
Abstract
Background
Pulmonary hypertension (PH) is a severe hemodynamic condition with high morbidity and mortality. Approved targeted therapies are limited for pediatric subjects, and treatments are widely adopted from adult algorithms. Macitentan is a safe and effective drug used for adult PH, but data on pediatric patients are limited. In this prospective single-center study, we investigated mid- and long-term effects of macitentan in children with advanced pulmonary hypertensive vascular disease.
Methods
Twenty-four patients were enrolled in the study for treatment with macitentan. Efficacy was determined by echo parameters and brain natriuretic peptide levels (BNP) at 3 months and 1 year. For detailed analysis, the entire cohort was subgrouped into patients with congenital heart disease-related PH (CHD-PH) and non-CHD-PH patients, respectively.
Results
Mean age of the patients was 10.7 ± 7.6 years; median observation period was 36 months. Twenty of 24 patients were on additional sildenafil and/or prostacyclins. Two of 24 patients discontinued because of peripheral edema.
Within the entire cohort, BNP levels and all echo measures such as right ventricular systolic pressure (RVSP), right ventricular end-diastolic diameter (RVED), tricuspid annular plane systolic excursion (TAPSE), pulmonary velocity time integral (VTI), and pulmonary artery acceleration time (PAAT) improved significantly after 3 months (p ≤ 0.01), whereas in the long term significant improvement persisted for BNP levels (−16%), VTI (+14%) and PAAT (+11%) (p < 0.05). By subgroup analysis, non-CHD PH patients showed significant improvements in BNP levels (−57%) and all echo measures (TAPSE +21%, VTI +13%, PAAT +37%, RVSP −24%, RVED −12%) at 3 months (p ≤ 0.01), whereas at 12 months, improvements persisted (p < 0.05) except for RVSP and RVED (nonsignificant). In CHD-PH patients, none of the measures changed (nonsignificant). 6-MWD (distance walked in 6 minutes) slightly increased but was not statistically evaluated.
Conclusion
Data presented herein account for the largest cohort of severely affected pediatric patients receiving macitentan. Overall, macitentan was safe and associated with significant beneficial effects and sustained positive signals after 1 year, albeit in the long term disease progression remains a major concern. Our data suggest limited efficacy in CHD-related PH, whereas favorable outcomes were mainly driven by improvements in patients with PH not related to CHD. Larger studies are needed to verify these preliminary results and to prove efficacy of this drug in different pediatric PH entities.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
This study investigates the outcomes of children with pulmonary hypertension treated with macitentan, a second-generation endothelin receptor antagonist, which is currently part of adult pulmonary arterial hypertension (PAH) treatment algorithms. |
We used macitentan in pediatric patients with advanced disease progression and analyzed clinical course, echocardiographic changes, and BNP levels over a 1-year observational period. |
Treatment was safe and effective in attenuating disease progression; CHD-PH patients appear to benefit less than those with other PH entities. |
1 Introduction
Pulmonary hypertension (PH) is a severe condition caused by progressive functional and structural abnormalities in the pulmonary vasculature. The hemodynamic consequences include a continuous increase of right ventricular (RV) afterload by elevated pressure/resistance leading to right heart failure over time. In childhood, PH may be idiopathic or heritable (iPAH, hPAH) or may occur in the context of various conditions—such as lung disease, congenital heart defects, or other disorders [1,2,3,4].
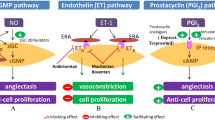
The term pulmonary hypertensive vascular disease (PHVD) was established in 2011 by the pediatric taskforce and highlights the common pathology of pulmonary vascular remodeling independent of associated underlying diseases [5]. Pathogenetically PHVD results from imbalances of the three main biological molecules nitric oxide (NO), prostacyclins (PGI), and endothelins (ET) leading to dysregulation of the pulmonary vascular tone defined by a lack of vasodilation and an increase of proliferation and local inflammation [6, 7]. Current treatment options exert their beneficial effects by influencing the pathways of these substances and have led to improved quality of life and survival of adult and pediatric patients with PAH [3, 8, 9]. Throughout the last two decades several newer substances became available and have been approved for PAH treatment, mainly based on larger studies in adults. Although these substances have been shown to be as beneficial in children, controlled pediatric studies are limited due to the smaller number of patients and the higher heterogeneity of PHVD [10]. Therefore, most pediatric treatment algorithms are currently adopted from adult guidelines and/or based on expert opinions [3, 11].
Macitentan, a second-generation endothelin receptor antagonist (ERA), is one of the promising newer substances and has been approved for adult PAH treatment in 2013 [12, 13].
Compared with its precursor bosentan it has improved biochemical properties favoring sustained receptor binding and higher tissue penetration [14, 15]. Clinically, it has the advantage of exhibiting fewer hepatic side effects and interactions with other drugs [14]. It has become a part of standard treatment in adults [4, 12].
Its use in children is currently investigated by a large randomized multicenter open-label study (TOMORROW study AC-055-312); its results will hopefully lead to approval of macitentan. However, the character of life-limiting disease often forces pediatricians to use non-approved drugs such as prostacyclins and other substances commonly used in adults to attenuate disease progression and to save time before more invasive treatment strategies such as reversed Potts shunt, balloon atrioseptostomy (BAS), or lung transplantation become necessary. During the past few years, there has been increasing promising experience with macitentan in children [16,17,18,19]. We recently reported on practical aspects and safety issues when using macitentan in pediatric patients and now present mid-term effects in children and adolescents with severe PHVD [20].
2 Methods
2.1 Study Design
This is a prospective single-center observational study conducted at the Division of Pediatric Cardiology at the Medical University of Vienna. Macitentan was used as compassionate therapy in 24 pediatric patients with PHVD. Informed consent was obtained from all patients and their respective caregivers before treatment initiation. The study was approved by the local ethics committee (local ethics number 1619/2018).
2.2 Study Endpoints
Primary endpoints of the study were changes of echo parameters such as estimated right ventricular systolic pressure (RVSP), right ventricular end diastolic diameter (RVED), tricuspid annular plane systolic excursion (TAPSE), pulmonary artery acceleration time (PAAT), and pulmonary velocity time integral (VTI), as well as measurement of brain natriuretic peptide (BNP) levels at baseline (before treatment initiation), 3 months and 1 year after treatment initiation (Fig. 1).

Flowchart of the study design with an overview on number of included patients, endpoints, macitentan treatment, and follow-up. BNP brain natriuretic peptide, BPD bronchopulmonary dysplasia, CHD congenital heart disease, PAH pulmonary arterial hypertension, NYHA FC New York Heart Association functional class, PAAT pulmonary artery acceleration time, RVED right ventricular end-diastolic diameter, RVSP right ventricular systolic pressure, 6-MWT 6-min walking, TAPSE tricuspid annular plane systolic excursion, VTI velocity time integral
To meet the pathophysiological differences resulting from different PAH entities, we performed additional subgroup analysis by separating patients with CHD-related PHVD (n = 10) from the others (non-CHD group, n = 12).
Secondary endpoints were changes of heart failure functional class (FC) according to New York Heart Association (NYHA) or modified Ross classification in younger children [21]. In patients able to perform the 6-min walking test (6-MWT), we assessed change of walking distance and saturation levels at the beginning and the end of walking as an additional efficacy parameter.
Efficacy was determined at 3 months and after 1 year. In patients with clinical deterioration and subsequent treatment escalation or further interventions during the study period, the most recent measures prior to escalation of therapy were taken for analysis. The first follow-up visit for assessment of efficacy was 3 months after treatment initiation. Safety was evaluated after 4 weeks and then every 3 months (Fig. 1).
2.3 Patients and Inclusion/Exclusion Criteria
Patients diagnosed with PHVD on the basis of measurements by transthoracic echocardiography and/or invasive catheterization were included. Classification of pulmonary hypertension (PH) was based on the consensus paper reported by the pediatric taskforce of the Pulmonary Vascular Research Institute (PVRI) (Panama Paper) and the updated classification proposed by the 6th World Symposium on Pulmonary Hypertension [5, 12]. Demographic data, PHVD entity, and ongoing treatment were assessed in all patients.
Neonates (< 1 month of age) as well as patients with postcapillary PH were excluded from the study.
2.4 Treatment
Different starting doses depending on body weight were applied as a single daily dose.
-
Infants with a body weight below 10 kg received a starting dose of 1 mg of macitentan, which was increased daily in 1 mg steps up to a maximum dose of 3 mg per day or a decrease of blood pressure, defined as a drop of > 15 % of systolic value and > 10 % of diastolic value compared with baseline [22].
-
Children with a body weight between 10 and 15 kg started with 3 mg of macitentan to reach a target dose of 5 mg (daily uptitration mode: 3–4–5 mg).
-
In children with a body weight between 15 and 20 kg, the starting dose was 3 mg, and this was increased to a maximum of 7.5 mg (uptitration mode: 3–5–7.5 mg).
-
Adolescents and all children with a body weight above 20 kg received 5 mg as starting dose. While older children were rapidly increased to a target dose of 10 mg (5–7.5–10 mg), children with a body weight between 20 and 30 kg remained on 7.5 mg until the first follow-up. They were then increased to the target dose of 10 mg, provided there was no significant drop of blood pressure or other side effects [22].
For dose adjustment in children with lower body weight, we used macitentan capsules containing lactose monohydrate as carrier substance. Older patients requiring higher doses received the original tablets (10 mg). Patients were titrated to target doses according to weight within a maximum of 1 week. If patients were switched from bosentan to macitentan, the latter was initiated 24 h after the last bosentan intake.
The first ten patients and all infants below 10 kg of body weight or under 2 years of age were started on macitentan in hospital. Measurement of saturation, blood pressure, and heart rate monitoring were performed daily for the first five days, either during hospital stay or in our outpatient clinic or by a caring nurse or physician and at each follow-up visit at our outpatient clinic.
2.5 Outcome Parameters
For assessment of efficacy, transthoracic echocardiography was performed by an experienced pediatric cardiologist, using a commercially available echocardiographic system (GE Vivid™ Cardiovascular Ultrasound System). Images were recorded digitally and analyzed using off-line software (Echo PAC software™). To obtain reliable values, we determined the average of three measurements for each echo variable. All parameters were determined by two blinded certified pediatric cardiologists.
-
Tricuspid annular plane systolic excursion (TAPSE, mm) as a surrogate for RV function was measured by M-mode in the apical four-chamber view [23, 24].
-
Pulmonary velocity time integral (VTI) and PA acceleration time (PAAT) as surrogates for pulmonary artery (PA) compliance and resistance were obtained from the RVOT pulse wave Doppler profile in the parasternal short-axis view [25,26,27]. The transducer position was adjusted to open the RVOT, with the sample volume placed proximal to the pulmonic valve. PAAT is the interval in milliseconds from the onset of ejection to the peak flow velocity. VTI (cm) is obtained by manually tracing the obtained Doppler spectrum. The pulmonary VTI values are measured by the built-in calculation package of the ultrasound unit and are equal to the area enclosed by the baseline and Doppler spectrum.
-
Right ventricular end-diastolic diameter (RVED, mm) reflecting RV load was obtained by M-mode at the parasternal long-axis view [28].
-
Right ventricular systolic pressure (RVSP) as an estimate for systolic PA pressure (sPAP) in the absence of right ventricular outflow tract obstruction or pulmonary valve/artery stenosis was assessed by measuring tricuspid regurgitation (TR) jet in the apical four-chamber view and was calculated by applying the modified Bernoulli equation. In the absence of TR, RVSP was derived from estimated mean PA pressure (mPAP) assessed by pulmonary regurgitation (PR) jet (measured from the short-axis view) using the following calculation: mean pulmonary artery pressure (mPAP) = 0.61 sPAP + 2 mmHg [29, 30]. RA pressure was not obtained and must be added to the RVSP values for estimation of systolic PA pressure. RVSP was not analyzed for the entire patient cohort as this measure was not reliably obtainable in patients with CHD-associated PHVD.
Furthermore, we assessed BNP levels as a surrogate for heart function in all patients. FC was determined by two independent pediatric cardiologists. 6-MWT—as an additional efficacy parameter—was performed in all patients capable to walk. As to our institutional protocol, oxygen saturation (SpO2) is continuously measured while walking until the end of the test.
For assessment of safety, laboratory tests including liver function parameters [serum glutamic oxaloacetic transaminase (SGOT), serum glutamate-pyruvate transaminase (SGPT), gamma glutamyl transferase (GGT), alkaline phosphatase (AP)], kidney parameters [creatinine (Crea) and uric acid (UA)], and blood cell count [hemoglobin (Hb), hematocrit (Hkt), white blood cells (WBC), and platelets (PLT)] were measured before, 4 weeks after initiation of treatment, and subsequently every three months on the targeted dosage of the drug.
2.6 Statistics
Data are reported as descriptive analysis. Continuous parameters were described as median with range. Categorical variables were displayed as frequencies. Differences between baseline, 3-month, and 1-year echo and laboratory parameters were tested with the nonparametric Wilcoxon test, and mixed-model regression analysis was done. Correlation was evaluated with Spearman’s rank correlation coefficient. Statistical significance was determined as p < 0.05, and the Holm–Bonferroni was used when applicable. Statistical tests were performed by using IBM SPSS Statistics for Windows, version 28 (IBM Corp., Armonk, NY, USA).
3 Results
Twenty-four pediatric patients (14 male, 10 female) were enrolled for treatment with macitentan. PHVD was classified according to PANAMA criteria [5] as either isolated idiopathic PAH (n = 7), associated with congenital heart defects (CHD) (n = 10), as developmental pulmonary vascular hypertensive disease (n = 2), associated with bronchopulmonary dysplasia (n = 2) and associated with other systemic disorders (n = 2) or CTEPH (n = 1). Detailed demographic data are listed in Table 1.
Mean age of the included patients was 10.7 ± 7.6 years (range 0.1–23 years). Median observation period was 36 months (interquartile range 15–43 months).
Macitentan was started as monotherapy in six patients; two out of six patients were switched from bosentan, while the remaining four received macitentan as first pharmacological agent. In ten patients, macitentan was used as part of a dual therapy in addition to an ongoing treatment with either PDE-Is, soluble guanylate cyclase stimulator, or PGI. Eight patients were on a combined treatment with PDE-Is in addition to either inhalative or intravenous prostacyclines, when macitentan was initiated (triple therapy). Eighteen of 24 patients received conventional baseline therapy with diuretics, iron supplements, anticoagulation, and/or digoxin (Table 1).
As to NYHA/modified Ross score, 12 patients (pts) were assigned to FC 2, 10 pts to FC 3, and 2 pts to FC 4.
The median estimated sPAP at treatment initiation was 78 mmHg (44–155 mmHg), the median BNP value was 375 pg/ml (30–14,078 pg/ml). Accordingly, 18/24 patients presented with suprasystemic (9) and systemic PA pressures (9) respectively, while the remaining 6 patients had subsystemic PA pressures (Table1).
Two of 24 discontinued the study because of side effects with symptomatic peripheral edema during the first two weeks after initiation, whereas macitentan was well tolerated in the remaining patients. These two patients were excluded from efficacy analysis. Tolerability and side effects of this substance was reported in detail in our previous work [20].
3.1 BNP levels
After initiation of macitentan median BNP levels (pg/ml) dropped from 375 (30–14,078) at baseline to 152 (32–6456; 22%, p = 0.03) after 3 months and to 153 (40–3920) after 1 year (baseline to 1 year: −16%; p < 0.05).
Subgroup analysis revealed significant improvement of BNP levels only for the non-CHD group with a median baseline value of 408 (30–14,078) decreasing to 143 (32–376) after 3 months (p = 0.03) and to 145 (40–934) after 1 year (p = 0.03). Among patients with CHD no significant changes were found (Fig. 2, Table 2).

a Median BNP levels (pg/ml) for the entire cohort before treatment (baseline), after 3 months and after 1 year of treatment. Decrease of median BNP levels from 375 to 152 at 3 months (p = 0.03) and to 153 at 1 year (p < 0.05). b Percent change of BNP levels according to patient groups (CHD-related PH, non-CHD-related PH): from baseline to 3 months (Δbl-3mo), from 3 months to 1 year (Δ3mo-1y) and from baseline to 1 year (Δbl-1y). Note improvement between bl-3mo and bl-1y with no change/worsening between 3 months and 1 year. BNP, brain natriuretic peptide; CHD, congenital heart disease; PH, pulmonary hypertension
3.2 Echo Parameters
TAPSE After initiation of macitentan, the longitudinal systolic RV function, as assessed by TAPSE (mm) improved from median 13 (7–21) to 16 (7–24) after 3 months (+11%; p = 0.001) and to 15 mm (9–24 mm) after 1 year (baseline to 1 year: ns). Similarly, z-scores changed from −4.2 to −3.4 after 3 months (bl-3mo p = 0.0002) and to −3.66 after 1 year (ns). In the non-CHD group, TAPSE values increased significantly from median 14 (9–19) to 16 (10–21; p = 0.005) after 3 months and to 15 after 1 year (10–18; bl-1y: p < 0.05). Similar improvements were found for calculated z-scores (bl-3mo: p = 0.0004, bl-1y: p = 0.03). In the CHD group none of the values changed (Fig. 3, Table 2).

a Median TAPSE levels (cm) for the entire cohort before treatment (baseline), after 3 months and after 1 year of treatment. Increase from median 13 to 16 after 3 months (p = 0.001) and to 15 mm (9–24 mm) at 1 year (bl-1y: ns). b Percent change of TAPSE as to patient groups (CHD-related PH, non-CHD-related PH): from baseline to 3 months (Δbl-3mo), from 3 months to 1 year (Δ3mo-1y) and from baseline to 1 year (Δbl-1y). Significant increase in the non-CHD group (bl-3mo; bl-1y) with worsening between 3 months and 1 year. CHD congenital heart disease, TAPSE tricuspid annular plane systolic excursion, PH pulmonary hypertension
Pulmonary VTI Within the entire cohort there was a significant increase of median pulmonary VTI (cm) from 15 (9–51) at baseline to 18 (10–59) after 3 months (+13%; p = 0.002) and to 16 (11–57) after 1 year (bl-1y: +14%; p = 0.03).
Whereas in the non-CHD group significant improvement was demonstrated between baseline (14; 9–18) and 3 months (17; 5–22, p = 0.005) and 1 year (15; 11–22, p = 0.04), no significant changes could be demonstrated for the CHD group (Fig. 4, Table 2).

a Median pulmonary VTI levels (cm) for the entire cohort before treatment (baseline), after 3 months and after 1 year of treatment. Increase from median 15 at baseline to 18 after 3 months (p = 0.002) and to 16 at 1 year (p = 0.03). b Percent change of VTI from baseline to 3 months (Δbl-3mo), from 3 months to 1 year (Δ3mo-1y) and from baseline to 1 year (Δbl-1y) as to patient groups (CHD related PH, non-CHD-related PH). Note improvement between bl-3mo and bl-1y with no change/worsening between 3 months and 1 year. CHD congenital heart disease, VTI velocity time integral, PH pulmonary hypertension
PAAT Overall, PAAT (ms) increased significantly from median 56 (38–90) at baseline to 66 (43–90; +9%; p = 0.003) after 3 months and to 69 (41–104) after 1 year (bl-1y: +11%; p = 0.01).
In the non-CHD group, there was a similar significant increase of median PAAT from 43 (38–81) at baseline to 64 (43–90) after 3 months (+37%; p = 0.002) and to 63 (41–92) after 1-year follow up (+25%; p = 0.02). Again, in the CHD group no significant changes were observed (Fig. 5, Table 2).

a PAAT levels (ms) for the entire cohort before treatment (baseline), after 3 months and after 1 year of treatment. Increase from median 56 at baseline to 66 after 3 months (p = 0.003) and to 69 after 1 year (p = 0.01). b Percent change of PAAT from baseline to 3 months (Δbl-3mo), from 3 months to 1 year (Δ3mo-1y) and from baseline to 1 year (Δbl-1y) as to patient groups (CHD-related PH, non-CHD-related PH). Note improvement between bl-3mo and bl-1y with no change/worsening between 3 months and 1 year. CHD congenital heart disease, PAAT pulmonary artery acceleration time, PH pulmonary hypertension
RVED RVED (mm) decreased from median 26 (16–43) at baseline to 24 (12–45) after 3 months (p = 0.02); after 1 year, RVED returned to a median of 25 (13–50; bl-1y: ns). These results were mirrored by the changes of z-scores (z-bl: +1.6, z-3mo: +1.2, z-1y: +1.2). In the non-CHD group, there was significant reduction from baseline (24; 12–35) to 3 months (22; 12–35, p = 0.01), whereas after 1 year reduction did not reach statistical significance (22; 13–42). In contrast, z-score calculation showed statistically significant improvements at both timepoints (z-bl: +1.4, z-3mo: +0.8; z-1y:+0.9; p < 0.02). In the CHD group neither absolute values nor z-scores showed significant changes (Table 2).
RVSP RVSP as a non-invasive estimate of systolic PA pressure was assessed only in the non-CHD group. There was a decrease from median 69 mmHg (47–101 mmHg) at baseline to 61 mmHg (33–85 mmHg) after 3 months (p = 0.01), which equals 24% change of difference. Decrease of RVSP to 53 mmHg (29–107 mmHg) after 1 year was not significant (Fig. 6, Table 2).

a Median RVSP levels (mmHg) in patients with non-CHD related PH before treatment (baseline), after 3 months and after 1 year of treatment. Decrease from median 69 at baseline to 61 after 3 months (p = 0.01) and to 53 at 1 year (ns). b Percent change of RVSP from baseline to 3 months (Δbl-3mo), from 3 months to 1 year (Δ3mo-1y) and from baseline to 1 year (Δbl-1y) (non-CHD-related PH). Note improvements between bl-3mo and bl-1y but worsening between 3 months and 1 year. CHD congenital heart disease, PH pulmonary hypertension, RVSP right ventricular systolic pressure
3.3 6-MWT and Oxygen Saturation
Twelve of 22 pts were old enough and mentally able to perform 6-MWT (5 non-CHD/7 CHD group). The mean change of walking distance was 21 m after 3 months and 28 m after 1 year for the entire cohort.
In the CHD group the walked distance was less, with 390 m at baseline (versus 430 m in the non-CHD group). After 3 months it increased by 13 m and dropped again to 388 m after 1 year.
In these patients mean oxygen saturation levels at rest were 87% and increased to 89% after 3 months but dropped again to 87% after 1 year. We also assessed “exercise” oxygen saturation levels, measured at the end of 6-MWT, levels were 66% at baseline and increased to 73% after 3 months but finally dropped again to 70%. In the non-CHD group only five patients performed 6-MWT and distance increased from 430 m at baseline to 475 m after 1 year. Measured saturation levels at rest and remained unchanged in the long term.
3.4 NYHA/Modified Ross Functional Class and Clinical Course
During the observational period of 1 year, 15/22 patients remained unchanged in terms of their functional classes. Three of 22 patients improved from FC class 3 to 2, and 1 patient improved from FC class 2 to 1. FC worsened in four patients (1 pt changed from FC 3 to 4 and 3 pts from FC 2 to 3). In 7/22 patients, treatment had to be escalated with either conventional therapy or targeted treatment (such as PGIs or PDE-l) in the further course of follow-up.
4 Discussion
Our efficacy data showed that macitentan had highly significant beneficial effects regarding echo parameters, BNP levels, and NYHA class at 3 months follow-up. However, for the entire cohort, these effects persisted only for PAAT, VTI, and BNP levels over the 1-year observational period, whereas improvements of the remaining parameters persisted but failed to reach statistical significance. Our findings suggest that the mid-term effects were mainly produced by the initial improvements. Between 3 months and 1 year all parameters remained either unchanged or even showed a trend of worsening towards baseline values (Table2).
Notably, our treated patients presented with rather advanced disease progression, as more than 50% had at least systemic PA pressures and were therefore receiving combination therapy including prostacyclins at the time of introduction of macitentan. Especially in those, we felt it was clinically important to take advantage of the better pharmacological properties of the second-generation ERA. Many of them were treated before initiation of the TOMORROW trial and would not have been eligible due to concomitant therapies or disease entity.
However, about one-third of our patients (7/22) had to receive therapy escalation (either targeted or conventional treatment) because of disease progression during the further follow-up period. This fact is an important finding and underlines the fatality of disease and that, in some patients, disease will continue to proceed over time despite initial response to treatment. Therefore, careful monitoring and timely escalation of therapy is essential for long-term outcome. Our findings and the fact that within the entire cohort not all measures showed sustained improvement are somehow disappointing and partially contrast the available data in children, although none of the published studies is completely comparable to ours [16, 19, 32]. Schweintzger et al. demonstrated significant improvements in echo parameters (PAAT, TAPSE, RVSP) as well as in BNP levels in a heterogeneous cohort of 18 pediatric patients with PAH after 6 months; notably, those patients had a lesser degree of PHVD [19].
Aypar et al. studied a cohort of 27 pediatric and adult patients between 12 and 35 years of age; two-thirds of them had CHD-PAH. They found significant improvement in 6-MWT, BNP, and saturation after 6 months, but these effects were no longer significant after 24 months; echo assessment was not performed [32].
The differences of outcomes in our study could be explained by the fact that our patient cohort presented with more advanced disease progression and that severely affected patients might, especially in the longer term, not benefit as much as patients with lesser degree of disease severity.
On the other hand, the decreasing effects after 1 year could also be explained by tachyphylaxis, which is known from other targeted substances such as PGIs. Whether increased dosage is necessary and how to adjust must be answered in future.
Finally, faced with a disease that is continuously progressing, incremental long-term improvement might simply not be achievable; its lack therefore does not necessarily mean treatment failure. Slowing down the process and gaining time to more invasive treatment alternatives—especially in pediatric patients—might also be a defined treatment goal and endpoint of future studies.
Overall, our data showed that VTI and PAAT were the only measures on echo with sustained significant improvement. Both are considered surrogates of pulmonary artery stiffness and resistance that inversely correlate with pulmonary blood flow [25, 26, 33]. VTI was not yet investigated in pediatric studies for assessment of efficacy, though it is rather easy to obtain. Recent studies have suggested VTI or VTI/RVSP as a simple non-invasive surrogate for PVR, which represents an important prognostic factor in patients with PAH [25,26,27]. Particularly for pediatric patients this parameter might be of interest as invasive testing to calculate PVR is not without risk. Macitentan is considered highly effective in modulating pulmonary vascular tone and stiffness resulting in improved pulmonary blood flow [15, 34]. Our findings of persisting improvements of VTI and PAAT as non-invasive surrogates are in accordance with this phenomenon, which is also supported by several adult studies showing a decrease of PVR by macitentan [13, 31].
Decrease of PVR in patients with PH is generally associated with an increase of cardiac output and RV function [35]. Whether improvement of RV function is directly achieved by macitentan or represents a consequence of lowered PVR is currently unclear [36,37,38]. As to our study, patients showed profound improvements in flow parameters, which were mirrored by improved RV function as assessed by TAPSE and BNP levels (Supplementary Fig. 1). However, in the long term the latter was less pronounced and only changes of BNP levels remained statistically significant (Table 2).
To rule out uncertainties due to heterogeneous entities and to address the fact that the commonly used echo and laboratory parameters might not account as well for patients with PHVD due to complex CHD, those patients were investigated separately in our subgroup analysis. Thereby we found that in patients with CHD none of the assessed parameters changed significantly at any time. In contrast, non-CHD patients showed highly significant improvements of all assessed parameters at 3 months, indicating that overall efficacy was mainly driven by beneficial effects in this subgroup. These effects accounted not only for ERA-naïve patients but also for those who had previously been treated with bosentan, suggesting a possible superiority of macitentan when compared with its precursor.
After 1 year, non-CHD patients (in contrast to the entire cohort) demonstrated statistically significant improvements regarding RV function (BNP and TAPSE) and flow parameters (VTI and PAAT). Changes of RVED absolute values did not reach statistical significance. However, the broad range of RVED in pediatric age might not reliably allow correct analysis of changes; we therefore additionally calculated z-scores, which showed (in contrast to the absolute values) significant improvement after 1 year (p = 0.02).
In the non-CHD group RVSP did not show significant improvement after 1 year, though it decreased by a median of 24% at 3 months and 22% at 1-year follow-up. This finding contrasts the persisting beneficial changes of the remaining functional parameters such as TAPSE, VTI, PAAT, and BNP. However, the lack of persisting significant decrease of RVSP might indicate disease progression earlier compared with other markers, all of which remained statistically significant despite a lack of further improvement between 3 months and 1 year. As such, one-third of the patients received therapy escalation due to clinical worsening during the further follow-up period. On the other hand, estimation of PA pressure by measuring RVSP may be subjected to numerous technical errors leading to over- or underestimation of RVSP. Though measurements were always obtained by the same echocardiographer, invasive assessment by catheter would have been more accurate.
About one-third of our patients had PHVD associated with complex CHD and/or Eisenmenger (ES). The beneficial role of targeted therapies in this patient cohort has been proven in many studies investigating different substances, mainly bosentan and sildenafil [39,40,41,42,43]. However, the role of macitentan in these patients remains controversial. Whereas some studies reported on positive clinical signals, a large controlled trial (clinical study to evaluate the effects of macitentan on exercise capacity in subjects with Eisenmenger syndrome, the MAESTRO study) did not reach its primary endpoint of change in 6-MWT [44,45,46,47].
In children, studies on targeted therapies restricted to PHVD associated with CHD are limited so far [48]. In most of the specifically pediatric cohorts, patients with CHD–PAH were investigated together with patients with PAH with other entities such as IPAH, CLD, and others [3, 49, 50, 51] . Given the huge variability of associated diseases and the relatively small numbers of pediatric patients, it might be difficult to conduct studies on each pediatric PHVD entity.
Furthermore, some of our patients in this group had segmental PH associated with complex CHD, such as pulmonary atresia, ventricular septal defect, and major aortopulmonary collaterals (PA/VSD and MAPCAs); all of them had pulmonary hemodynamics not suitable for immediate complete surgical repair or they had persisting PH after corrective surgery. Given the severity of this disease and the lack of curative treatments, the use of targeted therapies to improve hemodynamics—mostly in addition to transcatheter interventions or surgical procedures—seems reasonable in some of these patients. Several case series in adults and children have reported clinical improvements of patients receiving pharmacological therapy such as bosentan or sildenafil [52,53,54,55]. However, the role of targeted therapy currently remains under debate among experts. Similarly, patients in our study were managed according to a combined strategy consisting of medical treatment as adjunct to various interventions. To subtract/eliminate the effect of certain procedures in these patients, our data were always obtained in time intervals between two transcatheter or surgical interventions. Nevertheless, the used variables might not be fully suitable for assessment of efficacy in this setting of anatomy, as they may be subjected to factors other than hypertensive vascular disease. Our findings with only limited effects in these patients therefore need to be interpreted with caution.
6-MWT was performed in 12/22 patients (7 CHD, 5 non-CHD patients). The mean change of walking distance was 21 m after 3 months and 28 m after 1 year for the entire cohort. Among patients with CHD, associated PHVD walking distance and saturation levels remained essentially unchanged after 1 year. We did not perform statistical calculation of these findings as the number of patients was too small for analysis.
Routine cardiac catheterization to determine hemodynamic efficacy was not performed, although almost all our patients underwent invasive hemodynamic assessment at least once at the time of diagnosis of PHVD. We considered that sedation or general anesthesia for routine cardiac catheterization would make them susceptible to unjustified increased risk, while on the other hand, assessment of PA pressure and RV dysfunction was possible non-invasively by echo and other parameters in nearly all patients. Whereas this strategy stands in contrast to adult approach, which includes regular hemodynamic assessment to determine efficacy of treatments, regular cardiac catheterization in children remains a matter of discussion. Pediatric experts recommend this procedure at least once, whereas repeated catheterization should be performed on the basis of individual risk stratification given the rather high postprocedural complication rates ranging from 2% to 6% [1, 56]. Therefore, and in contrast to adults, many pediatric studies are meanwhile performed without invasive hemodynamic assessment as a primary endpoint.
4.1 Limitations
Limitations include the small sample size and the heterogeneity of the study population, which in fact reflects the shortcomings of a single-center observational study in pediatric patients with PAH. We addressed this drawback by performing subgroup analysis, which on the other hand further reduced the analyzed patient numbers.
However, whether all the parameters used offer an appropriate tool to monitor treatment efficacy in the different PH entities might be debatable.
The open-label nature of our study and the lack of a control group constitutes a further limitation. Moreover, macitentan doses used were empiric and extrapolated from adult studies. We did not perform a pharmacokinetic analysis, which is an important limitation of the study; however, this is part of the currently ongoing trial (TOMORROW). Therefore, and due to the overall small patient numbers, careful interpretation of our findings is warranted.
5 Conclusion
Data presented herein currently comprise the largest cohort of severely affected patients receiving macitentan at a pediatric tertiary care center. Overall, macitentan was safe and associated with beneficial mid-term effects, which were most pronounced during the first three months. In the long term, progression of this disease remains a major challenge and therapy escalation should be considered early. Our data suggest only limited efficacy in CHD-PH and that favorable outcomes were mainly driven by improvements in patients with PH not related to CHD. Larger studies are needed to verify these preliminary results and to prove our novel aspects regarding efficacy of this drug in different pediatric PHVD entities.
References
Rosenzweig EB, Abman SH, Adatia I, Beghetti M, Bonnet D, Haworth S, et al. Paediatric pulmonary arterial hypertension: updates on definition, classification, diagnostics and management. Eur Respir J [Internet]. 2019;53:1801916. https://doi.org/10.1183/13993003.01916-2018.
Berger RM, Beghetti M, Humpl T, et al. Clinical features of paediatric pulmonary hypertension: a registry study. Lancet. 2012;379(9815):537–546. https://doi.org/10.1016/S0140-6736(11)61621-8. Accessed 30 Sept 2022.
Hansmann G, Koestenberger M, Alastalo T-P, Apitz C, Austin ED, Bonnet D, et al. 2019 updated consensus statement on the diagnosis and treatment of pediatric pulmonary hypertension: The European Pediatric Pulmonary Vascular Disease Network (EPPVDN), endorsed by AEPC, ESPR and ISHLT. J Heart Lung Transplant Off Publ Int Soc Heart Transplant. 2019;38:879–901.
Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: Developed by the task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Endorsed by the International Society for Heart and Lung Transplantation (ISHLT) and the European Reference Network on rare respiratory diseases (ERN-LUNG). Eur Heart J [Internet]. 2022. https://doi.org/10.1093/eurheartj/ehac237.
del Cerro MJ, Abman S, Diaz G, Freudenthal AH, Freudenthal F, Harikrishnan S, et al. A consensus approach to the classification of pediatric pulmonary hypertensive vascular disease: report from the PVRI Pediatric Taskforce, Panama 2011. Pulm Circ. 2011;1:286–98. https://doi.org/10.4103/2045-8932.83456.
Tuder RM, Archer SL, Peter D, Erzurum SC, Christophe G, Evangelos M, et al. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D4-12. https://doi.org/10.1016/j.jacc.2013.10.025.
Lang IM, Bonderman D, Kneussl M, Marx M. Paediatric pulmonary vascular disease. Paediatr Respir Rev [Internet]. 2004 [cited 2019 Apr 13];5:238–48. https://linkinghub.elsevier.com/retrieve/pii/S152605420400048X.
Zijlstra WMH, Douwes JM, Rosenzweig EB, Schokker S, Krishnan U, Roofthooft MTR, et al. Survival differences in pediatric pulmonary arterial hypertension: clues to a better understanding of outcome and optimal treatment strategies. J Am Coll Cardiol. 2014;63:2159–69.
Moledina S, Hislop AA, Foster H, Schulze-Neick I, Haworth SG. Childhood idiopathic pulmonary arterial hypertension: a national cohort study. Heart [Internet]. 2010;96:1401–6. https://doi.org/10.1136/hrt.2009.182378.
Beghetti M. Current treatment options in children with pulmonary arterial hypertension and experiences with oral bosentan. Eur J Clin Invest. 2006;36(Suppl 3):16–24.
Avitabile CM, Vorhies EE, Ivy DD. Drug treatment of pulmonary hypertension in children. Paediatr Drugs. 2020;22:123–47.
Galiè N, Humbert M, Vachiery J-L, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J [Internet]. 2016;37:67–119. https://doi.org/10.1093/eurheartj/ehv317.
Pulido T, Adzerikho I, Channick RN, Delcroix M, Galiè N, Ghofrani H-A, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med [Internet]. 2013;369:809–18. https://doi.org/10.1056/NEJMoa1213917.
Dingemanse J, Sidharta PN, Maddrey WC, Rubin LJ, Mickail H. Efficacy, safety and clinical pharmacology of macitentan in comparison to other endothelin receptor antagonists in the treatment of pulmonary arterial hypertension. Expert Opin Drug Saf [Internet]. 2014;13:391–405. https://doi.org/10.1517/14740338.2014.859674.
Iglarz M, Binkert C, Morrison K, Fischli W, Gatfield J, Treiber A, et al. Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist. J Pharmacol Exp Ther [Internet]. 2008;327:736–45. https://doi.org/10.1124/jpet.108.142976.
Aypar E, Alehan D, Karagöz T, Aykan HH, Ertugrul İ. Clinical efficacy and safety of switch from bosentan to macitentan in children and young adults with pulmonary arterial hypertension. Cardiol Young [Internet]. 2018;28:542–7. https://www.cambridge.org/core/product/identifier/S1047951117002542/type/journal_article. Accessed 20 April 2022.
Flores M, Caro AT, Mendoza A. Initial experience in children with the use of macitentan in pulmonary arterial hypertension after side effects with other endothelin receptor antagonists. Prog Pediatr Cardiol [Internet]. 2019;52:55–6. http://www.sciencedirect.com/science/article/pii/S1058981318300341. Accessed 20 April 2022
Safdar Z, Thakur A, Frost A. Tolerability of switch to macitentan from bosentan in pulmonary arterial hypertension. South Med J [Internet]. 2017;110:223–8. http://sma.org/southern-medical-journal/article/tolerability-switch-macitentan-bosentan-pulmonary-arterial-hypertension. Accessed 20 April 2022
Schweintzger S, Koestenberger M, Schlagenhauf A, Grangl G, Burmas A, Kurath-Koller S, et al. Safety and efficacy of the endothelin receptor antagonist macitentan in pediatric pulmonary hypertension. Cardiovasc Diagn Ther. 2020;10:1675–85.
Albinni S, Pavo I, Kitzmueller E, Michel-Behnke I. Macitentan in infants and children with pulmonary hypertensive vascular disease. Feasibility, tolerability and practical issues – a single-centre experience. Pulm Circ. 2021;11:2045894020979503. https://doi.org/10.1177/2045894020979503.
Ross RD. The Ross classification for heart failure in children after 25 years: a review and an age-stratified revision. Pediatr Cardiol. 2012;33:1295–300.
Nafiu OO, Voepel-Lewis T, Morris M, Chimbira WT, Malviya S, Reynolds PI, et al. How do pediatric anesthesiologists define intraoperative hypotension? Paediatr Anaesth. 2009;19:1048–53.
Forfia PR, Fisher MR, Mathai SC, Housten-Harris T, Hemnes AR, Borlaug BA, et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med [Internet]. 2006;174:1034–41. https://doi.org/10.1164/rccm.200604-547OC.
Koestenberger M, Ravekes W, Everett AD, Stueger HP, Heinzl B, Gamillscheg A, et al. right ventricular function in infants, children and adolescents: reference values of the tricuspid annular plane systolic excursion (TAPSE) in 640 healthy patients and calculation of z score values. J Am Soc Echocardiogr [Internet]. 2009;22:715–9. https://linkinghub.elsevier.com/retrieve/pii/S0894731709003083. Accessed 20 Oct 2022
Abbas AE, Franey LM, Marwick T, Maeder MT, Kaye DM, Vlahos AP, et al. Noninvasive assessment of pulmonary vascular resistance by Doppler echocardiography. J Am Soc Echocardiogr [Internet]. 2013;26:1170–7. https://www.sciencedirect.com/science/article/pii/S0894731713004458. Accessed 20 Oct 2022
Arkles JS, Opotowsky AR, Ojeda J, Rogers F, Liu T, Prassana V, et al. Shape of the right ventricular Doppler envelope predicts hemodynamics and right heart function in pulmonary hypertension. Am J Respir Crit Care Med. 2011;183:268–76.
Opotowsky AR, Clair M, Afilalo J, Landzberg MJ, Waxman AB, Moko L, et al. A Simple echocardiographic method to estimate pulmonary vascular resistance. Am J Cardiol [Internet]. 2013;112:873–82. https://linkinghub.elsevier.com/retrieve/pii/S0002914913011296. Accessed 20 Oct 2022
Lammers AE, Apitz C, Michel-Behnke I, Koestenberger M. A guide to echocardiographic assessment in children and adolescents with pulmonary hypertension. Cardiovasc Diagn Ther. 2021;11:1160–77.
Chemla D, Castelain V, Humbert M, Hébert J-L, Simonneau G, Lecarpentier Y, et al. New formula for predicting mean pulmonary artery pressure using systolic pulmonary artery pressure. Chest [Internet]. 2004 [cited 2022 Apr 10];126:1313–7. https://linkinghub.elsevier.com/retrieve/pii/S001236921531312X.
Steckelberg RC, Tseng AS, Nishimura R, Ommen S, Sorajja P. Derivation of mean pulmonary artery pressure from noninvasive parameters. J Am Soc Echocardiogr Off Publ Am Soc Echocardiogr. 2013;26:464–8.
Galiè N, Jansa P, Pulido T, Channick RN, Delcroix M, Ghofrani H-A, et al. SERAPHIN haemodynamic substudy: the effect of the dual endothelin receptor antagonist macitentan on haemodynamic parameters and NT-proBNP levels and their association with disease progression in patients with pulmonary arterial hypertension. Eur Heart J. 2017;38:1147–55. Accessed 12 Dec 2022
Aypar E, Alehan D, Karagöz T, Aykan H, Ertugrul İ. Clinical efficacy and safety of switch from bosentan to macitentan in children and young adults with pulmonary arterial hypertension: extended study results. Cardiol Young [Internet]. 2020;30:681–5. https://www.cambridge.org/core/product/identifier/S1047951120000773/type/journal_article.
López-Candales A, Edelman K. Shape of the right ventricular outflow Doppler envelope and severity of pulmonary hypertension. Eur Heart J Cardiovasc Imaging [Internet]. 2012;13:309–16. https://doi.org/10.1093/ejechocard/jer235.
Nadeau V, Potus F, Boucherat O, Paradis R, Tremblay E, Iglarz M, et al. Dual ETA/ETB blockade with macitentan improves both vascular remodeling and angiogenesis in pulmonary arterial hypertension. Pulm Circ. 2018;8:2045893217741429.
Langleben D, Orfanos SE, Fox BD, Messas N, Giovinazzo M, Catravas JD. The paradox of pulmonary vascular resistance: restoration of pulmonary capillary recruitment as a sine qua non for true therapeutic success in pulmonary arterial hypertension. J Clin Med [Internet]. 2022;11:4568. https://www.mdpi.com/2077-0383/11/15/4568.
Brittain EL, Pugh ME, Wheeler LA, Robbins IM, Loyd JE, Newman JH, et al. Prostanoids but not oral therapies improve right ventricular function in pulmonary arterial hypertension. JACC Heart Fail [Internet]. 2013;1:300–7. https://www.sciencedirect.com/science/article/pii/S2213177913001893. Accessed 12 Dec 2022
Iglarz M, Landskroner K, Bauer Y, Vercauteren M, Rey M, Renault B, et al. Comparison of macitentan and bosentan on right ventricular remodeling in a rat model of non-vasoreactive pulmonary hypertension. J Cardiovasc Pharmacol. 2015;66:457.
Anton VN, Richard C, Emmanuelle C, Kiely David G, Tim MJ, Nicolas M, et al. The REPAIR Study. JACC Cardiovasc Imaging [Internet]. 2022;15:240–53. https://doi.org/10.1016/j.jcmg.2021.07.027.
Rosenzweig EB, Ivy DD, Widlitz A, Doran A, Claussen LR, Yung D, et al. Effects of long-term bosentan in children with pulmonary arterial hypertension. J Am Coll Cardiol [Internet]. 2005 [cited 2019 Apr 13];46:697–704. https://linkinghub.elsevier.com/retrieve/pii/S0735109705011964.
Gatzoulis MA, Beghetti M, Galiè N, Granton J, Berger RMF, Lauer A, et al. Longer-term bosentan therapy improves functional capacity in Eisenmenger syndrome: results of the BREATHE-5 open-label extension study. Int J Cardiol. 2008;127:27–32. Accessed 10 April 2022
Apostolopoulou SC, Manginas A, Cokkinos DV, Rammos S. Long-term oral bosentan treatment in patients with pulmonary arterial hypertension related to congenital heart disease: a 2-year study. Heart [Internet]. 2007;93:350–4. https://doi.org/10.1136/hrt.2006.100388.
Arnott C, Strange G, Bullock A, Kirby AC, O’Donnell C, Radford DJ, et al. Pulmonary vasodilator therapy is associated with greater survival in Eisenmenger syndrome. Heart [Internet]. 2018;104:732–7. https://doi.org/10.1136/heartjnl-2017-311876.
Barst RJ, Ivy D, Dingemanse J, Widlitz A, Schmitt K, Doran A, et al. Pharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertension. Clin Pharmacol Ther. 2003;73:372–82.
Wacker J, Weintraub RG. Macitentan in pulmonary arterial hypertension associated with congenital heart defects. Heart Lung Circ [Internet]. 2017;26:1006–7. https://linkinghub.elsevier.com/retrieve/pii/S1443950617313318.
Herbert S, Gin-Sing W, Howard L, Tulloh RMR. Early Experience of macitentan for pulmonary arterial hypertension in adult congenital heart disease. Heart Lung Circ [Internet]. 2017;26:1113–6. https://linkinghub.elsevier.com/retrieve/pii/S1443950617300318.
Blok IM, van Riel ACMJ, van Dijk APJ, Mulder BJM, Bouma BJ. From bosentan to macitentan for pulmonary arterial hypertension and adult congenital heart disease: Further improvement? Int J Cardiol [Internet]. 2017;227:51–2. https://linkinghub.elsevier.com/retrieve/pii/S0167527316336907
Gatzoulis MA, Landzberg M, Beghetti M, Berger RM, Efficace M, Gesang S, et al. Evaluation of macitentan in patients with Eisenmenger syndrome: results from the randomized, controlled MAESTRO Study. Circulation [Internet]. 2019;139:51–63. https://doi.org/10.1161/CIRCULATIONAHA.118.033575.
Beghetti M, Berger RMF. The challenges in paediatric pulmonary arterial hypertension. Eur Respir Rev [Internet]. 2014;23:498–504. https://doi.org/10.1183/09059180.00007714. Accessed 08 May 2022
Beghetti M. Bosentan in pediatric patients with pulmonary arterial hypertension. Curr Vasc Pharmacol. 2009;7:225–33. Accessed 08 May 2022
Ivy DD, Abman SH, Barst RJ, Berger RMF, Bonnet D, Fleming TR, et al. Pediatric pulmonary hypertension. J Am Coll Cardiol [Internet]. 2013;62:D117–26. https://linkinghub.elsevier.com/retrieve/pii/S0735109713058713. Accessed 18 Mar 2023
Hislop AA, Moledina S, Foster H, Schulze-Neick I, Haworth SG. Long-term efficacy of bosentan in treatment of pulmonary arterial hypertension in children. Eur Respir J [Internet]. 2011;38:70–7. https://doi.org/10.1183/09031936.00053510.
Yamamura K, Nagata H, Ikeda K, Ihara K, Hara T. Efficacy of bosentan therapy for segmental pulmonary artery hypertension due to major aortopulmonary collateral arteries in children. Int J Cardiol [Internet]. 2012;161:e1–3. https://linkinghub.elsevier.com/retrieve/pii/S0167527312002264.
Grant EK, Berger JT. Use of pulmonary hypertension medications in patients with tetralogy of fallot with pulmonary atresia and multiple aortopulmonary collaterals. Pediatr Cardiol [Internet]. 2016;37:304–12. https://doi.org/10.1007/s00246-015-1278-2.
Schuuring MJ, Bouma BJ, Cordina R, Gatzoulis MA, Budts W, Mullen MP, et al. Treatment of segmental pulmonary artery hypertension in adults with congenital heart disease. Int J Cardiol [Internet]. 2013;164:106–10. https://linkinghub.elsevier.com/retrieve/pii/S0167527311006243. Accessed 04 Nov 2022
Apostolopoulou SC, Vagenakis G, Rammos S. Pulmonary vasodilator therapy in tetralogy of Fallot with pulmonary atresia and major aortopulmonary collaterals: case series and review of literature. Cardiol Young. 2017;27:1861–4.
Cerro MJ, Moledina S, Haworth SG, Ivy D, Dabbagh MA, Banjar H, et al. Cardiac catheterization in children with pulmonary hypertensive vascular disease: consensus statement from the pulmonary vascular research institute, pediatric and congenital heart disease task forces. Pulm Circ [Internet]. 2016;6:118–25. https://doi.org/10.1086/685102. Accessed 20 Nov 2022
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open access funding provided by Medical University of Vienna.
Conflict of Interest
Sulaima Albinni, Julian Heno, Imre Pavo, Erwin Kitzmueller, Manfred Marx, and Ina Michel-Behnke declare that they have no potential conflicts of interest that might be relevant to the contents of this manuscript.
Ethics Approval
The study was approved by the local Ethics Comittee of the Medical University of Vienna (local ethics number 1619/2018).
Consent to Participate
Oral and written informed consent was obtained from all patients and caregivers before treatment initiation.
Data Availability Statement
Data analyzed during this study are included in this published article as supplementary information files. Additional patients’ data are available from the corresponding author on request.
Authors’ Contributions
Sulaima Albinni wrote the manuscript and was responsible for study design and conduction. Julian Heno performed the statistical analysis and contributed to the final manuscript. Imre Pavo was responsible for material preparation and data collection. Erwin Kitzmueller did the hemodynamic measurements and participated in research coordination. Manfred Marx contributed to interpretation and analysis of the data and reviewed the manuscript. Ina Michel-Behnke was responsible for study conduct and hemodynamic measurements and contributed to the final writing of this manuscript. All authors read and approved the final version.
Consent for Publication
Not applicable.
Code Availability
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Albinni, S., Heno, J., Pavo, I. et al. Macitentan in the Young—Mid-term Outcomes of Patients with Pulmonary Hypertensive Vascular Disease treated in a Pediatric Tertiary Care Center. Pediatr Drugs 25, 467–481 (2023). https://doi.org/10.1007/s40272-023-00573-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40272-023-00573-y