory review of medicines: identifying biases and best practices. Ther Innov Regul Sci. 2017;51(2):250–6. https://doi.org/10.1177/2168479016662681.
Acknowledgements
The authors would like to thank the Gulf Health Council in Riyadh for providing access to the data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This research and its open access publication was supported by an unrestricted grant from the Bill and Melinda Gates Foundation.
Declaration of conflicting interests
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Data are available upon request to corresponding author.
Code availability
Not applicable.
Author Contributions
HH: Provided the data and critically reviewed the manuscript. SA: Provided data and completed some of the analyses. IA: Provided the data and critically reviewed the manuscript. OA: Provided data and completed some of the analyses. ZA: Provided data and completed some of the analyses. NM: Devised the study, analysed the data and critically reviewed the manuscript. SW: Devised the study, analysed the data and wrote the manuscript. All the authors confirm that they have read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Hashan, H.M., Al-Muteb, S.K., Alismail, I.A. et al. Evaluation of the Performance of the Gulf Cooperation Council Centralised Regulatory Review Process: Strategies to Improve Product Authorisation Efficiency and Quality. Pharm Med 36, 223–231 (2022). https://doi.org/10.1007/s40290-022-00432-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40290-022-00432-0