
Abstract
Objective
The aim of this study was to identify factors affecting blood concentrations of voriconazole following letermovir coadministration using population pharmacokinetic (PPK) analysis in allogeneic hematopoietic stem cell transplant (allo-HSCT) recipients.
Methods
The following data were retrospectively collected: voriconazole trough levels, patient characteristics, concomitant drugs, and laboratory information. PPK analysis was performed with NONMEM® version 7.4.3, using the first-order conditional estimation method with interaction. We collected data on plasma voriconazole steady-state trough concentrations at 216 timepoints for 47 patients. A nonlinear pharmacokinetic model with the Michaelis–Menten equation was applied to describe the relationship between steady-state trough concentration and daily maintenance dose of voriconazole. After stepwise covariate modeling, the final model was evaluated using a goodness-of-fit plot, case deletion diagnostics, and bootstrap methods.
Results
The maximum elimination rate (Vmax) of voriconazole in patients coadministered letermovir and methylprednisolone was 1.72 and 1.30 times larger than that in patients not coadministered these drugs, respectively, resulting in decreased voriconazole trough concentrations. The developed PPK model adequately described the voriconazole trough concentration profiles in allo-HSCT recipients. Simulations clearly showed that increased daily doses of voriconazole were required to achieve an optimal trough voriconazole concentration (1–5 mg/L) when patients received voriconazole with letermovir and/or methylprednisolone.
Conclusions
The development of individualized dose adjustment is critical to achieve optimal voriconazole concentration, especially among allo-HSCT recipients receiving concomitant letermovir and/or methylprednisolone.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
We sought to assess the effects of letermovir coadministration on voriconazole trough concentrations in allogeneic hematopoietic stem cell transplant recipients using a robust population pharmacokinetic analysis. |
Coadministration of letermovir or methylprednisolone was related to a larger maximum elimination rate of voriconazole and consequently decreased the voriconazole plasma trough concentrations. |
The simulations clearly showed that increased daily doses of voriconazole were required to obtain an optimal trough voriconazole concentration (1–5 mg/L) when letermovir and/or methylprednisolone were coadministered. |
1 Introduction
Voriconazole, a triazole antifungal agent, inhibits enzymes involved in the synthesis of ergosterol, a primary component of fungal cell membranes. Voriconazole is used as a first-line agent for the prevention and treatment of invasive fungal infection (IFI) in allogeneic hematopoietic stem cell transplant (allo-HSCT) recipients [1,2,3,4,5]. However, the in vivo pharmacokinetics of voriconazole are complex and exhibit remarkable inter- and intra-individual variability [6]. Furthermore, the blood concentrations of voriconazole show a nonlinear response to dose adjustments, and excessive voriconazole concentration is reportedly associated with an increased risk of liver dysfunction [7, 8] and neurological adverse effects [9]. Thus, therapeutic drug monitoring is generally recommended for patients receiving voriconazole [10,11,12].
Cytomegalovirus reactivation was a common complication after allo-HSCT until the approval of letermovir in Japan in March 2018 [13]. This novel anti-cytomegalovirus drug, used for prophylaxis of cytomegalovirus reactivation until approximately 100 days after transplantation, effectively reduced the risk of clinically significant cytomegalovirus infection [13, 14]. Given the substantial efficacy of letermovir and voriconazole against microorganisms, their coadministration is widely used after allo-HSCT. However, letermovir is a well-known inducer of cytochrome P450 (CYP)-2C19, the major metabolic enzyme for voriconazole [15,16,17,18], as well as CYP2C9 enzymes [19]. This drug–drug interaction is estimated to reduce the blood concentrations of voriconazole [20], leading to an increased risk of IFI in allo-HSCT recipients. Various factors have been identified as affecting the in vivo pharmacokinetics of voriconazole, including age and CYP2C19 polymorphisms [21,22,23,24], C-reactive protein (CRP) [25,26,27], serum albumin levels [28], concomitant drugs (e.g., proton pump inhibitors [PPIs] [29,30,31,32] or steroids [33, 34]), among others [35,36,37].
Population pharmacokinetic (PPK) analysis with nonlinear mixed effect modeling is a conventional method for the development of an appropriate pharmacokinetic model with limited sampling data; it aids in the assessment of the potential impact of covariates on the pharmacokinetics of certain drugs. In PPK analysis, fixed and random effects can be obtained, including the population-averaged pharmacokinetic parameters and interindividual and residual variabilities. Given that frequent collection of blood samples is not feasible in clinical practice, PPK analysis is useful for the assessment of clinical data [38].
Although the drug–drug interaction clinical trial between voriconazole and letermovir has been reported previously [20], to the best of our knowledge, no PPK studies have been undertaken regarding the coadministration of letermovir and voriconazole using real-world clinical data. Therefore, in this study, we used PPK analysis to investigate the effects of concomitant letermovir use and other covariates on the blood concentrations of voriconazole.
2 Methods
2.1 Patients
We retrospectively analyzed 47 patients who underwent allo-HSCT for hematological disorders between April 2016 and March 2020 at the Kyushu University Hospital. Patients who received tacrolimus as prophylaxis for graft-versus-host disease and voriconazole as prophylaxis or treatment for IFI with therapeutic drug monitoring were included. Letermovir 480 mg/day was generally initiated intravenously or orally from the day of stem cell infusion. The exclusion criteria were as follows: (1) voriconazole initiated over 100 days after HSCT, (2) age < 20 years, and (3) liver dysfunction (aspartate aminotransferase and/or alanine aminotransferase level more than fivefold the upper normal range or total bilirubin level > 2.0 mg/dL). This study was conducted in accordance with the Declaration of Helsinki and its amendments and was approved by the Ethics Committee of Kyushu University Graduate School and Faculty of Medicine (approval no. 652-03).
2.2 Plasma Voriconazole Concentration
The plasma voriconazole concentration was measured using ultra-performance liquid chromatography or ultra-performance liquid chromatography-tandem mass spectrometry method, which was outsourced to SRL, Inc. (Tokyo, Japan) or LSI Medience Corporation (Tokyo, Japan). The limits of quantification of voriconazole were 0.1 mg/L for SRL and 0.3 mg/L for LSI. The calibration ranged between 0.1 and 40.0 mg/L for SRL and between 0.3 and 15.0 mg/L for LSI. The coefficients of variation were always < 15%. The voriconazole concentrations obtained within 5 days of voriconazole initiation were excluded from this analysis because the concentrations did not reach steady state [39].
2.3 Data Collection and Statistical Analysis
The following data were collected from patients’ electronic medical records: plasma voriconazole concentrations, daily voriconazole dose, liver function test values (aspartate aminotransferase, alanine aminotransferase, and total bilirubin), stem cell sources, conditioning regimens, and underlying diseases. Information about putative covariates, including age, sex, body weight, laboratory findings (CRP and albumin), and concomitant drugs (letermovir, PPI, and steroids), was also collected. Comparisons of voriconazole concentrations with or without letermovir coadministration were examined using the Mann–Whitney U test. Statistical analyses were performed using GraphPad Prism version 9 (GraphPad Software; San Diego, CA, USA).
2.4 Population Pharmacokinetic (PPK) Analysis
Nonlinear mixed effect modeling was used to construct the PPK model. The data were analyzed using the NONMEM® version 7.4.3 software package, and the first-order conditional estimation method with η–ε interaction was used as the estimation algorithm.
2.5 Base Model
A nonlinear pharmacokinetic model based on the Michaelis–Menten equation was applied to describe the relationship between the steady-state trough concentration and the daily maintenance dose of voriconazole. The base model is expressed as per Eq. 1. Because the bioavailability of voriconazole is known to be essentially 100% [19], the F-value was set to 1.
where daily dose is the amount of voriconazole administered daily, F is bioavailability, Km is Michaelis–Menten constant, Vmax is the maximum elimination rate, and Css trough is the steady-state trough concentration.
The Vmax and Km correspond to the capacity and affinity for the metabolic enzymes of voriconazole, respectively. The interindividual variability was modeled using an exponential error model, and residual variability was modeled using an additional error model.
2.6 Final Model
The following variables were selected as putative covariates potentially affecting the steady-state trough concentration of voriconazole: sex, age, body weight, albumin, CRP, and coadministration of letermovir, PPIs (rabeprazole, esomeprazole, lansoprazole, omeprazole, and vonoprazan), and steroids (prednisolone, methylprednisolone, and dexamethasone).
The covariate model was constructed based on differences in objective function values (OFVs) estimated by NONMEM between hierarchical models. The final model was established using a stepwise approach with forward inclusion followed by backward elimination. The significance levels in the likelihood ratio test using OFVs were set at 5% and 1% for forward inclusion and backward elimination, respectively.
An exponential model was used to evaluate the effects of continuous covariates on the parameters (Eq. 2).
where \(P_{ij}\) is the parameter i scaled for subject j, \(P_{{i{\text{pop}}}}\) is the typical value for population parameter i, \({\text{COV}}_{{\text{j}}}\) is the value of the covariate for subject j, \({\text{COV}}_{{{\text{med}}}}\) is the median value of the covariate in the study population, and \({\uptheta }\) is the exponential coefficients of the covariate.
The effect of categorical covariates was evaluated as shown in Eq. 3.
where \(\theta\) is the effect of the covariate on the parameter and X is an indicator variable of each factor (e.g., male = 0, female = 1).
2.7 Model Evaluation
During model development, the validity of the model was assessed using goodness-of-fit (GOF) plots. Once the final model was constructed, the validity and robustness of the model were evaluated using GOF plots, case deletion diagnostics (CDD), and the bootstrap method. GOF plots were used to evaluate the following relationships: observations versus population prediction (PRED) and individual prediction (IPRED), conditional weighted residuals [40] versus PRED, and absolute values of individual weighted residuals versus IPRED.
CDD was used to investigate the impact of each patient in the study population. Each patient was removed individually from the dataset, and the model parameters were re-estimated. In the bootstrap method, 1000 bootstrap replicates were generated by random sampling with replacement. The final model was fitted to these replicate datasets, and parameter estimates were obtained. The median and 95% confidence intervals (calculated using the percentile method) were compared with the parameter estimates obtained from the original dataset. GOF plots, CDD, and the bootstrap method were performed using R version 3.6.1, Xpose4 version 4.6.1, and Perl-speaks-NONMEM version 4.9.0 [41, 42].
2.8 Simulation
Dosing simulations were conducted using the final PPK model. The influence of significant covariates in the final model on the voriconazole trough concentrations was investigated. Dosing schedules were set at 300, 400, 500, and 600 mg/day as maintenance doses. In total, 1000 voriconazole steady-state trough concentrations were simulated in each scenario using a Monte Carlo simulation with the estimated final model parameters. The target therapeutic range for the steady-state trough concentration of voriconazole was set to 1–5 mg/L [11]. Simulations and data processing were performed using the R version 3.6.1.
3 Results
3.1 Patient Characteristics
The patient characteristics are shown in Table 1. A total of 216 voriconazole trough concentration samples were collected from the data on 47 allo-HSCT recipients. The median age and body weight were 51 years (range 22–69) and 55.0 kg (range 31.3–90.3), respectively, without sex-related bias. The median albumin and CRP levels were 3.8 g/dL (range 2.5–5.7) and 0.32 mg/dL (range 0.01–19.60), respectively. Letermovir was concomitantly administered to 19 patients. PPIs were initiated as follows: rabeprazole in 16, esomeprazole in 13, lansoprazole in seven, omeprazole in six, and vonoprazan in five patients.
In addition, prednisolone (n = 19), methylprednisolone (n = 18), and dexamethasone (n = 2) were used at the median daily doses of 17.5 mg (range 2.5–50.0), 40.0 mg (range 25.0–80.0), and 16.5 mg (range 13.2–19.8) at the first time of voriconazole plasma concentration measurement, respectively.
3.2 Effect of Letermovir Coadministration on Trough Concentration and Concentration Per Dose Ratio of Voriconazole
We examined the effect of letermovir coadministration on the trough concentration and concentration per dose (C/D) ratio of voriconazole. The median trough concentration in the letermovir (+) group was significantly lower than that in the letermovir (−) group (1.03 vs. 1.85 mg/L; P < 0.0001) (Fig. 1a). Similarly, the median C/D ratio in the letermovir (+) group was significantly lower than that in the letermovir (−) group (2.50 vs. 4.84 mg/L; P < 0.0001) (Fig. 1b).

Comparison of (a) VRCZ concentration and (b) C/D ratio between patients without LMV (LMV(−); 145 samples) and those with LMV (LMV(+); 71 samples). The bars show the median values; boxes represent the 25th and 75th percentiles of the data. C/D ratio concentration per dose ratio, LMV letermovir, VRCZ voriconazole. ****P < 0.0001
3.3 PPK Analysis in Final Model
The relationships among tested covariates are shown in electronic supplementary material (ESM)-1. After stepwise covariate modeling, coadministration of letermovir (change in OFV [ΔOFV] − 74.349; P < 0.01) and methylprednisolone (ΔOFV − 22.528; P < 0.01) were significant covariates for Vmax, suggesting that the coadministration of these drugs may affect the capacity of the metabolic enzymes of voriconazole. The estimated Vmax was 72% larger in patients with than without letermovir coadministration and was 30% larger in patients with than without methylprednisolone coadministration. The parameter estimates of the final model are presented in Table 2. The relative standard errors of the estimated parameters were acceptable (relative standard errors < 30%), suggesting that all parameters were precisely estimated. The regression equation for Vmax in the final model was as follows:
where Vmax,i is the Vmax in subject i, LMVi is concomitant letermovir use in subject i (used = 1, not used = 0), and mPSLi is concomitant methylprednisolone use in subject i (used = 1, not used = 0). The plots of individual ETAs versus continuous and categorical covariates in the final PPK model are shown in ESM 2 and 3, respectively.
3.4 Model Evaluation
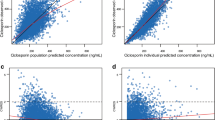
The GOF plot for the final model (Fig. 2) exhibited a high predictive performance, and no systemic deviations were observed. However, a slight overprediction was observed in the PRED versus observations plot. The CDD results are shown in Fig. 3. CDD did not reveal any patients with a significant influence on the estimation of covariate effects. The results of the bootstrap method are listed in Table 2. The median value of each estimated parameter from bootstrapping matched the original population estimates in the final model, and the significance of the covariates was further verified because the confidence intervals of their estimates did not include 1.

Goodness-of-fit plots of the final model. The DV represents the observed steady-state trough concentration of voriconazole. PRED values were obtained using population parameters. IPRED values were obtained using individual empirical Bayesian estimated parameters. Straight solid lines represent line of identity (a, b) and y = 0 (c–f). CWRES conditional weighted residuals, DV dependent variable, IPRED individual prediction, |IWRES| absolute individual weighted residuals, PRED population prediction

Percentage changes in estimated parameters of covariate effect (cotreatment with (a) letermovir and (b) methylprednisolone) for maximum elimination rate in case deletion diagnostic analysis. Grey shaded bars represent estimated parameter using all data and data with each ID excluded
3.5 Simulation
Figure 4 shows the simulation results of the steady-state trough concentrations of voriconazole. Coadministration of letermovir and methylprednisolone was associated with large and moderate reductions in voriconazole exposure, respectively. The appropriate voriconazole trough concentrations were obtained at doses of 300 or 400 mg/day in patients without coadministration of letermovir and methylprednisolone. In contrast, appropriate voriconazole concentrations were achieved at higher doses in patients treated with letermovir (voriconazole 500 or 600 mg/day) or methylprednisolone (voriconazole 400 or 500 mg/day).

Simulated steady-state trough concentrations of VRCZ. Dosing schedules were set at 300, 400, 500, and 600 mg/day. Boxes indicate the 25th, 50th, and 75th percentiles; whiskers indicate the 10th–90th percentiles. The gray concentration range represents 1–5 mg/L, which is the target trough concentration range. DDI drug–drug interaction, LMV letermovir, mPSL methylprednisolone, VRCZ voriconazole
4 Discussion
In the current study, we sought to assess the effects of letermovir coadministration on voriconazole trough concentrations in allo-HSCT recipients using a robust PPK analysis. Coadministration of letermovir or methylprednisolone was related to a larger Vmax of voriconazole and consequently decreased the plasma voriconazole trough concentrations. The simulations clearly showed that increased daily doses of voriconazole were required to obtain an optimal voriconazole trough concentration (1–5 mg/L) when letermovir and/or methylprednisolone were coadministered.
We observed that the median voriconazole trough concentration with letermovir coadministration was 44% lower than that without letermovir coadministration because of the 1.7-fold increase in the Vmax of voriconazole. Our simulation indicated that, when treated with letermovir, a voriconazole dose of 500 or 600 mg/day was optimal to obtain an appropriate voriconazole trough concentration. Our findings were consistent with those of some previous studies. In a phase I clinical trial among healthy individuals, coadministration of letermovir reduced the area under the curve and maximum plasma drug concentration of voriconazole by 44 and 39%, respectively [20]. Hikasa et al. [43] revealed that the voriconazole blood concentrations of allo-HSCT recipients treated with prophylactic letermovir were 33.2% lower than those not treated with letermovir. Nakashima et al. [44] previously reported that the median fold change of the voriconazole trough concentration during letermovir administration was 0.40. Voriconazole is primarily metabolized by CYP2C9 and CYP2C19 [15,16,17]. Letermovir is assumed to increase the expression of CYP2C9/CYP2C19 by activating the pregnane X receptor, a transcriptional regulator involved in regulating the expression of drug-metabolizing enzymes [20, 45]. Further investigation of the letermovir-mediated induction of CYP2C9/CYP2C19 is warranted; however, letermovir-induced increases in the capacity of the metabolic enzymes of voriconazole play a critical role in the remarkable reduction of plasma voriconazole concentrations in allo-HSCT recipients. To the best of our knowledge, this is the first study to identify the effect of letermovir on voriconazole concentrations through a PPK analysis.
Of the three types of corticosteroids analyzed in this study, only methylprednisolone remained a significant influencing factor in the final model. Coadministration of methylprednisolone could increase the Vmax of voriconazole by 1.3-fold, leading to a requirement for a dosage of voriconazole 400 or 500 mg/day to achieve an appropriate voriconazole trough concentration among allo-HSCT recipients. Dolton et al. [34] reported that glucocorticoids significantly reduced voriconazole blood concentrations; among the glucocorticoids, methylprednisolone and dexamethasone exhibited stronger effects on voriconazole blood concentrations than prednisolone. In addition, under in vitro conditions, dexamethasone induced CYP2C19 through interaction of the glucocorticoid receptor with a glucocorticoid-responsive element in the promoter region of CYP2C19 [46]. However, we could not confirm the effects of dexamethasone on voriconazole trough concentrations, presumably because of the very small number of patients treated with dexamethasone. In allo-HSCT settings, high-dose (1–2 mg/kg) methylprednisolone is usually initiated for the prophylaxis and/or treatment of acute graft-versus-host disease [47]. Czock et al. [48] reported that high doses of glucocorticoids induced CYP3A4, which was involved, albeit with a minor contribution, in voriconazole metabolism [49], suggesting that high-dose methylprednisolone may also reduce voriconazole trough concentrations by inducing CYP3A4. Imataki et al. [50] reported a reduction in voriconazole blood concentration in a steroid dose-dependent manner in patients with hematological malignancies. Based on these findings, the induction of both CYP2C19 and CYP3A4 by high-dose methylprednisolone may have contributed to the decreased voriconazole concentrations in the current study.
In the present PPK analysis, we used an equation expressing the relationship between steady-state trough concentration and daily dose. Because the voriconazole pharmacokinetics were characterized by nonlinear elimination, a nonlinear pharmacokinetic model with the Michaelis–Menten equation was selected as the base model. Previous PPK analyses often described voriconazole concentration profiles using one-compartment or two-compartment models [8, 51,52,53,54]. All 216 points obtained in this study were trough levels, without points in the absorption and distribution phases. These conditions contributed to the construction of a simplified equation model in this study.
4.1 Limitations
This study had several limitations. First, we did not estimate the absolute oral bioavailability of voriconazole. Although we assumed the bioavailability of voriconazole was 100%, several studies have reported a reduced bioavailability of voriconazole when used at low doses or in special populations such as transplant patients or extensive metabolizers [18, 55, 56]. Therefore, we conducted a sensitivity analysis assuming oral bioavailability of 45.9% [55]. Even in this case, letermovir and methylprednisolone were still identified as significant covariates. The simulation results for dose adjustment during oral administration were also similar to those assuming that the bioavailability was 100% (ESM-4). Therefore, we believe that this assumption did not result in a bias. However, since the dataset of this study included only 28 voriconazole concentrations during intravenous administration (about 13% of the total), it is difficult to recommend an optimal dose for intravenous administration of voriconazole based on the present analysis. Second, we did not evaluate the effects of genetic polymorphisms of drug-metabolizing enzymes, especially CYP2C19 polymorphisms, on voriconazole concentrations in this PPK analysis. The CYP2C19 poor metabolizer phenotype is known to exist in approximately 20% of the Japanese population [57,58,59]. A previous PPK analysis indicated that this phenotype was associated with a remarkable reduction in voriconazole clearance [52]. Because the present study was retrospective, we could not conduct genotyping of CYP2C19. Therefore, a prospective study including information about CYP2C19 polymorphisms and more patients is warranted to confirm our findings. Another limitation of this study was that we did not evaluate the absorption and distribution of voriconazole because we used only trough concentrations for this analysis. However, since the main purpose of this analysis was to evaluate concomitant medications that could change the elimination of voriconazole, we believe that the analysis with only the trough concentration was reasonable for this study.
5 Conclusion
The voriconazole trough concentration profiles were quantitatively described using the developed PPK model among allo-HSCT recipients. Coadministration of letermovir and/or methylprednisolone, frequently used during the posttransplant period, led to a remarkable decrease in voriconazole trough concentrations. Therefore, individualized dosage regimens of voriconazole are important to ensure its efficacy against IFI and to minimize the risk of adverse effects, especially in the setting of allo-HSCT.
References
Fukuda T, Boeckh M, Guthrie KA, Mattson DK, Owens S, Wald A, et al. Invasive aspergillosis before allogeneic hematopoietic stem cell transplantation: 10-year experience at a single transplant center. Biol Blood Marrow Transplant. 2004;10(7):494–503.
Wingard JR, Carter SL, Walsh TJ, Kurtzberg J, Small TN, Baden LR, et al. Randomized, double-blind trial of fluconazole versus voriconazole for prevention of invasive fungal infection after allogeneic hematopoietic cell transplantation. Blood. 2010;116(24):5111–8.
Patterson TF, Thompson GR 3rd, Denning DW, Fishman JA, Hadley S, Herbrecht R, et al. Practice Guidelines for the Diagnosis and Management of Aspergillosis: 2016 Update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;63(4):e1–60.
Tissot F, Agrawal S, Pagano L, Petrikkos G, Groll AH, Skiada A, et al. ECIL-6 guidelines for the treatment of invasive candidiasis, aspergillosis and mucormycosis in leukemia and hematopoietic stem cell transplant patients. Haematologica. 2017;102(3):433–44.
Walsh TJ, Anaissie EJ, Denning DW, Herbrecht R, Kontoyiannis DP, Marr KA, et al. Treatment of aspergillosis: clinical practice guidelines of the Infectious Diseases Society of America. Clin Infect Dis. 2008;46(3):327–60.
Wang T, Xie J, Wang Y, Zheng X, Lei J, Wang X, et al. Pharmacokinetic and pharmacodynamic properties of oral voriconazole in patients with invasive fungal infections. Pharmacotherapy. 2015;35(9):797–804.
Matsumoto K, Abematsu K, Shigemi A, Kanazawa N, Watanabe E, Yokoyama Y, et al. Therapeutic drug monitoring of voriconazole in Japanese patients: analysis based on clinical practice data. J Chemother. 2016;28(3):198–202.
Suzuki Y, Tokimatsu I, Sato Y, Kawasaki K, Sato Y, Goto T, et al. Association of sustained high plasma trough concentration of voriconazole with the incidence of hepatotoxicity. Clin Chim Acta. 2013;23(424):119–22.
Hamada Y, Seto Y, Yago K, Kuroyama M. Investigation and threshold of optimum blood concentration of voriconazole: a descriptive statistical meta-analysis. J Infect Chemother. 2012;18(4):501–7.
Park WB, Kim NH, Kim KH, Lee SH, Nam WS, Yoon SH, et al. The effect of therapeutic drug monitoring on safety and efficacy of voriconazole in invasive fungal infections: a randomized controlled trial. Clin Infect Dis. 2012;55(8):1080–7.
Hamada Y, Tokimatsu I, Mikamo H, Kimura M, Seki M, Takakura S, et al. Practice guidelines for therapeutic drug monitoring of voriconazole: a consensus review of the Japanese Society of Chemotherapy and the Japanese Society of Therapeutic Drug Monitoring. J Infect Chemother. 2013;19(3):381–92.
Hanai Y, Hamada Y, Kimura T, Matsumoto K, Takahashi Y, Fujii S, et al. Optimal trough concentration of voriconazole with therapeutic drug monitoring in children: a systematic review and meta-analysis. J Infect Chemother. 2021;27(2):151–60.
Marty FM, Ljungman P, Chemaly RF, Maertens J, Dadwal SS, Duarte RF, et al. Letermovir prophylaxis for cytomegalovirus in hematopoietic-cell transplantation. N Engl J Med. 2017;377(25):2433–44.
Mori Y, Jinnouchi F, Takenaka K, Aoki T, Kuriyama T, Kadowaki M, et al. Efficacy of prophylactic letermovir for cytomegalovirus reactivation in hematopoietic cell transplantation: a multicenter real-world data. Bone Marrow Transplant. 2021;56(4):853–62.
Murayama N, Imai N, Nakane T, Shimizu M, Yamazaki H. Roles of CYP3A4 and CYP2C19 in methyl hydroxylated and N-oxidized metabolite formation from voriconazole, a new anti-fungal agent, in human liver microsomes. Biochem Pharmacol. 2007;73(12):2020–6.
Mikus G, Schöwel V, Drzewinska M, Rengelshausen J, Ding R, Riedel KD, et al. Potent cytochrome P450 2C19 genotype-related interaction between voriconazole and the cytochrome P450 3A4 inhibitor ritonavir. Clin Pharmacol Ther. 2006;80(2):126–35.
Hyland R, Jones BC, Smith DA. Identification of the cytochrome P450 enzymes involved in the N-oxidation of voriconazole. Drug Metab Dispos. 2003;31(5):540–7.
Schulz J, Kluwe F, Mikus G, Michelet R, Kloft C. Novel insights into the complex pharmacokinetics of voriconazole: a review of its metabolism. Drug Metab Rev. 2019;51(3):247–65.
Inc. P. Vfend Prescribing Information in the U.S.: voriconazole tablets, injection suspension: LAB-0271-39.0; January 2021.
Marshall WL, McCrea JB, Macha S, Menzel K, Liu F, van Schanke A, et al. Pharmacokinetics and tolerability of letermovir coadministered with azole antifungals (Posaconazole or Voriconazole) in healthy subjects. J Clin Pharmacol. 2018;58(7):897–904.
Liu Y, Qiu T, Liu Y, Wang J, Hu K, Bao F, et al. Model-based voriconazole dose optimization in Chinese adult patients with hematologic malignancies. Clin Ther. 2019;41(6):1151–63.
Li X, Frechen S, Moj D, Lehr T, Taubert M, Hsin CH, et al. A Physiologically based pharmacokinetic model of voriconazole integrating time-dependent inhibition of CYP3A4, genetic polymorphisms of CYP2C19 and predictions of drug-drug interactions. Clin Pharmacokinet. 2020;59(6):781–808.
Lee J, Ng P, Hamandi B, Husain S, Lefebvre MJ, Battistella M. Effect of therapeutic drug monitoring and cytochrome P450 2C19 genotyping on clinical outcomes of voriconazole: a systematic review. Ann Pharmacother. 2021;55(4):509–29.
Zhang Y, Hou K, Liu F, Luo X, He S, Hu L, et al. The influence of CYP2C19 polymorphisms on voriconazole trough concentrations: Systematic review and meta-analysis. Mycoses. 2021;64(8):860–73.
van Wanrooy MJ, Span LF, Rodgers MG, van den Heuvel ER, Uges DR, van der Werf TS, et al. Inflammation is associated with voriconazole trough concentrations. Antimicrob Agents Chemother. 2014;58(12):7098–101.
Veringa A, Ter Avest M, Span LF, van den Heuvel ER, Touw DJ, Zijlstra JG, et al. Voriconazole metabolism is influenced by severe inflammation: a prospective study. J Antimicrob Chemother. 2017;72(1):261–7.
Gautier-Veyret E, Truffot A, Bailly S, Fonrose X, Thiebaut-Bertrand A, Tonini J, et al. Inflammation is a potential risk factor of voriconazole overdose in hematological patients. Fundam Clin Pharmacol. 2019;33(2):232–8.
Dote S, Sawai M, Nozaki A, Naruhashi K, Kobayashi Y, Nakanishi H. A retrospective analysis of patient-specific factors on voriconazole clearance. J Pharm Health Care Sci. 2016;2:10.
Huang Q, Liu Q, Yin T, Hu L, Ding H, Liu S, et al. Effect of proton pump inhibitors on voriconazole concentrations in Chinese patients with malignant hematological diseases. Eur J Clin Pharmacol. 2020;76(6):833–42.
Yan M, Wu ZF, Tang D, Wang F, Xiao YW, Xu P, et al. The impact of proton pump inhibitors on the pharmacokinetics of voriconazole in vitro and in vivo. Biomed Pharmacother Biomed Pharmacother. 2018;108:60–4.
Wood N, Tan K, Purkins L, Layton G, Hamlin J, Kleinermans D, et al. Effect of omeprazole on the steady-state pharmacokinetics of voriconazole. Br J Clin Pharmacol. 2003;56 Suppl 1(Suppl 1):56–61.
Qi F, Zhu L, Li N, Ge T, Xu G, Liao S. Influence of different proton pump inhibitors on the pharmacokinetics of voriconazole. Int J Antimicrob Agents. 2017;49(4):403–9.
Li M, Zhu L, Chen L, Li N, Qi F. Assessment of drug-drug interactions between voriconazole and glucocorticoids. J Chemother. 2018;30(5):296–303.
Dolton MJ, Ray JE, Chen SC, Ng K, Pont LG, McLachlan AJ. Multicenter study of voriconazole pharmacokinetics and therapeutic drug monitoring. Antimicrob Agents Chemother. 2012;56(9):4793–9.
Chen C, Yang T, Li X, Ma L, Liu Y, Zhou Y, et al. Population pharmacokinetics of voriconazole in Chinese patients with hematopoietic stem cell transplantation. Eur J Drug Metab Pharmacokinet. 2019;44(5):659–68.
Dolton MJ, Mikus G, Weiss J, Ray JE, McLachlan AJ. Understanding variability with voriconazole using a population pharmacokinetic approach: implications for optimal dosing. J Antimicrob Chemother. 2014;69(6):1633–41.
Shi C, Xiao Y, Mao Y, Wu J, Lin N. Voriconazole: a review of population pharmacokinetic analyses. Clin Pharmacokinet. 2019;58(6):687–703.
Sheiner LB, Beal SL. Evaluation of methods for estimating population pharmacokinetic parameters. III. Monoexponential model: routine clinical pharmacokinetic data. J Pharmacokinet Biopharm. 1983;11(3):303–19.
Imamura CK, Furihata K, Okamoto S, Tanigawara Y. Impact of cytochrome P450 2C19 polymorphisms on the pharmacokinetics of tacrolimus when coadministered with voriconazole. J Clin Pharmacol. 2016;56(4):408–13.
Hooker AC, Staatz CE, Karlsson MO. Conditional weighted residuals (CWRES): a model diagnostic for the FOCE method. Pharm Res. 2007;24(12):2187–97.
Jonsson EN, Karlsson MO. Xpose–an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58(1):51–64.
Lindbom L, Pihlgren P, Jonsson EN. PsN-Toolkit–a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241–57.
Hikasa S, Shimabukuro S, Osugi Y, Ikegame K, Kaida K, Fukunaga K, et al. Decrease in voriconazole concentration-to-dose ratio after letermovir initiation: a retrospective, observational study. Bone Marrow Transplant. 2021;56(4):949–51.
Nakashima T, Inamoto Y, Fukushi Y, Doke Y, Hashimoto H, Fukuda T, et al. Drug interaction between letermovir and voriconazole after allogeneic hematopoietic cell transplantation. Int J Hematol. 2021;113(6):872–6.
Sun H, Lou XY, Wu XY, Wang H, Qu Q, Tan SL, et al. Up-regulation of CYP2C19 expression by BuChang NaoXinTong via PXR activation in HepG2 cells. PLoS ONE. 2016;11(7):e0160285.
Chen Y, Ferguson SS, Negishi M, Goldstein JA. Identification of constitutive androstane receptor and glucocorticoid receptor binding sites in the CYP2C19 promoter. Mol Pharmacol. 2003;64(2):316–24.
Murata M. Prophylactic and therapeutic treatment of graft-versus-host disease in Japan. Int J Hematol. 2015;101(5):467–86.
Czock D, Keller F, Rasche FM, Haussler U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin Pharmacokinet. 2005;44(1):61–98.
Theuretzbacher U, Ihle F, Derendorf H. Pharmacokinetic/pharmacodynamic profile of voriconazole. Clin Pharmacokinet. 2006;45(7):649–63.
Imataki O, Yamaguchi K, Uemura M, Fukuoka N. Voriconazole concentration is inversely correlated with corticosteroid usage in immunocompromised patients. Transpl Infect Dis. 2018;20(4):e12886.
Hope WW. Population pharmacokinetics of voriconazole in adults. Antimicrob Agents Chemother. 2012;56(1):526–31.
McDougall DA, Martin J, Playford EG, Green B. Determination of a suitable voriconazole pharmacokinetic model for personalised dosing. J Pharmacokinet Pharmacodyn. 2016;43(2):165–77.
Pascual A, Csajka C, Buclin T, Bolay S, Bille J, Calandra T, et al. Challenging recommended oral and intravenous voriconazole doses for improved efficacy and safety: population pharmacokinetics-based analysis of adult patients with invasive fungal infections. Clin Infect Dis. 2012;55(3):381–90.
Matsumoto K, Ikawa K, Abematsu K, Fukunaga N, Nishida K, Fukamizu T, et al. Correlation between voriconazole trough plasma concentration and hepatotoxicity in patients with different CYP2C19 genotypes. Int J Antimicrob Agents. 2009;34(1):91–4.
Han K, Capitano B, Bies R, Potoski BA, Husain S, Gilbert S, et al. Bioavailability and population pharmacokinetics of voriconazole in lung transplant recipients. Antimicrob Agents Chemother. 2010;54(10):4424–31.
Scholz I, Oberwittler H, Riedel KD, Burhenne J, Weiss J, Haefeli WE, et al. Pharmacokinetics, metabolism and bioavailability of the triazole antifungal agent voriconazole in relation to CYP2C19 genotype. Br J Clin Pharmacol. 2009;68(6):906–15.
Nakamura K, Goto F, Ray WA, McAllister CB, Jacqz E, Wilkinson GR, et al. Interethnic differences in genetic polymorphism of debrisoquin and mephenytoin hydroxylation between Japanese and Caucasian populations. Clin Pharmacol Ther. 1985;38(4):402–8.
Jurima M, Inaba T, Kadar D, Kalow W. Genetic polymorphism of mephenytoin p(4’)-hydroxylation: difference between Orientals and Caucasians. Br J Clin Pharmacol. 1985;19(4):483–7.
Horai Y, Nakano M, Ishizaki T, Ishikawa K, Zhou HH, Zhou BI, et al. Metoprolol and mephenytoin oxidation polymorphisms in Far Eastern Oriental subjects: Japanese versus mainland Chinese. Clin Pharmacol Ther. 1989;46(2):198–207.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by JSPS KAKENHI Grant number JP20K16078.
Conflict of interest
Kimitaka Suetsugu, Shota Muraki, Junshiro Fukumoto, Ryosuke Matsukane, Yasuo Mori, Takeshi Hirota, Toshihiro Miyamoto, Nobuaki Egashira, Koichi Akashi, and Ichiro Ieiri have no conflicts of interest that are directly relevant to the content of this article.
Ethics approval
This study was conducted in accordance with the Declaration of Helsinki and its amendments; the study was approved by the Ethics Committee of Kyushu University Graduate School and Faculty of Medicine (approval no. 652–03).
Consent to participate
No informed consent from patients was required as this as a retrospective study.
Consent for publication
All authors approved this manuscript for submission.
Availability of data and material
The datasets generated and/or analyzed during the current study are not publicly available because of privacy issues but are available from the corresponding author upon reasonable request.
Code availability
Not applicable.
Author contributions
KS, SM, and II contributed to the study design. Data collection and analysis was performed by KS, SMki, and JF. The first draft of the manuscript was written by KS, and the final version of the manuscript was approved by all authors.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Suetsugu, K., Muraki, S., Fukumoto, J. et al. Effects of Letermovir and/or Methylprednisolone Coadministration on Voriconazole Pharmacokinetics in Hematopoietic Stem Cell Transplantation: A Population Pharmacokinetic Study. Drugs R D 21, 419–429 (2021). https://doi.org/10.1007/s40268-021-00365-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-021-00365-0