
Abstract
Background
Cutaneous neurofibromas cause disfigurement and discomfort in individuals with neurofibromatosis type 1 (NF-1).
Methods
The primary objective of this phase II, open-label, single-arm trial was to assess whether orally administered everolimus reduced the surface volume of cutaneous neurofibromas in patients with NF-1.
Results
Of 22 patients who took the study drug, 17 completed the trial; 5 patients withdrew due to adverse events. Sixteen patients had photographs of sufficient quality for assessment of the primary outcome. A significant reduction in lesion surface volume, defined as an end of trial volume > 2 standard errors (SE) less than baseline volume, was observed for 4/31 lesions (13%) from 3/16 patients (19%). Additionally, a statistically significant absolute change in average height for paired lesions was observed (p = 0.048). Although not a prespecified outcome measure, a dramatic reduction in the size of 3 large plexiform neurofibromas with a cutaneous component was also noted and documented by measurement of maximum circumference or magnetic resonance imaging-based volumetric analysis. Adverse events were common in this trial, but no serious adverse events occurred.
Conclusions
Although this was a small, exploratory trial that was not powered for significance, the reduction in surface volume observed in this study is noteworthy assuming that the natural course for untreated lesions is to maintain or increase in volume. Future studies are needed with larger study populations that incorporate longer durations of treatment and better standardization of volumetric measurements.
Trial Registration ClinicalTrials.gov Identifier: NCT02332902
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Statistically significant reductions in lesion surface volume and absolute change in average height for paired lesions were observed with the use of orally administered everolimus as a treatment for neurofibromatosis type 1-associated cutaneous neurofibromas |
Dramatic reductions in the size of three large plexiform neurofibromas with a cutaneous component were also observed by measurement of maximum circumference or magnetic resonance imaging-based volumetric analysis |
Adverse events were common, but no serious adverse events occurred |
1 Introduction
Neurofibromatosis type 1 (NF-1) is an autosomal dominant genetic disorder resulting from pathogenic variants in the NF1 gene encoding the protein neurofibromin. Neurofibromatosis type 1 affects ~ 1 in 3000 births and is characterized by cutaneous, subcutaneous, plexiform, and deep neurofibromas, café-au-lait macules, axillary and inguinal freckling, optic glioma, iris hamartomas (Lisch nodules), and neurologic, musculoskeletal, cardiac, vascular, and cerebrovascular abnormalities. The occurrence, number, and size of neurofibromas in individuals with NF-1 increase with age, causing disfigurement and discomfort. Plexiform neurofibromas occur in ~ 50% of individuals with NF-1, may have a cutaneous component, and cause significant disfigurement, morbidity, and mortality. Options for medical management of cutaneous and plexiform neurofibromas in patients with NF-1 are lacking [1,2,3,4,5].
Loss of neurofibromin function in patients with NF-1 leads to activation of Ras and downstream signaling pathways, including the mammalian target of rapamycin (mTOR) signaling pathway. Disinhibition of mTOR is associated with unregulated protein synthesis, cell overgrowth, and tumor formation. Everolimus, a derivative of the compound rapamycin, inhibits activity of the mTOR protein. Prior studies have demonstrated that rapamycin and everolimus effectively inhibit proliferation of neurofibromin-deficient cells in vitro and NF1-associated tumors in vivo including astrocytes, malignant peripheral nerve sheath tumor cells, genetically engineered neurofibromin-deficient mouse embryonic fibroblasts, malignant peripheral nerve sheath tumors xenografts, and genetically engineered mouse models [1, 6,7,8,9,10,11,12,13,14,15,16,17,18].
In 2014, Hua et al. reported the use of sirolimus (rapamycin) for the management of NF1-associated plexiform neurofibromas in three patients whose plexiform neurofibromas remained stable in size with treatment [19]. In 2014 and 2015, Weiss et al. reported results of a phase II study in which treatment with sirolimus increased the time to progression of progressive plexiform neurofibromas in patients with NF-1 but did not reduce the size of non-progressive plexiform neurofibromas [20, 21].
The purpose of this exploratory trial was to assess, in a small patient population, the potential clinical utility and safety of everolimus as a treatment for NF1-associated cutaneous neurofibromas. The primary objective of this trial was to determine whether orally administered everolimus reduced the surface volume of cutaneous neurofibromas in patients with NF-1. Safety analyses included monitoring of specific serum analytes and assessment of adverse events.
2 Methods
2.1 Study Design
This was a phase II, open-label, single-arm trial [trial protocol available in the Electronic Supplementary Material (ESM)]. Patients were recruited from two clinical sites in Houston, TX, USA: The University of Texas MD Anderson Cancer Center and the McGovern Medical School, The University of Texas Health Science Center at Houston. This study was approved by the University of Texas McGovern Medical School Committee for the Protection of Human Subjects (HSC-14-0758). This trial was registered with ClinicalTrials.gov Identifier NCT02332902. The trial duration was 6 months.
2.2 Study Population
Eligible patients were at least 18 years of age, had a clinical diagnosis of NF-1, disfiguring cutaneous neurofibromas in a region amenable to photography, satisfactory results on baseline safety evaluations including bone marrow, liver and renal function, and lipid profile, and were willing and able to comply with the requirements of the trial. Female patients of childbearing potential were confirmed to not be pregnant prior to starting the trial by assessment of blood beta-human chorionic gonadotropin levels and were required to use highly effective methods of contraception throughout the duration of the trial. Male patients with partners of childbearing potential were required to use adequate contraception for the duration of the trial and for 8 weeks after conclusion of the trial.
Patients were excluded if they were currently receiving anticancer treatments or had received anticancer treatments within the previous 4 weeks, had known intolerance or hypersensitivity to everolimus or other rapamycin analogs, had known impairment of gastrointestinal function or gastrointestinal disease, had uncontrolled diabetes mellitus, had any severe or uncontrolled heart, liver, lung, or bleeding condition, were receiving systemic treatment with corticosteroids or other immunosuppressive agents, had a history of seropositivity for human immunodeficiency virus, had received live attenuated vaccines within the previous week, had a history of primary malignancy (except for non-melanoma skin cancer or carcinoma in situ of the cervix, uterus, or breast from which the patient has been disease free for 3 or more years), were pregnant or lactating, had participated in any clinical trial with an investigational drug within the previous month, or had a history of non-compliance with medical regimens or were considered unlikely to comply with the study protocol. Written informed consent was obtained from all patients included in this study.
2.3 Interventions
Enrolled patients were provided a starting 10-mg dose of everolimus (Novartis Pharmaceuticals, Basel, Switzerland) and instructed to take the medication daily, by mouth, in the morning, consistently with or without food. Serum everolimus concentrations were assessed 2 weeks after trial initiation; the target trough concentration for all patients was 5–15 ng/mL. If needed, the daily dose of everolimus was adjusted up or down by 2.5 mg, at 2-week intervals, until the target trough concentration was achieved. Serum everolimus concentrations were also assessed in all patients 2 weeks after any dose change and at 3 and 6 months following enrollment. Complete blood counts, bilirubin, alanine aminotransferase, aspartate aminotransferase, international normalized ratio, total cholesterol, fasting triglycerides and glucose, serum creatinine, vascular endothelial growth factor (VEGF)-A, VEGF-D, soluble VEGF receptor (sVEGFR)-1, sVEGFR-2, phosphatidylinositol glycan anchor biosynthesis class F, c-Kit, collagen type IV, and beta-human chorionic gonadotropin, as appropriate, were measured at baseline and 3 and 6 months. Protocol guidelines for dose reduction and interruption or discontinuation of the study drug were defined according to the type, severity, and recurrence of adverse events, including hematologic and non-hematologic toxicities, and the diagnosis of infection (ESM).
At enrollment, four target cutaneous neurofibromas for each patient were identified by the principal investigators. All four target lesions were photographed and lesion 1 was biopsied. At 3 months, lesions 2–4 were photographed and lesion 2 was biopsied. At the end of the trial, lesions 3 and 4 were photographed and lesion 3 was biopsied.
Two- and three-dimensional photographs were taken of each lesion with a three-dimensional LifeViz™ Micro photography system (QuantifiCare, Valbonne, France), with a split lens and polarized flash illumination to capture simultaneously two images of the same surface, at different viewing angles. A stereovision algorithm reconstructed and quantitatively analyzed the skin surface three dimensionally, permitting measurement of the surface volume and depth of the lesions.
Three patients had large plexiform fibromas with a cutaneous component. These lesions were not amenable to assessment with three-dimensional photography. Lesion circumference was measured by the investigators pre- and post-treatment, where possible. Otherwise, magnetic resonance images taken clinically of these patients were analyzed pre- and post-treatment to evaluate for any response.
2.4 Outcome Measures
All patients still enrolled at the end of the trial were included in the analysis of outcomes, regardless of whether interruptions of the study drug had occurred during the course of the trial. The primary outcome measure was any reduction in cutaneous neurofibroma lesion volume assuming the natural course for untreated lesions is to maintain or increase volume. Secondary outcomes were reductions in lesion average and maximum height.
Safety analyses consisted of laboratory tests at baseline and 3 and 6 months and the documentation and assessment of adverse events. Laboratory tests included the assessment of serum everolimus cooncentrations, complete blood counts, bilirubin, alanine aminotransferase, aspartate aminotransferase, international normalized ratio, total cholesterol, fasting triglycerides and glucose, serum creatinine, VEGF-A, VEGF-D, sVEGFR-1, sVEGFR-2, phosphatidylinositol glycan anchor biosynthesis class F, c-Kit, collagen type IV, and beta-hCG, as appropriate. Adverse-event monitoring continued for at least 30 days after completion of the trial. Adverse events were described clinically, enumerated as categorical traits, and evaluated for seriousness, duration, study relatedness, action taken, whether therapy was given, and outcome.
2.5 Statistical Analysis
The paired difference between baseline and 6-month measurements was calculated for each cutaneous neurofibroma lesion. A significant reduction in size was defined as a 6-month volume that was 2 standard errors (SE) less than the baseline volume. Standard error for the volume measurement was 1.46 mm3 as determined by the Lifeviz Micro validation study. Means (95% confidence intervals) were calculated by an intercept-only random-effects model with a robust sandwich variance estimator to account for repeated lesions measured within each subject. All analysis was completed with Stata 13 SE (College Station, TX, USA). A two-sided p value of 0.05 was used to determine statistical significance.
3 Results
Patients were recruited between February and September 2015. As this was an open-label, single-arm trial, all patients were assigned to a single treatment group. Twenty-five patients enrolled in the study. One was found to be pregnant on screening laboratory studies and was considered a screen failure. Two patients withdrew consent immediately and never took the study drug, leaving 22 patients entered into the interventional phase of the trial. Baseline characteristics of patients are shown in Table 1.
Of the 22 patients who took the study drug, dose interruptions of the study drug occurred for eight patients (36%) as a result of adverse events. Five patients (23%) withdrew secondary to adverse events. Adverse events are detailed in Table 2. Protocol deviations included one or more missed doses of the study medication (eight patients, 36%), need for repeat biopsy (three patients, 14%), missing or lost laboratory tests (six patients, 27%), missed vital signs (two patients, 9%), and ingestion of the study drug prior to a review of the results of screening laboratory tests by the principal investigator (two patients, 9%).
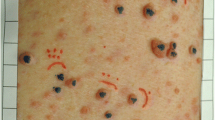
Seventeen patients completed the trial, but only 16 had photographs of sufficient quality for analysis of the primary outcome. Baseline and end-of-trial volumetric and height measurements were obtained for 31 cutaneous neurofibromas: 15 patients had complete data for two lesions; one patient had complete data for only one lesion. Representative two-dimensional photographs of patient lesions are shown in Fig. 1.

Two-dimensional photographs of cutaneous neurofibromas pre- (a, c) and post- (b, d) treatment with everolimus. The photographs illustrate visible reductions in the surface volume and height of lesions post-treatment
A significant reduction in lesion surface volume, defined as an end-of-trial volume > 2 SE less than baseline volume, was observed for 4/31 lesions (13%) from 3/16 patients (19%). In total, a reduction in surface volume of any amount was observed for 21/31 lesions (68%) from 13/16 patients (81%), including three lesions (10%) from two patients (13%) that showed a reduction in surface volume of 25% or more, of which one lesion showed a reduction in surface volume of greater than 50%. Overall, however, neither the absolute change in surface volume for paired lesions (p = 0.582) nor the percent change in surface volume for paired lesions (p = 0.425) was statistically significant.
A reduction in average lesion height was observed for 19/31 lesions (61%) from 13/16 patients (81%), including three lesions (10%) from three patients (19%) that showed a reduction in average height of 25% or more, of which one lesion showed a reduction in average height of greater than 50%. Overall, a statistically significant absolute change in average height for paired lesions was observed (p = 0.048), but the percent change in average height for paired lesions was not statistically significant (p = 0.080).
A reduction in maximum lesion height was observed for 19/31 lesions (61%) from 13/16 patients (81%) including two lesions (6%) from two patients (13%) that showed a reduction in maximum height of 25% or more, of which one lesion showed a reduction in maximum height of greater than 50%. Overall, however, neither the absolute change in maximum height for paired lesions (p = 0.948) nor the percent change in maximum height for paired lesions (p = 0.574) was statistically significant.
Of the three patients with large plexiform lesions with a cutaneous component, one had a lesion amenable to circumferential measurement. This lesion demonstrated a marked reduction in maximal circumference from 46 to 38 cm over the 6-month treatment period (Fig. 2). The other two patients with large plexiform lesions with a cutaneous component had pre- and post-treatment magnetic resonance imaging scans performed clinically and consented to volumetric analysis of these films. One lesion decreased 35% from 1.93 to 1.26 cm3 and the other decreased 13% from 61.25 to 53.44 cm3 during the course of this trial (Fig. 3).

Circumferential measurement of the left foot neurofibroma of one patient pre- (a) and post- (b) treatment with everolimus. The photographs illustrate a reduction in maximal circumference from 46 to 38 cm over the 6-month treatment period

Magnetic resonance images of the right lower leg and tibia neurofibroma of one patient pre- (a) and post- (b) treatment with everolimus and the left-sided face neurofibroma of another patient pre- (c) and post- (d) treatment with everolimus. The images illustrate a 35% reduction in volume of the leg lesion, from 1.93 to 1.26 cm3, and a 13% reduction in volume of the facial lesion, from 61.25 to 53.44 cm3
No serious adverse events occurred. Two patients never took the study drug and were excluded from adverse-event analysis. Of the 22 patients included in the assessment of adverse events, all patients (100%) experienced at least one adverse event over the course of this trial. Adverse events occurring in > 5% of patients included stomatitis, upper respiratory infections, gastrointestinal upset, skin disorders, decreased oral intake/weight loss, headache, tooth infection, urinary tract infection, fatigue, and vaginitis. A detailed list of all adverse events is provided in Table 2.
4 Discussion
Prior studies have investigated whether mTOR inhibitors such as rapamycin or everolimus can inhibit the proliferation of neurofibromin-deficient cells in vitro and in vivo, and limited studies have evaluated the effect of these compounds on patients with NF-1-associated plexiform neurofibromas [6,7,8,9, 12, 17,18,19,20,21]. This trial is, to the best of our knowledge, the first to date to evaluate the efficacy and safety of everolimus for reducing the surface volume of cutaneous neurofibromas in patients with NF-1.
Although this was a small exploratory trial that was not powered for significance, there were several noteworthy observations. A significant reduction in the lesion surface volume of cutaneous neurofibromas, defined as an end-of-trial volume > 2 SE less than baseline volume, was observed for 4/31 lesions (13%) from 3/16 patients (19%). Additionally, a statistically significant absolute change in average height for paired lesions was observed (p = 0.048). Assuming the natural course for untreated lesions is to maintain or increase volume, these observations in a trial of such short duration are intriguing.
Neither the percent change in average lesion height nor the absolute or percent change in maximum lesion height achieved statistical significance. Nonetheless, efficacy was suggested, raising the question as to whether a longer duration of treatment or a larger sample size might produce more compelling effects. During the course of this trial, investigators also noticed that surface lesions appeared to become softer after treatment with everolimus, but no objective measurement of this observation was made.
In addition to the pre-specified outcome measures, investigators noted a dramatic reduction in the size of three large plexiform neurofibromas with a cutaneous component by measurement of the maximum circumference or magnetic resonance imaging-based volumetric analysis. These data also suggest that further trials might prove mTOR inhibitors to be beneficial for plexiform lesions with a cutaneous component in patients with NF-1.
Adverse events were common in this trial. All patients who took the study medication experienced at least one adverse event; however, no serious adverse events occurred. Similar to prior studies, [22, 23] the adverse events that occurred in this study affected a wide variety of organ systems. The most common adverse events observed in this trial included stomatitis, upper respiratory disorders, skin irritation, gastrointestinal upset, and decreased oral intake/weight loss.
Chief limitations of this study are the small size of the study population and the short duration of the trial. The small size of the study population limited statistical power. The short duration of the trial could have minimized changes in lesion surface volume and height and restricted the occurrence of adverse events.
5 Conclusions
A significant reduction in lesion surface volume of cutaneous neurofibromas, defined as an end-of-trial volume > 2 SE less than baseline volume, was observed for 4/31 lesions (13%) from 3/16 patients (19%). A statistically significant absolute change in average height for paired lesions was also observed (p = 0.048). Changes in percent average height and maximum height of target lesions were not statistically significant. Adverse events were common in this trial, but no serious adverse events occurred. Future studies are needed to more rigorously evaluate the efficacy and safety of everolimus for the treatment of NF1-associated cutaneous neurofibromas. Future studies will need to include larger study populations and incorporate longer durations of treatment and better standardization of volumetric measurements and methods for measuring lesion density.
References
Franz DN, Weiss BD. Molecular therapies for tuberous sclerosis and neurofibromatosis. Curr Neurol Neurosci Rep. 2012;12(3):294–301.
Friedman JM. Neurofibromatosis 1. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews®. Seattle (WA): GeneReviews; 1993. https://www.ncbi.nlm.nih.gov/books/NBK1109/. Accessed 24 Apr 2018.
Gutmann DH, Ferner RE, Listernick RH, et al. Neurofibromatosis type 1. Nat Rev Dis Primers. 2017;3:17004.
Adams L, Marshall L. Neurofibromatosis type 1. Nat Rev Dis Primers. 2017;3:17005.
Roth TM, Petty EM, Barald KF. The role of steroid hormones in the NF1 phenotype: focus on pregnancy. Am J Med Genet A. 2008;146A(12):1624–33.
Dasgupta B, Yi Y, Chen DY, et al. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 2005;65(7):2755–60.
Endo M, Yamamoto H, Setsu N, et al. Prognostic significance of AKT/mTOR and MAPK pathways and antitumor effect of mTOR inhibitor in NF1-related and sporadic malignant peripheral nerve sheath tumors. Clin Cancer Res. 2013;19(2):450–61.
Johannessen CM, Reczek EE, James MF, et al. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci USA. 2005;102(24):8573–8.
Johansson G, Mahller YY, Collins MH, et al. Effective in vivo targeting of the mammalian target of rapamycin pathway in malignant peripheral nerve sheath tumors. Mol Cancer Ther. 2008;7(5):1237–45.
Rosner M, Hanneder M, Siegel N, et al. The mTOR pathway and its role in human genetic diseases. Mutat Res. 2008;659(3):284–92.
Sandsmark DK, Pelletier C, Weber JD, et al. Mammalian target of rapamycin: master regulator of cell growth in the nervous system. Histol Histopathol. 2007;22(8):895–903.
Zou CY, Smith KD, Zhu QS, et al. Dual targeting of AKT and mammalian target of rapamycin: a potential therapeutic approach for malignant peripheral nerve sheath tumor. Mol Cancer Ther. 2009;8(5):1157–68.
Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4(5):335–48.
Narayanan V. Tuberous sclerosis complex: genetics to pathogenesis. Pediatr Neurol. 2003;29(5):404–9.
Houghton PJ. Everolimus. Clin Cancer Res. 2010;16(5):1368–72.
Paghdal KV, Schwartz RA. Sirolimus (rapamycin): from the soil of Easter Island to a bright future. J Am Acad Dermatol. 2007;57(6):1046–50.
Hegedus B, Banerjee D, Yeh TH, et al. Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res. 2008;68(5):1520–8.
Johannessen CM, Johnson BW, Williams SM, et al. TORC1 is essential for NF1-associated malignancies. Curr Biol. 2008;18(1):56–62.
Hua C, Zehou O, Ducassou S, et al. Sirolimus improves pain in NF1 patients with severe plexiform neurofibromas. Pediatrics. 2014;133(6):e1792–7.
Weiss B, Widemann BC, Wolters P, et al. Sirolimus for progressive neurofibromatosis type 1-associated plexiform neurofibromas: a neurofibromatosis Clinical Trials Consortium phase II study. Neuro Oncol. 2015;17(4):596–603.
Weiss B, Widemann BC, Wolters P, et al. Sirolimus for non-progressive NF1-associated plexiform neurofibromas: an NF clinical trials consortium phase II study. Pediatr Blood Cancer. 2014;61(6):982–6.
Bissler JJ, McCormack FX, Young LR, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358(2):140–51.
Franz DN, Belousova E, Sparagana S, et al. Long-term use of everolimus in patients with tuberous sclerosis complex: final results from the EXIST-1 study. PLoS One. 2016;11(6):e0158476.
Acknowledgements
We are indebted to the patients and families who participated in this trial. We also thank Matthew J. Scheible of the Graduate School of Biomedical Sciences, The University of Texas Health Science Center at Houston for assistance with data verification, and Raye L. Alford, Ph.D., of Genetics Genomics Consulting LLC for assistance with the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported by Novartis Pharmaceuticals (CRAD001CUS232T) and a research grant from the Texas Neurofibromatosis Foundation.
Conflict of Interest
John M. Slopis, Joshua A. Samuels, and Mary Kay Koenig report grants and personal fees from Novartis Pharmaceuticals. Keri C. Smith reports a grant from the Texas Neurofibromatosis Foundation. Joshua A. Samuels reports personal fees from MedStudy, Inc. Adelaide A. Hebert, Hope Northrup, and Mary Kay Koenig report provisional patents pending. John M. Slopis is employed by The University of Texas MD Anderson Cancer Center. Octavio Arevalo, Adelaide A. Hebert, Hope Northrup, Riascos, Joshua A. Samuels, Keri C. Smith, Mary Kay Koenig, Cynthia S. Bell, and Patti Tate are or were employed by The University of Texas Health Science Center at Houston at the time of this trial.
Data Availability
All data generated or analyzed during this study are included in this published article and the Electronic Supplementary Material.
Ethics Approval
The study protocol along with consent forms were reviewed and approved by the Committee for the Protection of Human Subjects at the University of Texas Health Science Center at Houston (HSC-MS-14-0758).
Consent to Participate
All subjects consented to participate in this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Slopis, J.M., Arevalo, O., Bell, C.S. et al. Treatment of Disfiguring Cutaneous Lesions in Neurofibromatosis-1 with Everolimus: A Phase II, Open-Label, Single-Arm Trial. Drugs R D 18, 295–302 (2018). https://doi.org/10.1007/s40268-018-0248-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-018-0248-6