
Abstract
Background
Infigratinib is a fibroblast growth factor receptor (FGFR)-specifc tyrosine kinase inhibitor indicated for the treatment of patients with previously treated, unresectable, locally advanced or metastatic cholangiocarcinoma. However, few studies have been conducted to evaluated the safety of infigratinib in the real world. In this study, we conducted a pharmacovigilance study to evaluate the adverse events (AEs) of infigratinib by using the Food and Drug Administration Adverse Event Reporting System (FAERS) database.
Methods
OpenVigil 2.1 was employed to extract the FAERS database. Descriptive analysis was used to describe the characteristics of infigratinib-associated AE reports. Disproportionality analysis was performed by calculating the proportional reporting ratio (PRR), reporting odds ratios (ROR), and Bayesian analysis confidence propagation neural network (BCPNN) to detect positive signals.
Results
Our findings revealed 149 AE reports, among which 36 significant signals were identified. These significant AE signals were mainly observed in gastrointestinal disorders (N = 26, ROR = 26.03, PRR = 8.44, information component [IC] = 3.08) and skin and subcutaneous tissue disorders (N = 21, ROR = 92.13, PRR = 40.41, IC = 5.34). Notably, dehydration and skin exfoliation were unexpected AEs, but had relatively high signal intensities (ROR = 29.75, PRR = 26.64, IC = 4.74; ROR = 50.61, PRR = 45.24, IC = 5.50, respectively) despite not being listed on the drug label. Furthermore, our analysis showed that infigratinib dose differed statistically between severe and non-severe reports (113.82 ± 16.13 mg vs 125 ± 0.00 mg, t = − 4.28; p < 0.001). However, there were no significant differences in sex, age, and types of AEs between the two groups (p = 0.06, p = 0.86, and p = 0.93, respectively).
Conclusions
These findings suggest that gastrointestinal and skin toxicities are the most common adverse reactions for infigratinib. It is important to recognize skin exfoliation and dehydration in clinical practice, as they are unexpected AEs. Additionally, our study indicates that infigratinib dose may correlate with an increased risk of AE severity, highlighting the need for dose adjustment of infigratinib when exposure to the drug is increased due to internal or external factors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Gastrointestinal and skin toxicities are the most common adverse reactions for infigratinib. |
Skin exfoliation and dehydration are unexpected adverse events (AEs) for infigratinib, which need to be close monitored in practice. |
The infigratinib dose may correlate with an increased risk of AE severity. |
1 Introduction
The fibroblast growth factor receptor (FGFR) signaling pathway plays a crucial role in the proliferation and survival of malignant cancer cells, specifically cholangiocarcinoma [1, 2]. Previous studies have shown that FGFR2 fusions or rearrangements are present in 10–16% of patients with intrahepatic cholangiocarcinoma [3]. Consequently, FGFR becomes a key target for cholangiocarcinoma therapy. Infigratinib, an orally bioavailable inhibitor of FGFRs, has demonstrated selective binding and inhibition of FGFR1, FGFR2, and FGFR3 [4]. Through this mechanism, infigratinib reduces cell proliferation in tumors with activating FGFR amplifications, mutations, or fusions [5]. Several clinical trials have provided evidence of infigratinib's promising clinical activity and manageable safety profile in previously treated locally advanced or metastatic cholangiocarcinoma patients with FGFR2 gene fusions or rearrangements [6,7,8]. As a result, infigratinib received approval on May 28 2021 from the US Food and Drug Administration (FDA) for the treatment of previously treated, unresectable, locally advanced or metastatic cholangiocarcinoma with a FGFR2 fusion or other rearrangement. However, it is important to acknowledge the potential adverse reactions associated with infigratinib [9, 10]. Retinal pigment epithelial detachment, hyperphosphatemia, soft tissue mineralization, and embryo-fetal toxicity are among the adverse effects observed in previous studies [7, 8]. Although most of the safety data comes from clinical trials, these studies are limited by strict inclusion criteria, small sample sizes, and reduced follow-up. Therefore, a post-marketing surveillance investigation is necessary to explore potential adverse events (AEs) further. The Food and Drug Adverse Event Reporting System (FAERS), a public database of spontaneously reported AEs, is widely utilized for post-marketing surveillance [11, 12]. The FAERS database compensates for the limitations of clinical trials by providing AE data for detection potential positive signals. Consequently, we conducted a real-world pharmacovigilance study to examine the safety of infigratinib using the FAERS database.
2 Material and Methods
2.1 Data Sources and Data Collection
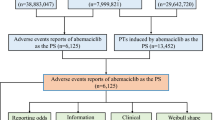
Data collection for AEs related to infigratinib was conducted between May 28, 2021 and September 30, 2022. The FAERS database and OpenVigil 2.11 were utilized to retrieve AE records associated with both the generic name and brand name of the drug (‘infigratinib’ and ‘Truseltiq’). In each FAERS AE report, reporters assigned role codes to reported drugs, designating them as primary suspect, secondary suspect, concomitant, or interacting. This study included primary and secondary suspect drugs that were responsible for AEs. Relevant information, such as individual safety reports (ISR), outcome, drug name, role code, dosage, indication, event, case ID, and clinical characteristics (sex, reporter country, age) of the patient, were gathered. To ensure data accuracy, duplicate and conflicting records were excluded, with only the latest case ID retained. In cases where the case ID was the same, the ISR with the largest number was selected. Preferred terms (PTs) associated with indication, off-label use, and product use issues were removed from the analysis to minimize confounding effects.
2.2 Definition of Adverse Events
The FAERS database codes AEs using Medical Dictionary for Regulatory Activities (MedDRA) terminology at a PT level. We used MedDRA version 26.0 to categorize AEs in each report into system organ class (SOC) levels.
2.3 Statistical Analysis
Descriptive analysis was employed to present the clinical characteristics of AE reports, including event, outcome, sex, age, and reporting country. Proportions were compared using Pearson's chi-squared (χ2) or Fisher's exact test, while independent samples t-test and Kruskal-Wallis test were applied for continuous data and skewed distribution data, respectively. A statistical significance level of p < 0.05 was considered significant. To identify AE signals, disproportionality analysis was conducted through the calculation of reporting odds ratio (ROR), proportional reporting ratio (PRR), and Bayesian analysis confidence propagation neural network (BCPNN). The algorithm equations and criteria for the disproportionality analysis are provided in Tables 1 and 2. A signal was deemed significant only if it fulfilled all three algorithm criteria simultaneously. Furthermore, based on FDA classification, AE cases were categorized as serious or non-serious. AEs were considered ‘serious’ if they resulted in death, hospitalization (initial or prolonged), disability or permanent damage, life-threatening situations, or other serious medical events. Sex distribution, types of AEs, infigratinib dosage, and age were compared between AEs with serious and non-serious outcomes. All data processing and statistical analyses were performed using Microsoft Excel version 2019 and SPSS 23.0 statistical software.
3 Results
3.1 Descriptive Analysis
A total of 149 AE reports were retrieved from the FAERS database using OpenVigil 2.1. After data cleaning, 113 records (75.84%) were removed, comprising 72 AE reports associated with other drugs and 41 duplicate records. This resulted in 36 AE reports (24.16%) remaining for inclusion in this study. In terms of age distribution, patients aged 45–64 years (22.22%) and 65–74 years (19.44%) accounted for a higher proportion compared with other age groups, with a median age of 62.00 years. Regarding sex, there were more female patients (58.33%) than male patients (30.56%). The majority of the reports were from the United States (75.00%), followed by China (8.33%), Spain (5.56%), Belgium (5.56%), Puerto Rico (2.78%), and Australia (2.78%). The most reported outcomes associated with infigratinib-related AEs were hospitalization (33.33%), followed by other outcomes (25.00%) and death (16.67%). The characteristics of AE reports for infigratinib are listed in Table 3.
3.2 Disproportionality Analysis
3.2.1 Preferred Term (PT) Analysis
A total of 133 AE signals related to infigratinib were extracted from the FAERS database, and 19 signals were considered significant according to the disproportionality analysis. In terms of signal intensity, blood phosphorus increased (ROR = 2095.95, PRR = 1812.85, IC = 10.78), hyperphosphatemia (ROR = 1476.99, PRR = 1357.32, IC = 10.37), onychomadesis (ROR = 1045.72, PRR = 932.78, IC = 9.84) had relatively strong signal intensities. As to the frequencies of AE reports, fatigue (N = 10), alopecia (N = 6), and stomatitis (N = 6) had relatively high frequencies. The significant AE signals of infigratinib at the PT level are listed in Table 4.
3.2.2 System Organ Class (SOC) Analysis
The significant AE signals of infigratinib were distributed into eight SOCs. Gastrointestinal disorders involved six PTs, skin and subcutaneous tissue disorders involved five PTs, and metabolism and nutrition disorders involved three PTs. Each of the other five SOCs involved one PT. Investigation (ROR = 2095.95, PRR = 1812.85, IC = 10.78), skin and subcutaneous tissue disorders (ROR = 92.13, PRR = 40.41, IC = 5.34), eye disorders (ROR = 40.25, PRR = 37.07, IC = 5.21), metabolism and nutrition disorders (ROR = 35.37, PRR = 24.22, IC = 4.60), gastrointestinal disorders (ROR = 26.03, PRR = 8.44, IC = 3.08), and neoplasms benign, malignant and unspecified (including cysts and polyps) (ROR = 23.49, PRR = 21.05, IC = 4.40) had relatively strong signal intensities. The significant AE signals at the SOC level are listed in Table 5.
3.2.3 Serious Versus Non-Serious AEs
Infigratinib dose differed statistically between severe and non-severe AEs (113.82 ± 16.13 mg vs 125 ± 0.00 mg, respectively; t = − 4.28; p < 0.001), as shown in Table 6. However, sex, age, and types of AEs did not differ significantly between the two groups (p = 0.06, p = 0.86, and p = 0.93, respectively).
4 Discussion
The study revealed that in the real world, the most common adverse reactions of infigratinib were gastrointestinal and skin toxicities. Furthermore, unexpected AEs including skin exfoliation and dehydration require more attention in clinical practice. Frequentist analysis and Bayes analysis are the two disproportionality analysis methods used to detect positive AE signals [13, 14]. The frequentist analysis, which includes ROR and PRR, is simple and sensitive [15]. However, its accuracy is excessively dependent on the number of AE reports, and a small number of AE reports in the database can easily result in false-positive signals [15]. On the other hand, Bayes analysis, which includes BCPNN and multi-item gamma poisson shrinker (MGPS), provides robust calculation results and a strong ability to predict adverse reactions [14]. Therefore, to minimize bias caused by using a single algorithm, both frequentist analysis and Bayes analysis were employed for signal detection.
Patients aged 45–64 years old and 65–74 years old accounted for a greater proportion compared with other age groups, which is consistent with the higher incidence rate of cholangiocarcinoma in these age groups [16]. The results also showed that AEs were more likely to occur in females, suggesting that significant attention should be paid to female patients. Additionally, most of the AE reports were from the United States, which may be attributed to the fact that infigratinib was first marketed in the United States [5].
The most common drug-related adverse reactions listed in the infigratinib label were nail toxicity, stomatitis, dry eye, fatigue, alopecia, arthralgia, dysgeusia, constipation, dry mouth, diarrhea, dry skin, decreased appetite, vomiting, and hyperphosphatemia [17]. These findings were consistent with the results of AE signals analysis, indicating the credibility of this study. Although the number of AE reports associated with increased blood phosphorus, hyperphosphatemia, and onychomadesis was small, the signal intensities were high, indicating a strong relationship with infigratinib. Gastrointestinal disorders and skin and subcutaneous tissue disorders had the highest number of PT and AE reports among all SOCs, suggesting that gastrointestinal and skin toxicities are the most common adverse reactions for infigratinib. Therefore, close monitoring of gastrointestinal and skin toxicities is necessary during infigratinib treatment. Investigations had the highest signal intensity at the SOC level, including only one PT, namely nausea blood phosphorus. Thus, attention should be paid to blood phosphorus in patients receiving infigratinib treatment.
Unexpected AEs were defined as significant AEs that were not listed in the drug label. Dehydration (ROR = 29.75, PRR = 26.64, IC = 4.74) and skin exfoliation (ROR = 50.61, PRR = 45.24, IC = 5.50) were considered unexpected AEs as they were not included in the drug label. In the phase II study of infigratinib in patients with FGFR-altered advanced cholangiocarcinoma, four patients developed grade 1 or 2 dehydration, but none experienced grade 3 or 4 dehydration [7]. Hence, dehydration was found to be highly correlated with infigratinib, and caution should be exercised by clinicians regarding this AE. Moreover, the results also suggested that the FDA should consider adding dehydration to the drug label. As for skin exfoliation, there were no relevant reports on skin exfoliation induced by infigratinib. However, based on the disproportionality analysis, skin exfoliation showed a high correlation with infigratinib, indicating the need for caution in clinical practice.
The study further revealed that the infigratinib dose may correlate with an increased risk of AE severity. In a phase I trial, many AEs were found to be dose-dependent, with higher doses being more prone to developing serious AEs [8, 18]. Therefore, in order to reduce the toxicity of the drug, the infigratinib dose should be modified when the exposure of infigratinib is increased by some internal or external factors, such as patients with hepatic or renal impairment, co-administered with strong or moderate CYP3A inhibitors, or co-administered with food [17,18,19].
Despite the comprehensive analysis of AE signals of infigratinib based on real-world data, this study also has certain limitations that should be considered. Firstly, the FAERS database, being a spontaneous AE reporting system, cannot provide the total number of patients receiving infigratinib treatment, making it impossible to estimate the incidence of adverse reactions. Secondly, the data obtained from the FAERS database are random and incomplete, which may reduce the accuracy of analysis results. Thirdly, due to missing information, the causal relationship between AEs and drugs cannot be determined. Lastly, although disproportionality analysis is commonly used for AE mining, it lacks a gold standard for assessing the significance of suspected adverse drug reactions, potentially leading to false-positive signals. Nonetheless, the FAERS database remains one of the largest databases of adverse drug events in the world, providing valuable drug safety information and serving as an important tool for post-marketing safety assessments.
5 Conclusion
This study utilized the FAERS data to mine and analyze the AE signals of infigratinib. The findings indicated that gastrointestinal and skin toxicities were the most common infigratinib-related adverse reactions. Skin exfoliation and dehydration were identified as unexpected AEs, highlighting the importance for clinicians to be aware of these two AEs. Due to the potential risk of increased AE severity with infigratinib dose, dose adjustment is recommended when the infigratinib exposure is increased by some internal and external factors. Although this study has limitations, it provides valuable insights into the AEs of infigratinib in the real-world setting, warranting further clinical studies for validation.
References
Lee PC, Hendifar A, Osipov A, Cho M, Li D, Gong J. Targeting the fibroblast growth factor receptor (FGFR) in advanced cholangiocarcinoma: clinical trial progress and future considerations. Cancers (Basel). 2021;13(7):1706.
Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014;383(9935):2168–79.
Kongpetch S, Jusakul A, Lim JQ, Ng CCY, Chan JY, Rajasegaran V, Lim TH, Lim KH, Choo SP, Dima S, Popescu I, Duda DG, Kukongviriyapan V, Khuntikeo N, Pairojkul C, Rozen SG, Tan P, Teh BT. Lack of Targetable FGFR2 Fusions in Endemic Fluke-Associated Cholangiocarcinoma. JCO Glob Oncol. 2020;6:628–38.
Botrus G, Raman P, Oliver T, Bekaii-Saab T. Infigratinib (BGJ398): an investigational agent for the treatment of FGFR-altered intrahepatic cholangiocarcinoma. Expert Opin Investig Drugs. 2021;30(4):309–16.
Kang C. Infigratinib: first approval. Drugs. 2021;81(11):1355–60.
Javle M, Roychowdhury S, Kelley RK, Sadeghi S, Macarulla T, Weiss KH, Waldschmidt DT, Goyal L, Borbath I, El-Khoueiry A, Borad MJ, Yong WP, Philip PA, Bitzer M, Tanasanvimon S, Li A, Pande A, Soifer HS, Shepherd SP, Moran S, Zhu AX, Bekaii-Saab TS, Abou-Alfa GK. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol Hepatol. 2021;6(10):803–15.
Javle M, Lowery M, Shroff RT, Weiss KH, Springfeld C, Borad MJ, Ramanathan RK, Goyal L, Sadeghi S, Macarulla T, El-Khoueiry A, Kelley RK, Borbath I, Choo SP, Oh DY, Philip PA, Chen LT, Reungwetwattana T, Van Cutsem E, Yeh KH, Ciombor K, Finn RS, Patel A, Sen S, Porter D, Isaacs R, Zhu AX, Abou-Alfa GK, Bekaii-Saab T. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol. 2018;36(3):276–82.
Nogova L, Sequist LV, Perez Garcia JM, Andre F, Delord JP, Hidalgo M, Schellens JH, Cassier PA, Camidge DR, Schuler M, Vaishampayan U, Burris H, Tian GG, Campone M, Wainberg ZA, Lim WT, LoRusso P, Shapiro GI, Parker K, Chen X, Choudhury S, Ringeisen F, Graus-Porta D, Porter D, Isaacs R, Buettner R, Wolf J. Evaluation of BGJ398, a fibroblast growth factor receptor 1–3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: results of a global phase I, dose-escalation and dose-expansion study. J Clin Oncol. 2017;35(2):157–65.
Kommalapati A, Tella SH, Borad M, Javle M, Mahipal A. FGFR inhibitors in oncology: insight on the management of toxicities in clinical practice. Cancers (Basel). 2021;13(12):2968.
Lacouture ME, Sibaud V, Anadkat MJ, Kaffenberger B, Leventhal J, Guindon K, Abou-Alfa G. Dermatologic adverse events associated with selective fibroblast growth factor receptor inhibitors: overview, prevention, and management guidelines. Oncologist. 2021;26(2):e316–26.
Shu Y, Ding Y, He X, Liu Y, Wu P, Zhang Q. Hematological toxicities in PARP inhibitors: a real-world study using FDA adverse event reporting system (FAERS) database. Cancer Med. 2023;12(3):3365–75.
Ogura T, Shiraishi C. Comparison of adverse events occurred during administration of dipeptidyl peptidase-4 inhibitor in patients with diabetes using FDA adverse event reporting system. Clin Drug Investig. 2023;43(2):129–40.
Huang L, Guo T, Zalkikar JN, Tiwari RC. A review of statistical methods for safety surveillance. Ther Innov Regul Sci. 2014;48(1):98–108.
Lu Z, Suzuki A, Wang D. Statistical methods for exploring spontaneous adverse event reporting databases for drug-host factor interactions. BMC Med Res Methodol. 2023;23(1):71.
Böhm R, von Hehn L, Herdegen T, Klein HJ, Bruhn O, Petri H, Höcker J. OpenVigil FDA—inspection of U.S. American adverse drug events pharmacovigilance data and novel clinical applications. PLoS ONE. 2016;11(6): e0157753.
Khan SA, Tavolari S, Brandi G. Cholangiocarcinoma: epidemiology and risk factors. Liver Int. 2019;39(Suppl 1):19–31.
US Food and Drug Administration. Label. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214622s000lbl.pdf. Accessed 25 Mar 2023.
Kelly CM, Shoushtari AN, Qin LX, D’Angelo SP, Dickson MA, Gounder MM, Keohan ML, Mcfadyen C, Sjoberg A, Singer S, DeMatteo RP, Hwang S, Heinemann MH, Francis JH, Antonescu CR, Chi P, Tap WD. A phase Ib study of BGJ398, a pan-FGFR kinase inhibitor in combination with imatinib in patients with advanced gastrointestinal stromal tumor. Invest New Drugs. 2019;37(2):282–90.
Yu J, Wang Y, Ragueneau-Majlessi I. Strong pharmacokinetic drug-drug interactions with drugs approved by the US Food and Drug Administration in 2021: mechanisms and clinical implications. Clin Ther. 2022;44(11):1536–44.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
No funding was received.
Availability of Data and Material
Data are available on the FAERS database.
Competing Interests
The authors declare that they have no competing interests.
Ethics Approval and Consent to Participate
Not applicable.
Patient Consent for Publication
Not applicable.
Authors' Contributions
DZ and XL conceived the study and performed the data analysis. DZ drafted the manuscript. DZ and JW revised the manuscript. All authors have read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Zhao, D., Long, X., Zhou, J. et al. Pharmacovigilance Study of Infigratinib: A Safety Analysis of the FDA Adverse Event Reporting System. Drugs R D 23, 403–409 (2023). https://doi.org/10.1007/s40268-023-00439-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-023-00439-1