
Abstract
Triheptanoin (Dojolvi™), a synthetic medium-chain triglyceride, is being developed by Ultragenyx Pharmaceutical as a pharmaceutical-grade anaplerotic compound for use in the treatment of inherited metabolic disorders. In June 2020, triheptanoin received its first regulatory approval, in the USA, for use as a source of calories and fatty acids for the treatment of pediatric and adult patients with molecularly confirmed long-chain fatty acid oxidation disorders (LC-FAOD). Triheptanoin has also been investigated for use as a treatment in a range of other metabolic disorders or other diseases where energy deficiency is implicated. This article summarizes the milestones in the development of triheptanoin leading to this first regulatory approval for use in the treatment of pediatric and adult patients with LC-FAOD.
Similar content being viewed by others


A synthetic medium-chain triglyceride is being developed by Ultragenyx Pharmaceutical for the treatment of inherited metabolic disorders |
Received its first regulatory approval on 30 June 2020 in the USA |
Approved for use as a source of calories and fatty acids for the treatment of pediatric and adult patients with molecularly confirmed LC-FAOD |
1 Introduction
Triheptanoin (Dojolvi™), a synthetic medium-chain triglyceride (MCT), is being developed by Ultragenyx Pharmaceutical for use in the treatment of inherited metabolic disorders [1, 2]. On 30 June 2020, triheptanoin received its first regulatory approval, in the USA, for use as a source of calories and fatty acids for the treatment of pediatric and adult patients with molecularly confirmed long-chain fatty acid oxidation disorders (LC-FAOD) [1, 3].
LC-FAOD are a group of rare, life-threatening, genetic disorders caused by inherited autosomal recessive defects in transport proteins or metabolic enzymes in the mitochondrial long-chain fatty acid β-oxidation pathway, preventing the conversion of long-chain fatty acids into energy [4,5,6]. Clinical manifestations of LC-FAOD resulting from deficiencies in mitochondrial energy metabolism include cardiomyopathy, exercise intolerance, frequent episodes of myalgia, recurrent rhabdomyolysis, and hypoglycemia. Management of LC-FAOD involves the avoidance of fasting, the use of diets high in carbohydrates and restricted in long-chain fatty acids, and diet supplementation with MCTs (generally involving mixtures of even-chain 8-, 10- and 12-carbon fatty acids) as a source of calories and fatty acids [4,5,6,7,8]. It was hypothesized that triheptanoin, a synthetic medium-chain triglyceride consisting of three odd-chain 7-carbon fatty acids on a glycerol backbone, may have additional clinical benefit over even-chain fatty acid MCTs, acting as an anaplerotic compound, with propionyl-CoA produced during its metabolism [9, 10].

Key milestones in the development of triheptanoin. CACT carnitine-acylcarnitine translocase deficiency, FAD fatty acid disorders, GSD-II glycogen storage disease type II, GSD-V glycogen storage disease type V, IND Investigational New Drug, LC-FAOD long-chain fatty acid oxidation disorders, NDA New Drug Application
Triheptanoin, produced by Ultragenyx in a pharmaceutical-grade form and supplied as an oral liquid, should be administered at mealtimes or with snacks with a recommended target daily dosage of triheptanoin of up to 35% of the patient’s total prescribed daily calorie intake (DCI) divided into at least four doses [1]. For patients switching from another MCT product, the other MCT should be discontinued before initiating triheptanoin at the last tolerated daily dosage of the other MCT; the total daily dosage of triheptanoin can then be increased by ~ 5% DCI every 2–3 days until the target dosage of up to 35% DCI is achieved. For patients not already receiving an MCT product, triheptanoin should be initiated at a total daily dosage of ~ 10% DCI (divided into at least four doses), with an increase to the recommended total daily dosage of up to 35% DCI over a period of 2–3 weeks. Triheptanoin is to be mixed with semi-solid foods or liquid prior to administration orally or enterally via a silicone or polyurethane feeding tube. Use of more frequent smaller doses can be considered in the case of tolerability issues. If the target daily dosage of up to 35% DCI is not achieved due to tolerability, the patient should be maintained at the maximum tolerated dosage. For gastrointestinal adverse reactions, dosage reduction can be considered until symptoms resolve [1].
In addition to development in LC-FAOD, phase II trials have been conducted, are ongoing, or are planned (by Ultragenyx and/or other organizations/institutions), to investigate triheptanoin in a range of other indications, including glucose transporter type 1 deficiency syndrome (GLUT1DS; De Vivo disease) [11, 12], Huntington’s disease [12], glycogen storage disease type V (GSD-V; McArdle’s disease) (NCT02919631; NCT02432768), refractory epilepsy [13, 14], alternating hemiplegia of childhood [15], adult polyglucosan body disease [16] and Rett syndrome (NCT02696044). After a phase III crossover trial conducted by Ultragenyx to investigate triheptanoin in GLUT1DS failed to meet its primary and key secondary endpoints [17], development of triheptanoin by Ultragenyx in GLUT1DS was discontinued [although phase II trials (NCT03301532 and NCT03181399) by other parties are continuing in this indication].

Chemical structure of triheptanoin
The US FDA granted Ultragenyx Orphan Drug designation for triheptanoin for fatty acid disorders in May 2006 [18], for glycogen storage disease type II (Pompe disease) in February 2008 [19], for GLUT1DS in October 2014 (since withdrawn) [20] and for fatty acid oxidation disorders in April 2015 [21]. The European Commission granted Ultragenyx Orphan Drug designation for triheptanoin for very-long-chain 3-hydroxyacyl-CoA-dehydrogenase deficiency in December 2012 (withdrawn in February 2016) [22]; for GLUT1DS in May 2015 (withdrawn in December 2019) [23]; for very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency in June 2015 [24]; for three other subtypes of LC-FAOD [i.e. long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHAD) [25], mitochondrial trifunctional protein deficiency [26], and carnitine palmitoyltransferase II deficiency (CPT II) [27] ] in July 2015; for GSD-V in July 2016 [28]; and for carnitine-acylcarnitine translocase deficiency in July 2020 [29]. In addition, in March 2019, the US FDA granted triheptanoin Fast Track designation and Rare Pediatric Disease designation [30]. Triheptanoin was launched by Ultragenyx in July 2020 and is now commercially available in the USA [31]. Triheptanoin is also under regulatory review in Canada and Brazil [32].
1.1 Company Agreements and Patent Information
Pharmaceutical-grade triheptanoin was originated by the Baylor Research Institute (BRI). In September 2012, Ultragenyx entered into an exclusive license agreement with BRI for certain intellectual property related to triheptanoin (including multiple patents, for the USA and in other jurisdictions), under which Ultragenyx is granted the sole right to develop, manufacture, and commercialize licensed products for all human and animal uses of triheptanoin [33]. In January 2013, Ultragenyx licensed exclusive North American rights to triheptanoin for the treatment of LC-FAOD [34], expanding the license to worldwide rights in July 2013 [35]. In August 2014, Ultragenyx entered into a license agreement with UniQuest Pty Ltd. for intellectual property related to the treatment of refractory epilepsy and other seizure-related and neurologic disorders with triheptanoin [36]. In January 2015, Ultragenyx entered into a license agreement with the Institut National de la Santé Et de la Recherche Médicale (INSERM) Transfert SA and the Institut du Cerveau et de la Moelle Épinière for intellectual property related to the treatment of Huntington’s disease with triheptanoin [37].
2 Scientific Summary
2.1 Pharmacology
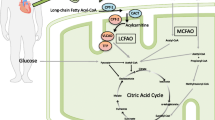
Triheptanoin is a synthetic, medium-chain triglyceride consisting of three odd-chain 7-carbon (heptanoate) fatty acids on a glycerol backbone [1, 9]. In LC-FAOD, triheptanoin is used as an anaplerotic compound, acting as a source of calories and fatty acids to bypass the LC-FAOD enzyme deficiencies [1, 9].
Following oral administration, triheptanoin undergoes hydrolysis by pancreatic lipases in the intestines, releasing the heptanoate molecules [1]. Multiple peak concentrations of heptanoate are observed following triheptanoin dosing. Furthermore, heptanoate pharmacokinetics exhibit a high level of inter-patient variability [1].
Heptanoate can traverse the double mitochondrial membrane independent of an active transport system or the carnitine carrier [8, 38]. Oxidation of heptanoate within the mitochondria yields two molecules of acetyl-CoA and one molecule of propionyl-CoA (an anaplerotic substrate) for each molecule of heptanoate [5, 8]. Heptanoate can also be metabolized to 4- and 5-carbon ketone bodies in the liver [1, 5, 9].
Pharmacologic effects of triheptanoin in patients with LC-FAOD (including improvements in cardiac function) are discussed in Sect. 2.2. An open-label clinical trial using brain magnetic resonance spectroscopy in patients with Huntington’s disease found that triheptanoin can improve brain energy metabolism (with apparent improvements in motor function) [39]. Other clinical trials found that triheptanoin may have some efficacy in reducing the frequency of focal seizures in adult patients with refractory epilepsy [14] but does not reduce the occurrence of paroxysmal events in alternating hemiplegia of childhood [15].
Triheptanoin pharmacodynamic effects have also been investigated in preclinical studies using mouse models of disease, notably in disorders relating to energy deficiencies. A study using a mouse model of Rett syndrome found that triheptanoin supplementation improved motor function, decreased adiposity, and increased glucose tolerance and insulin sensitivity [40]. In a study using a mouse model of amyotrophic lateral sclerosis, triheptanoin demonstrated neuroprotective properties, delaying the onset of motor symptoms [41]. Furthermore, triheptanoin supplementation to a ketogenic diet reduced memory impairment and astrogliosis in a mouse model of familial Alzheimer’s disease [42].
2.2 Therapeutic Efficacy
Triheptanoin supplementation increased left ventricular ejection fraction (LVEF) and reduced left ventricular wall mass at rest, and reduced heart rate during exercise compared with trioctanoin supplementation over 4 months in a double-blind, randomized controlled trial (NCT01379625) in patients with LC-FAOD [5]. In this pivotal phase II trial, 32 patients (aged 7–64 years, mean 25 years) with confirmed LC-FAOD were randomly assigned a diet containing 20% of their total daily energy from either triheptanoin (a 7-carbon fatty acid triglyceride) or trioctanoin (an 8-carbon fatty acid triglyceride). Primary outcomes, measured after 4 months of treatment, included changes from baseline in total energy expenditure, cardiac function (measured by echocardiogram), exercise tolerance, and phosphocreatine recovery following acute exercise [5].
Over the 4-month treatment period, LVEF increased 7.4% in patients who received triheptanoin supplementation relative to those who received trioctanoin (p = 0.046) for the 21 patients (10 on triheptanoin, 11 on trioctanoin) for whom echocardiogram data were available [5]. Triheptanoin recipients also had a 20% decrease in left ventricular wall mass relative to trioctanoin recipients (p = 0.041). Among all study participants, maximum heart rate during a moderate intensity exercise test after 4 months’ treatment was, on average, 7 bpm lower among triheptanoin recipients compared with trioctanoin recipients (p = 0.040). No other significant differences between the two treatment groups were observed for other efficacy outcomes in the study [5].
In the triheptanoin and trioctanoin groups, respectively, five and six patients had CPT II deficiency, four and five patients had VLCAD deficiency, and seven and five patients had LCHAD or trifunctional protein deficiency [5]. At study entry, all patients had had at least one significant episode of rhabdomyolysis. Patients with anemia, peripheral neuropathy limiting the ability to walk, or a history of myocardial infarction were excluded from the trial [5].
In addition to the pivotal double-blind trial, a 78-week single-arm phase II trial (NCT01886378) in 29 patients (aged 10 months to 59 years, mean 12 years) with severe LC-FAOD has provided evidence that triheptanoin (titrated to a target dose of 25–35% of total DCI) can reduce the frequency and duration of major clinical events, improve exercise endurance and tolerance, and increase health-related quality of life [7, 8]. Further data from a retrospective chart review [6] and case reports [43] have suggested that triheptanoin supplementation in patients with LC-FAOD can reduce hospitalization days/year [6], decrease the frequency of hypoglycemia events [6], stabilize cardiomyopathy [43], and improve cardiac function [43]. Triheptanoin has also been used in the treatment of patients with LC-FAOD under Expanded Access programs (NCT03773770, NCT03768817, NCT01461304).
Features and properties of triheptanoin
Alternative names | C7 fatty acid; C7 oil; Dojolvi; glycerol triheptanoate; glyceryl triheptanoate; UX-007 |
|---|---|
Class | Esters; small molecules; triglycerides |
Mechanism of action | Triglyceride replacement |
Route of administration | Oral, enteral |
Pharmacodynamics | Bypasses LC-FAOD enzyme deficiencies; acts as an anaplerotic compound to replenish tricarboxylic acid cycle intermediates |
Pharmacokinetics | Following oral administration, triheptanoin is extensively hydrolyzed to heptanoate and glycerol; heptanoate pharmacokinetics exhibit a high level of inter-patient variability |
Most common adverse events | Gastrointestinal disorders, including abdominal pain, diarrhea, vomiting, and nausea |
ATC codes | |
WHO ATC code | A16 (Other Alimentary Tract and Metabolism Products) A16A (Other Alimentary Tract and Metabolism Products) C10A (Lipid Modifying Agents, Plain) N04 (Anti-Parkinson Drugs) N07X (Other Nervous System Drugs) |
EphMRA ATC code | A16 (Other Alimentary Tract and Metabolism Products) A16A (Other Alimentary Tract and Metabolism Products) C10A (Cholesterol and Triglyceride Regulating Preparations) N4 (Anti-Parkinson Drugs) N7X (All other CNS drugs) |
Chemical name | 1,3-di(heptanoyloxy)propan-2-yl heptanoate |
2.3 Adverse Events
Triheptanoin is generally well tolerated [1]. The most common adverse reactions reported in clinical trials of triheptanoin were gastrointestinal disorders, including abdominal pain, diarrhea, vomiting, and nausea [5, 7, 8]. Gastrointestinal events were generally mild to moderate (grade 1 or 2) in severity and were managed by administering smaller (more frequent) doses of triheptanoin and/or mixing the product with food. Other common adverse events observed in clinical trials in patients with LC-FAOD included common symptoms or complications of the disease (e.g. rhabdomyolysis, myalgia, fatigue) or adverse events considered typical for a (largely) pediatric population (e.g. gastroenteritis/viral gastroenteritis, upper respiratory tract infection, pyrexia) [5, 7, 8].
Key clinical trials of triheptanoin
Identifier | Indication | Phase | Drug(s) | Location(s) | Sponsor(s)/Collaborator(s) | Status |
|---|---|---|---|---|---|---|
NCT01379625 | LC-FAOD | II | Triheptanoin; trioctanoin | USA | OHSU, University of Pittsburgh | Completed |
NCT01886378 | LC-FAOD | II | Triheptanoin | UK, USA | Ultragenyx | Completed |
NCT02214160 | LC-FAOD | II | Triheptanoin | UK, USA | Ultragenyx | Active |
NCT02599961 | GLUT1DS | III | Triheptanoin | Australia, Denmark, Spain, UK, USA | Ultragenyx | Completed |
NCT02453061 | Huntington’s disease | II | Triheptanoin; placebo | France, Netherlands | INSERM; Ultragenyx | Active |
NCT02432768 | GSD-V | II | Triheptanoin; placebo | Denmark | Rigshospitalet | Completed |
NCT02696044 | Rett syndrome | II | Triheptanoin | USA | CRND, Norcross; Ultragenyx | Recruiting |
2.4 Ongoing Clinical Trials
-
NCT02214160 is a single-arm, phase II extension study being conducted by Ultragenyx to evaluate the long-term safety and efficacy of triheptanoin in LC-FAOD subjects previously enrolled in triheptanoin clinical trials.
-
NCT02453061 is a randomized, double-blind, placebo-controlled phase II trial being conducted by INSERM (France), in collaboration with Ultragenyx, to evaluate the safety and efficacy of triheptanoin in the treatment of Huntington’s disease.
-
NCT02696044 is a single-arm, phase II trial being conducted by the Center for Rare Neurological Diseases (Norcross), in collaboration with Ultragenyx and the Rett Syndrome Research Trust, to evaluate the safety and tolerability of triheptanoin in subjects with Rett syndrome.
Change history
22 October 2020
The article Triheptanoin: First Approval, written by Matt Shirley, was originally published electronically in SpringerLink on 8 September 2020 without open access. After publication in volume 80, issue 15, pages 1595���1600 Ultragenyx Pharmaceutical Inc., requested that the article be Open Choice to make the article an open access publication.
References
US FDA. Dojolvi™ (triheptanoin) oral liquid: US prescribing information. 2020. http://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213687s000lbl.pdf. Accessed 31 Jul 2020.
Ultragenyx Pharmaceutical. Ultragenyx announces U.S. FDA approval of Dojolvi™ (UX007/triheptanoin), the first FDA-approved therapy for the treatment of long-chain fatty acid oxidation disorders [media release]. 2020. https://www.ultragenyx.com. Accessed 31 Jul 2020.
US FDA. Dojolvi (triheptanoin) oral liquid: NDA approval. 2020. http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2020/213687Orig1s000replacementltr.pdf. Accessed 31 Jul 2020.
Merritt JL, Norris M, Kanungo S. Fatty acid oxidation disorders. Ann of Transl Med. 2018;6(24):473.
Gillingham MB, Heitner SB, Martin J, et al. Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: a double blinded, randomized controlled trial. J Inherit Metab Dis. 2017;40(6):831–43.
Vockley J, Marsden D, McCracken E, et al. Long-term major clinical outcomes in patients with long chain fatty acid oxidation disorders before and after transition to triheptanoin treatment—a retrospective chart review. Mol Genet Metab. 2015;116(1–2):53–60.
Vockley J, Burton B, Berry GT, et al. Results from a 78-week, single-arm, open-label phase 2 study to evaluate UX007 in pediatric and adult patients with severe long-chain fatty acid oxidation disorders (LC-FAOD). J Inherit Metab Dis. 2019;42(1):169–77.
Vockley J, Burton B, Berry GT, et al. UX007 for the treatment of long chain-fatty acid oxidation disorders: safety and efficacy in children and adults following 24 weeks of treatment. Mol Genet Metab. 2017;120(4):370–7.
Roe CR, Mochel F. Anaplerotic diet therapy in inherited metabolic disease: therapeutic potential. J Inherit Metab Dis. 2006;29(2–3):332–40.
Roe CR, Sweetman L, Roe DS, et al. Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J Clin Invest. 2002;110(2):259–69.
Mochel F, Hainque E, Gras D, et al. Triheptanoin dramatically reduces paroxysmal motor disorder in patients with GLUT1 deficiency. J Neurol Neurosurg Psychiatry. 2016;87(5):550–3.
Hainque E, Gras D, Meneret A, et al. Long-term follow-up in an open-label trial of triheptanoin in GLUT1 deficiency syndrome: a sustained dramatic effect. J Neurol Neurosurg Psychiatry. 2019;90(11):1291–3.
Borges K, Kaul N, Germaine J, et al. Randomized trial of add-on triheptanoin vs medium chain triglycerides in adults with refractory epilepsy. Epilepsia Open. 2019;4(1):153–63.
Borges K, Kaul N, Germaine J, et al. Open-label long-term treatment of add-on triheptanoin in adults with drug-resistant epilepsy. Epilepsia Open. 2020;5(2):230–9.
Hainque E, Caillet S, Leroy S, et al. A randomized, controlled, double-blind, crossover trial of triheptanoin in alternating hemiplegia of childhood. Orphanet J Rare Dis. 2017;12(1):160.
Schiffmann R, Wallace ME, Rinaldi D, et al. A double-blind, placebo-controlled trial of triheptanoin in adult polyglucosan body disease and open-label, long-term outcome. J Inherit Metab Dis. 2018;41(5):877–83.
Ultragenyx Pharmaceutical. Ultragenyx announces negative topline results from Phase 3 study of UX007 in patients with Glut1 DS with disabling movement disorders [media release]. 2018. https://www.ultragenyx.com. Accessed 31 Jul 2020.
US FDA. Triheptanoin: Orphan Drug designation for treatment of fatty acid disorders. 2006. http://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=20062203. Accessed 31 Jul 2020.
US FDA. Triheptanoin: Orphan Drug designation for treatment of glycogen storage disorder II (Pompe disease). 2008. http://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=20062204. Accessed 31 Jul 2020.
US FDA. Triheptanoin: Orphan Drug designation for treatment of glucose transporter type-1 deficiency syndrome. 2014. http://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=20144264. Accessed 31 Jul 2020.
US FDA. Triheptanoin: Orphan Drug designation for treatment of fatty acid oxidation disorders. 2015. http://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=20154740. Accessed 31 Jul 2020.
European Medicines Agency. Triheptanoin: orphan designation for the treatment of very-long-chain 3-hydroxyacyl-CoA-dehydrogenase deficiency. 2016. http://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3121081. Accessed 31 Jul 2020.
European Medicines Agency. Triheptanoin: orphan designation for the treatment of glucose transporter type-1 deficiency syndrome. 2020. http://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3151495. Accessed 31 Jul 2020.
European Medicines Agency. Triheptanoin: orphan designation for the treatment of very long-chain acyl-CoA dehydrogenase deficiency. 2020. http://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3151508. Accessed 31 Jul 2020.
European Medicines Agency. Triheptanoin: orphan designation for the treatment of long-chain 3-hydroxyacyl-coA dehydrogenase deficiency. 2020. http://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3151524. Accessed 31 Jul 2020.
European Medicines Agency. Triheptanoin: orphan designation for the treatment of mitochondrial trifunctional protein deficiency. 2020. http://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3151525. Accessed 31 Jul 2020.
European Medicines Agency. Triheptanoin: orphan designation for the treatment of carnitine palmitoyltransferase II deficiency. 2020. http://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3151526. Accessed 31 Jul 2020.
European Medicines Agency. Triheptanoin: orphan designation for the treatment of McArdle’s disease. 2016. http://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3161710. Accessed 31 Jul 2020.
European Medicines Agency. Triheptanoin: orphan designation for the treatment of carnitine-acylcarnitine translocase deficiency. 2020. http://ec.europa.eu/health/documents/community-register/html/o2302.htm#mod_download. Accessed 25 Aug 2020.
US FDA. Triheptanoin: Center for Drug Evaluation and Research integrated review. 2020. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/213687Orig1s000IntegratedR.pdf. Accessed 31 Jul 2020.
Ultragenyx Pharmaceutical. Ultragenyx announces U.S. commercial launch of Dojolvi™ (triheptanoin), the first FDA-approved therapy for the treatment of long-chain fatty acid oxidation disorders [media release]. 2020. https://www.ultragenyx.com. Accessed 31 Jul 2020.
Ultragenyx Pharmaceutical. Ultragenyx reports second quarter 2020 financial results and corporate update [media release]. 2020. https://www.ultragenyx.com. Accessed 25 Aug 2020.
Ultragenyx Pharmaceutical. 10-K US Securities and Exchange filing [media release]. 2020. https://www.ultragenyx.com. Accessed 31 Jul 2020.
Ultragenyx Pharmaceutical. Ultragenyx announces in-licensing of clinical-stage product triheptanoin [media release]. 2013. https://www.ultragenyx.com. Accessed 31 Jul 2020.
Ultragenyx Pharmaceutical. Ultragenyx gains worldwide rights for triheptanoin (UX007) [media release]. 2013. https://www.ultragenyx.com. Accessed 31 Jul 2020.
Ultragenyx Pharmaceutical. Ultragenyx announces license of intellectual property for the treatment of epilepsy and other seizure-related disorders with triheptanoin [media release]. 2014. https://www.ultragenyx.com. Accessed 31 Jul 2020.
Ultragenyx Pharmaceutical. Ultragenyx announces license of intellectual property related to the treatment of Huntington’s disease with triheptanoin [media release]. 2015. https://www.ultragenyx.com. Accessed 31 Jul 2020.
Wehbe Z, Tucci S. Therapeutic potential of triheptanoin in metabolic and neurodegenerative diseases. J Inherit Metab Dis. 2020;43(3):385–91.
Adanyeguh IM, Rinaldi D, Henry P-G, et al. Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology. 2015;84(5):490–5.
Park MJ, Aja S, Li Q, et al. Anaplerotic triheptanoin diet enhances mitochondrial substrate use to remodel the metabolome and improve lifespan, motor function, and sociability in MeCP2-null mice. PLoS One. 2014;9(10):e109527.
Tefera TW, Wong Y, Barkl-Luke ME, et al. Triheptanoin protects motor neurons and delays the onset of motor symptoms in a mouse model of amyotrophic lateral sclerosis. PLoS One. 2016;11(8):e0161816.
Aso E, Semakova J, Joda L, et al. Triheptanoin supplementation to ketogenic diet curbs cognitive impairment in APP/PS1 mice used as a model of familial Alzheimer’s disease. Curr Alzheimer Res. 2013;10(3):290–7.
Vockley J, Charrow J, Ganesh J, et al. Triheptanoin treatment in patients with pediatric cardiomyopathy associated with long chain-fatty acid oxidation disorders. Mol Genet Metab. 2016;119(3):223–31.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from comments received were made by the author on the basis of scientific completeness and accuracy. Matt Shirley is a salaried employee of Adis International Ltd/Springer Nature, is responsible for the article content and declares no relevant conflicts of interest.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Additional information
The original version of this article was revised due to a retrospective Open Access order.
Enhanced material for this AdisInsight Report can be found at https://doi.org/10.6084/m9.figshare.12859181.
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Shirley, M. Triheptanoin: First Approval. Drugs 80, 1595–1600 (2020). https://doi.org/10.1007/s40265-020-01399-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-020-01399-5