
Abstract
Sparsentan (FILSPARI™) is an oral, dual endothelin angiotensin receptor antagonist that is being developed by Travere Therapeutics for the treatment of immunoglobulin A (IgA) nephropathy and focal segmental glomerulosclerosis (FSGS). In February 2023, sparsentan received accelerated approval in the USA for reducing proteinuria in adults with primary IgA nephropathy who are at risk of rapid disease progression. This article summarizes the milestones in the development of sparsentan leading to this first approval for IgA nephropathy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Digital Features for this AdisInsight Report can be found at https://doi.org/10.6084/m9.figshare.22313596 |
A dual endothelin angiotensin receptor antagonist is being developed by Travere Therapeutics for the treatment of IgA nephropathy and FSGS |
Received its first approval on 17 February 2023 in the USA under the Accelerated Approval pathway |
Approved for reducing proteinuria in adults with primary IgA nephropathy who are at risk of rapid disease progression |
1 Introduction
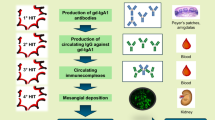
Immunoglobulin A (IgA) nephropathy, also known as Berger’s disease, is a rare but serious immune complex-mediated glomerulonephritis, characterized by mesangial deposition of abnormally structured IgA and a variety of histopathologic lesions [1,2,3]. The IgA deposition in the kidney causes inflammation and glomerular injury, leading to leaking of blood (haematuria) and protein (proteinuria) in the urine [1,2,3]. IgA nephropathy is the most common type of primary glomerulonephritis, with 30–40% of patients progressing to kidney failure within 20–30 years of diagnosis [3].
Historically, the mainstay of treatment for IgA nephropathy has been optimal supportive care, aimed at reducing blood pressure and proteinuria, and minimising lifestyle risk factors [4]. Immunosuppressive therapy with corticosteroids, with its own safety risks, may also have a place in the management of this disease. There is clearly a high unmet medical need for effective and well tolerated treatments for patients with IgA nephropathy who are at high risk for progressive kidney function decline [4]. Clinical studies have shown that dual inhibition of the renin-angiotensin-aldosterone system (RAAS) and endothelin-1 provided nephroprotection and reduced proteinuria in patients with chronic kidney disease [5].
Sparsentan (FILSPARI™) is a single, small-molecule, oral, dual endothelin angiotensin receptor antagonist (DEARA) that is being developed by Travere Therapeutics (previously Retrophin) for the treatment of IgA nephropathy and focal segmental glomerulosclerosis (FSGS). On 17 February 2023, sparsentan received accelerated approval in the USA for reducing proteinuria in adults with primary IgA nephropathy who are at risk of rapid disease progression, generally a urine protein-to-creatinine ratio (UP/C) ≥ 1.5 g/g [6, 7]. It is the first and currently the only approved non-immunosuppressive treatment for IgA nephropathy. The approval was based on interim results from the ongoing phase III PROTECT trial (Sect. 2.3.1). The recommended sparsentan dosage is initially 200 mg once daily for 14 days, then increased to 400 mg once daily, as tolerated [7]. Dosage adjustments/modifications are required for elevations in aminotransferase levels and when sparsentan is coadministered with a strong CYP3A inhibitor. Because of the potential increase in liver and embryo-foetal toxicity risks (Sect. 2.4), sparsentan is available in the USA only through a Risk Evaluation and Mitigation Strategy (REMS) programme, called FILSPARI REMS [7]. Sparsentan has been filed for approval in the EU for the treatment of IgA nephropathy [8].

Key milestones in the development of sparsentan. CMAA Conditional Marketing Authorization Application, est. estimated, FSGS focal segmental glomerulosclerosis, IgAN immunoglobulin A nephropathy, NDA New Drug Application, PDUFA Prescription Drug User Fee Act
Sparsentan is in phase III development for FSGS. The drug was also evaluated for hypertension in phase II trials by Ligand Pharmaceuticals in 2011, but the clinical development for this indication is discontinued. Sparsentan has orphan drug designation for IgA nephropathy [9, 10] and FSGS [11, 12] in the USA and EU.
1.1 Company Agreements
In September 2021, Vifor Pharma Group (now CSL Vifor) acquired exclusive commercialization rights to sparsentan in Europe, Australia and New Zealand from Travere Therapeutics in exchange for upfront, milestone and royalty payments [13].
In February 2012, Travere Therapeutics acquired rights to a DEARA compound from Ligand Pharmaceuticals in exchange for upfront, milestone and royalty payments [14].
In April 2006, Pharmacopeia (now Ligand Pharmaceuticals) acquired exclusive worldwide rights to certain lead and backup DEARA candidates from Bristol-Myers Squibb Company in exchange for milestone and royalty payments [15].

Chemical structure of sparsentan
2 Scientific Summary
2.1 Pharmacodynamics
Endothelin-1 and angiotensin II signalling pathways contribute to the progression of IgA nephropathy [16]. Sparsentan simultaneously blocks these pathways by binding to endothelin type A and angiotensin II type 1 receptors, with high affinity (Ki 12.8 and 0.36 nM, respectively) and selectivity (> 500-fold over endothelin type B and angiotensin II type 2 receptors) [7].
In a mouse model of IgA nephropathy, sparsentan was similar to losartan in reducing blood pressure, but provided a more rapid reduction in albuminuria and greater protection from glomerulosclerosis than losartan, which may be attributed to the dual mechanism of action of sparsentan [17]. Sparsentan also showed nephroprotective effects in rat [18] and mouse [19] models of FSGS.
2.2 Pharmacokinetics
The pharmacokinetics of sparsentan are best described by a two-compartment model with first order absorption with lag time, dose-dependent bioavailability and first order elimination [20]. Following oral administration of single 200–1600 mg doses, sparsentan maximum plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC) increased in a less than dose-proportional manner [7]. Sparsentan displays time-dependent pharmacokinetics, potentially by inducing its own metabolism over time [7].
Following a single 400 mg oral dose, the median time to Cmax was ≈ 3 h [7]. After multiple administrations of 400 mg once daily (recommended dosage), steady-state plasma concentrations are reached within 7 days, with no accumulation. At steady state with 400 mg once daily, geometric mean values for sparsentan were: Cmax 6.47 μg/mL, AUC 63.6 μg·h/mL, apparent volume of distribution 61.4 L and half-life 9.6 h. About 99% of sparsentan is bound to plasma protein. The drug is predominantly metabolized by CYP3A. The apparent clearance of sparsentan is 3.88 L/h after the initial 400 mg dose and 5.11 L/h at steady state. In healthy subjects receiving a single radiolabelled sparsentan dose of 400 mg, 80% of the administered dose was recovered in faeces (9% unchanged) and 2% in urine (negligible quantity unchanged). The majority (82%) of the radioactivity was recovered within a 10-day sample collection period [7].
When a single oral dose of sparsentan 800 mg was administered with a high-fat and -calorie meal, sparsentan Cmax and AUC were increased by 22% and 108%, respectively, although such a meal had no clinically significant effect on the pharmacokinetics of a single 200 mg dose [7]. Sparsentan should be taken before the morning or evening meal and the dosing pattern with respect to meals should be maintained. Age (18–73 years), sex, race, mild to moderate reduction in estimated glomerular filtration rate (eGFR; 30–89 mL/min/1.73 m2), or mild to moderate liver function impairment (Child-Pugh class A or B) had no clinically significant effect on sparsentan pharmacokinetics. The effects of severe liver function impairment (Child-Pugh class C) or eGFR < 30 mL/min/1.73 m2 have not been studied [7].
In vitro, sparsentan is a substrate, inhibitor and inducer of CYP3A, an inducer of CYP2B6, CYP2C9 and CYP2C19, a substrate of p-glycoprotein and BCRP, and an inhibitor of p-glycoprotein, BCRP, OATP1B3 and OAT3 [7]. Coadministration of sparsentan with angiotensin receptor blockers, endothelin receptor antagonists or aliskiren (a renin inhibitor) is contraindicated due to increased risks of hypotension, syncope, hyperkalaemia and changes in kidney function. Clinically relevant drug interactions may occur when sparsentan is coadministered with strong CYP3A inhibitors, moderate CYP3A inhibitors, strong CYP3A inducers, antacids, acid reducing agents, nonsteroidal anti-inflammatory drugs (including selective cyclooxygenase inhibitors), CYP2B6, CYP2C9 and CYP2C19 substrates, sensitive p-glycoprotein and BCRP substrates, and agents that increase serum potassium. Consult local prescribing information for specific recommendations [7]. There were no clinically relevant drug-drug interactions when sparsentan is coadministered with dapagliflozin, a sodium-glucose co-transporter 2 inhibitor [21].
Features and properties of sparsentan
Alternative names | DEARA; FILSPARI; PS-433540; RE-021 |
Class | Anti-inflammatories; antihypertensives; isoxazoles; nerve growth factors; small molecules; spiro compounds; sulfonamides; urologics; vascular disorder therapies |
Mechanism of action | Angiotensin II type 1 receptor antagonists; endothelin type A receptor antagonists |
Route of administration | Oral |
Pharmacodynamics | Shows nephroprotective effects in rodent models of IgA nephropathy and focal segmental glomerulosclerosis |
Pharmacokinetics | Less than dose-proportional, time-dependent pharmacokinetics; time to peak plasma concentration ≈ 3 h; volume of distribution 61.4 L; apparent clearance 5.11 L/h; half-life 9.6 h |
Most frequent adverse events | Peripheral oedema, hypotension (including orthostatic hypotension), dizziness, hyperkalaemia, anaemia |
ATC codes | |
WHO ATC code | G04B-X (Other Urologicals) |
EphMRA ATC code | G4X (All Other Urological Products) |
Chemical name | 4′-((2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl)-N-(4,5-dimethylisoxazol-3-yl)-2'-(ethoxymethyl)-[1,1'-biphenyl]-2-sulfonamide |
2.3 Therapeutic Trials
2.3.1 IgA Nephropathy
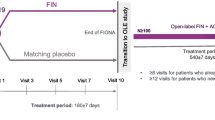
Sparsentan was more effective than irbesartan in reducing proteinuria in adult patients with IgA nephropathy in a randomized, double-blind, multinational phase III trial (NCT03762850; PROTECT) [7, 22, 23]. Eligible patients were aged ≥ 18 years and had biopsy-proven IgA nephropathy, eGFR ≥ 30 mL/min/1.73 m2 and total urine protein ≥ 1.0 g/day despite receiving a stable maximally tolerated dose of RAAS inhibitor at ≥ 50% of maximum labelled dose [22, 23]. Exclusion criteria included other glomerulopathies and recent treatment with systemic immunosuppressants [22, 23]. Stratified by eGFR and urine protein excretion values, patients were randomized to sparsentan (target dose 400 mg once daily) or irbesartan (target dose 300 mg once daily) for 114 weeks, followed by a sparsentan open-label extension period of 156 weeks [22]. At baseline, across the two treatment groups, mean eGFR was 57.0 mL/min/1.73 m2, median UP/C based on 24 h urine collection was 1.2 g/g and almost all patients were receiving a RAAS inhibitor [22]. In an interim analysis of the prespecified primary analysis set, mean percent change from baseline in UP/C at week 36 (primary endpoint) was significantly (p < 0.0001) higher in the sparsentan than in the irbesartan group (– 49.8% vs – 15.1%) [6]. In a post hoc sensitivity analysis (conducted at the request of the US FDA) that included the first 281 randomized patients, the geometric mean percent change from baseline in UP/C at week 36 was – 45% in the sparsentan group and – 15% in the irbesartan group (ratio 0.65; 95% CI 0.55–0.77; p < 0.0001) [7].
2.3.2 Focal Segmental Glomerulosclerosis
Sparsentan was more effective than irbesartan in reducing proteinuria in patients with FSGS in a randomized, double-blind, multinational phase II trial (NCT01613118; DUET) [24, 25]. Eligible patients were aged 8–75 years and had biopsy-proven FSGS or an FSGS-associated mutation in genes encoding podocyte proteins, UP/C ≥ 1.0 g/g and eGFR > 30 ml/min/1.73 m2. Chronic immunosuppressants were permitted if doses remained stable for ≥ 1 month before the trial [24]. After a 2-week washout period for RAAS inhibitors, patients were randomized to sparsentan 200, 400 or 800 mg once daily, or irbesartan 300 mg once daily for 8 weeks. Baseline demographic and disease characteristics were generally similar between the sparsentan and irbesartan groups; mean eGFR was 74.4 and 74.5 ml/min/1.73 m2, and median UP/C was 3.61 and 3.12 g/g, respectively. In the full analysis set, geometric mean percent reduction from baseline in UP/C at week 8 (primary endpoint) was significantly (p = 0.006) greater in the pooled (all doses combined) sparsentan group than in the irbesartan group (– 44.8% vs – 18.5%; n = 73 and 36, respectively). Similar results were seen for pooled sparsentan 400 and 800 mg groups versus irbesartan, although there was no significant difference between individual sparsentan dose groups versus irbesartan group. Significantly (p = 0.04) more sparsentan (all doses combined) than irbesartan recipients achieved FSGS partial remission end point (FPRE), defined as UP/C ≤ 1.5 g/g and a > 40% reduction in UP/C from baseline to week 8 (28% vs 9%; n = 64 and 32, respectively) [24]. In patients who continued sparsentan in an open-label extension of DUET, sustained proteinuria reduction was seen over up to 240 weeks [26].
Sparsentan showed promising interim efficacy in patients with FSGS in a randomized, double-blind, multinational, phase III trial (NCT03493685; DUPLEX) [27,28,29]. After a 2-week washout period, 371 patients aged 8–75 years with primary FSGS were randomized to sparsentan or irbesartan for 108 weeks, with final efficacy assessment planned at week 112 [27]. The primary endpoint is the slope of eGFR from week 6 to week 108 [27]. In an unblinded interim analysis in the first ≈ 190 patients reaching 36 weeks of treatment, significantly (p = 0.0094) more sparsentan than irbesartan recipients achieved FPRE (42.0% vs 26.0%) [29].
Key clinical trials of sparsentan sponsored by Travere Therapeutics
Drug(s) | Indication | Phase | Status | Location(s) | Identifier |
|---|---|---|---|---|---|
Sparsentan, irbesartan, dapagliflozin | IgA nephropathy | III | ANR | Multinational | NCT03762850; EudraCT 2017-004605-41; PROTECT |
Sparsentan, irbesartan | FSGS | III | ANR | Multinational | NCT03493685; EudraCT 2016-005141-23; DUPLEX |
Sparsentan, Irbesartan | FSGS | II | ANR | Multinational | NCT01613118; EudraCT 2014-002358-38; DUET |
Sparsentan | Proteinuric glomerular diseasesa | II | Recruiting | Multinational | NCT05003986; EudraCT 2021-000621-27; EPPIK |
2.4 Adverse Events
In the PROTECT trial in patients with IgA nephropathy, the most common (incidence ≥ 2%) adverse reactions with sparsentan over up to 110 weeks of treatment included peripheral oedema (14% vs 9% with irbesartan), hypotension (including orthostatic hypotension) [14% vs 6%], dizziness (13% vs 5%), hyperkalaemia (13% vs 10%), anaemia (5% vs 2%), acute kidney injury (4% vs 2%) and transaminase elevations (2.5% vs 2%) [7]. A decrease in haemoglobin (> 2 g/dL from baseline and below the lower limit of normal), potentially due to haemodilution, was seen in 11% of sparsentan and 5% of irbesartan recipients. However, no patient discontinued treatment because of anaemia or decreased haemoglobin in the PROTECT trial [7].
During the 8-week double-blind treatment period in the DUET trial in patients with FSGS, treatment-emergent adverse events (TEAEs) occurred in 76.7% of sparsentan and 72.2% of irbesartan recipients [24]. Drug-related TEAEs occurred in 43.8% and 36.1% of patient in the respective groups. The incidence of serious TEAEs (2.7% vs 2.8%) and TEAEs leading to discontinuation of treatment (2.7% vs 2.8%) were similar between the groups. The most common TEAEs with sparsentan (incidence ≥ 8% and difference ≥ 4% vs irbesartan) include hypertension (16.4% vs 8.3%), oedema (12.3% vs 2.8%), nausea (12.3% vs 8.3%, diarrhoea (8.2% vs 2.8%) and vomiting (8.2% vs 2.8%). TEAEs that were less common with sparsentan than with irbesartan included nasal congestion (2.7% vs 11.1%), fatigue (4.1% vs 11.1%), muscle spasms (0% vs 5.6%), hyperkalaemia (1.4% vs 5.6%) and upper respiratory tract infection (2.7% vs 5.6%) [24]. No new or unexpected TEAEs were seen over up to 240 weeks of treatment with sparsentan [26].
The US prescribing information for sparsentan carries a boxed warning about the risk of liver toxicity and embryo-foetal toxicity [7]. Sparsentan antagonizes the endothelin receptor. Endothelin receptor antagonists are associated with a risk of elevated aminotransferases and liver toxicity/failure. This potential risk with sparsentan should be managed by monitoring liver enzyme levels and dosage modifications. Based on animal studies, sparsentan administered during pregnancy may cause foetal harm; therefore, it is contraindicated during pregnancy. Patients should be advised not to breastfeed during sparsentan treatment. Other warnings and precautions pertaining to the use of sparsentan include hypotension, acute kidney injury, hyperkalaemia and fluid retention [7].
2.5 Ongoing Clinical Trials
The PROTECT trial in patients with IgA nephropathy, and the DUET and DUPLEX trials in those with FSGS are still ongoing. An open-label, single-arm, multinational phase II trial (NCT05003986; EPPIK) is investigating the long-term antiproteinuric efficacy, nephroprotective effects and safety of sparsentan in paediatric patients with FSGS, minimal change disease, IgA nephropathy, IgA vasculitis or Alport syndrome [30].
The University of Leicester is conducting an open-label, single-group, single-centre phase II trial (NCT04663204; SPARTAN) to determine the nephroprotective potential of sparsentan in patients with newly diagnosed IgA nephropathy. The University of Edinburgh is conducting a randomized, double-blind, active-controlled phase II trial to assess the effect of sparsentan on vascular function in antineutrophil cytoplasmic autoantibody-associated vasculitis (NCT05630612; SPARVASC).
3 Current Status
Sparsentan received its first approval on 17 February 2023 in the USA under the Accelerated Approval pathway to reduce proteinuria in adults with primary IgA nephropathy who are at risk of rapid disease progression, generally a UP/C ≥ 1.5 g/g [6].
Change history
06 June 2023
A Correction to this paper has been published: https://doi.org/10.1007/s40265-023-01900-w
References
Donadio JV, Grande JP. IgA nephropathy. N Engl J Med. 2002;347(10):738–48.
Fabiano RC, Pinheiro SV, Simoes ESAC. Immunoglobulin A nephropathy: a pathophysiology view. Inflamm Res. 2016;65(10):757–70.
Lai KN, Tang SC, Schena FP, et al. IgA nephropathy. Nat Rev Dis Primers. 2016;2:16001.
Floege J, Rauen T, Tang SCW. Current treatment of IgA nephropathy. Semin Immunopathol. 2021;43(5):717–28.
Komers R, Plotkin H. Dual inhibition of renin-angiotensin-aldosterone system and endothelin-1 in treatment of chronic kidney disease. Am J Physiol Regul Integr Comp Physiol. 2016;310(10):R877–84.
Travere Therapeutics. Travere Therapeutics announces FDA accelerated approval of FILSPARITM (sparsentan), the first and only non-immunosuppressive therapy for the reduction of proteinuria in IgA nephropathy [media release]. 17 Feb 2023. http://www.travere.com.
Travere Therapeutics Inc. FILSPARI™ (sparsentan) tablets, for oral use: US prescribing information. 2023. https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/216403s000lbl.pdf. Accessed 9 Mar 2023.
CSL Vifor. CSL Vifor and Travere Therapeutics announce EMA has accepted for review the Conditional Marketing Authorization application for sparsentan for the treatment of IgA Nephropathy [media release]. 22 Aug 2022. http://www.cslvifor.com.
Travere Therapeutics. Travere Therapeutics announces orphan drug designation for sparsentan for the treatment of IgA nephropathy [media release]. 12 Jan 2021. http://www.travere.com.
Travere Therapeutics. Travere Therapeutics announces European Commission has granted orphan designation to sparsentan for the treatment of IgA nephropathy [media release]. 18 Feb 2021. http://www.travere.com.
Retrophin. Retrophin receives european orphan drug designation for sparsentan for the treatment of focal segmental glomerulosclerosis [media release]. 16 Nov 2015. http://www.retrophin.com.
Ligand Pharmaceuticals. Ligand partner Retrophin receives orphan drug designation for sparsentan [media release]. 9 Jan 2015. http://www.ligand.com.
Vifor Pharma. Vifor Pharma and Travere Therapeutics announce licensing agreement for the commercialization of sparsentan in Europe, Australia and New Zealand [media release]. 16 Sep 2021. http://www.viforpharma.com.
Ligand Pharmaceuticals. Ligand licenses DARA program to Retrophin [media release]. 21 Feb 2012. http://www.ligand.com.
Pharmacopeia. Pharmacopeia licenses novel therapeutic candidates from Bristol-Myers Squibb [media release]. 3 Apr 2006. http://www.pharmacopeia.com.
Gesualdo L, Griffin S, Tharaux PL. The dual role of endothelin-1 and angiotensin II in disease progression of focal segmental glomerulosclerosis and IgA nephropathy. EMJ Nephrol. 2022. https://doi.org/10.33590/emjnephrol/22C0912.
Nagasawa H, Suzuki H, Jenkinson C, et al. Sparsentan, the dual endothelin angiotensin receptor antagonist (DEARA), attenuates albuminuria and protects from the development of renal injury to a greater extent than losartan in the GDDY mouse model of IGA nephropathy: a 16-week study [abstract no. MO261]. Nephrol Dial Transplant. 2022;37(Suppl 3):i183.
Bedard P, Jenkinson C, Komers R. Sparsentan protects the glomerular basement membrane and glycocalyx, and attenuates proteinuria in a rat model of focal segmental glomerulosclerosiS (FSGS) [abstract no. MO255]. Nephrol Dial Transplant. 2022;37(Suppl 3):i177.
Gyarmati G, Shroff U, Izuhara A, et al. Sparsentan improves glomerular blood flow and augments protective tissue remodeling in mouse models of focal segmental glomerulosclerosis (FSGS) [abstract no. FC 016]. Nephrol Dial Transplant. 2021;36(Suppl 1):i10.
Chen S, Wada R, Zhang L, et al. Population pharmacokinetic analysis of sparsentan in healthy volunteers and subjects with focal segmental glomerulosclerosis (FSGS) [abstract no. P-036]. Clin Pharmacol Ther. 2022;111(Suppl 1):S14.
Chen SC, Cai D, Winnett C, et al. Effect of multiple doses of sparsentan on the single-dose pharmacokinetics of dapagliflozin: an open-label drug–drug interaction study in healthy adults. Clin Pharmacol Drug Dev. 2023. https://doi.org/10.1002/cpdd.1231.
Wong M, Barratt J, Komers R, et al. Baseline characteristics of adults enrolled in the ongoing phase 3 randomized, double-blind, active-control trial of sparsentan for the treatment of immunoglobulin a nephropathy (PROTECT) [abstract no. MO209]. Nephrol Dial Transplant. 2022;37(Suppl 3):i142–4.
Barratt J, Rovin B, Diva U, et al. Implementing the kidney health initiative surrogate efficacy endpoint in patients with IgA nephropathy (the PROTECT trial). Kidney Int Rep. 2019;4(11):1633–7.
Trachtman H, Nelson P, Adler S, et al. DUET: a phase 2 study evaluating the efficacy and safety of sparsentan in patients with FSGS. J Am Soc Nephrol. 2018;29(11):2745–54.
Komers R, Gipson DS, Nelson P, et al. Efficacy and safety of sparsentan compared with irbesartan in patients with primary focal segmental glomerulosclerosis: randomized, controlled trial design (DUET). Kidney Int Rep. 2017;2(4):654–64.
Srivastava T, Tesar V, Campbell KN, et al. Long-term efficacy and safety of sparsentan in FSGS: 240-week analysis of the DUET open-label extension (OLE) [abstract no. FR-OR57]. J Am Soc Nephrol. 2022;33:33.
Komers R, Diva U, Inrig JK, et al. Study design of the phase 3 sparsentan versus irbesartan (DUPLEX) study in patients with focal segmental glomerulosclerosis. Kidney Int Rep. 2020;5(4):494–502.
Trachtman H, Radhakrishnan J, Komers R, et al. Baseline characteristics of patients enrolled in the ongoing phase 3 randomized, double-blind, active-control trial of sparsentan for the treatment of focal segmental glomerulosclerosis (DUPLEX) [abstract no. MO254]. Nephrol Dial Transplant. 2022;37(Suppl 3):i175–6.
Travere Therapeutics. Travere Therapeutics Announces achievement of interim proteinuria endpoint in the ongoing phase 3 DUPLEX study of sparsentan in focal segmental glomerulosclerosis [media release]. 2 Feb 2021. https://travere.com/.
Trachtman H, Saleem M, Coppo R, et al. Sparsentan for treatment of pediatric patients with selected proteinuric glomerular diseases: design of the phase 2 EPPIK study [abstract no. 927]. Arch Dis Child. 2022;107(Suppl 2):A96–7.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. Yahiya Y. Syed is a salaried employee of Adis International Ltd/Springer Nature, and declares no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
The original article has been revised due to retrospective open choice order.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Syed, Y.Y. Sparsentan: First Approval. Drugs 83, 563–568 (2023). https://doi.org/10.1007/s40265-023-01864-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-023-01864-x