
Abstract
Therapeutic approaches towards a functional cure or eradication of HIV have gained renewed momentum upon encouraging data emerging from studies in SIV monkey models and recent results from human clinical studies. However, a multitude of questions remain to be addressed, including how to deal with the latent viral reservoir, how to boost the host immune response to the virus and what the hurdles are to reach relevant viral compartments in the body. Advances have been made especially with regard to identifying agents that can reactivate the latent virus in vivo and boost the cellular and humoral immunity, but it remains largely unclear whether any of these strategies can awaken a sufficiently large fraction of the viral reservoir and whether the boosted immunity can prevent rapid viral replication once antiretroviral treatments are stopped.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
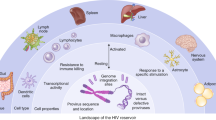
HIV cure remains a scientific challenge to this day. |
Viral control without antiretroviral treatment is the main goal of a functional cure for HIV. |
Combination approaches may be needed for successful virus control. |
1 Introduction
The Human Immunodeficiency Virus 1 (HIV-1) has infected more than 70 million individuals since its entry into the human population [1]. Thanks to the development and increasing availability of effective antiretroviral treatment (ART), the rate of new infections has slowed, and many million lives have been saved. Although ART has dramatically helped to render HIV infection into a generally well-managed chronic infection, issues related to cost, stigma and toxicity still need to be resolved. In response to these needs, the scientific community has aimed to develop strategies that could provide an “HIV cure”, to alleviate the need for life-long ART treatment, prevent forward infections, and ultimately, to remove the virus from the body. As such, a functional cure of HIV infection on one side and the complete eradication of the virus from infected individuals on the other, remain ambitious goals on the path towards controlling and ending the HIV pandemic. Progress has been made towards both, achieving a “functional cure” (defined as sustained virus control to undetectable/low levels in the absence of antiretroviral treatment) as well “sterilising cure”, indicating the complete eradication of the virus from its latent reservoir hiding places and from the infected individual. Although no effective and scalable strategy for either goal exists currently, there is scientific evidence that a functional cure and viral reservoir eradication could indeed be achieved. In fact, 10 years after the "Berlin patient" was first reported to be cured of HIV [2], a second individual known as the “London patient” has also been found to be free of any detectable traces of HIV for almost 2 years [3]. In these two individuals, both suffering from a haematological malignancy, HIV cure was achieved after a complex and risky procedure, including an allogenic stem-cell transplantation from healthy donors who were homozygous for the CCR5-Δ32 deletion. However, such interventions are associated with high morbidity and mortality rates. Thus, they are only justified in select individuals with haematologic disorders requiring stem-cell transplant and make them non-scalable for all HIV-infected individuals. Still, it is important that the observations with the Berlin patient could be repeated and that ongoing studies include a growing number of individuals to further test these strategies with less invasive and more widely scalable regimens. This will possibly help the field to design more widely applicable approaches including genetic modification “in vivo” and “suppression and protection strategies” that are beginning to be tested in pre-clinical models [4].
2 Viral Reservoir as a Major Obstacle for HIV Cure
To date, many HIV cure clinical trials have been conducted in small cohorts, testing different immune-based interventions such as therapeutic T cell vaccines, cytokine administrations, immune checkpoint inhibitors, and a plethora of drugs with variable safety profiles that are more or less effective in reactivating latent reservoir virus [5]. A major challenge for both, functional cure and approaches aiming at total viral eradication, is the existence of a latent viral reservoir which is formed shortly after acute HIV infection [6]. Over the last years, different aspects surrounding the establishment and maintenance of the viral reservoir have been intensively studied and mechanisms of viral latency, such as long-lived infected cells or residual ongoing replication have been described. Despite this, many features of the viral reservoir still remain unknown and pose major challenges for the development of successful HIV curative strategies.
The latent viral reservoir is established from the first moments of HIV infection. Animal models have demonstrated that within the first 72 h of SIV-infection, viral seeding in lymph nodes and gut-associated lymphoid tissue is occurring [7]. Recent data from cohorts with acute HIV infection support the notion that rapid reservoir seeding occurs also in humans early after HIV acquisition, even under early ART initiation [8]. In addition, as shown in the RV254/SEARCH 010 study, if ART treatment is not initiated early after infection signs of immune damage, gut T-cell depletion and gradual increases in reservoir size are known to occur, starting already within first Fiebig stages [8]. Once established, a large portion if not the vast majority of the latently infected cell population is constituted by memory CD4+ lymphocytes, which have HIV genomes integrated into the cellular DNA [9]. This latent state is maintained by different mechanisms including the production of low levels of Nf-kB and NFAT, which hinder transcriptional activity, as well as the presence of immune receptors such as PD-1, CTLA-4 and others, that limit T-cell activation, and thereby, prevent viral reactivation and effective T-cell-mediated killing [10]. Furthermore, homeostatic proliferation of latently infected cells may also be a major driver of the persistence of HIV DNA [11]. The viral reservoir is also thought to be sustained through ongoing residual viral replication, even in the presence of ART and despite appropriate viral suppression in the peripheral blood, although this is still a topic of ongoing debate. Replication is further believed to be aided by cell-to-cell spread and by insufficient levels of antiretroviral drugs in different tissues and ART sanctuaries [12]. Furthermore, aside from T-cell resident viral reservoirs, data from humanised mouse models and natural HIV infection indicate that myeloid cells may also contain an important portion of the viral reservoir, even in the brain [13]. This is further evidenced by the recent finding that in individuals who stop their ART, macrophage-tropic HIV-1 variants can be detected in recrudescing viraemia [14]. This clearly warrants further research to characterise myeloid reservoirs in different tissues, including compartments such as the central nervous system [15]. Regardless though, since a large fraction of CD4 + lymphocytes (and other potential reservoir cells) are spread throughout different body tissues and only about 2% are found in peripheral blood, it is challenging to precisely define the size and most relevant compartments of the viral reservoir [16].
Aside from uncertainties on the various compartments of the viral reservoirs, advances in the field are also complicated by the fact that reservoir measurement and methods are not well standardised, even though in the last decade different test and tools have been developed [17]. Among them, the determination of total and integrated HIV DNA by quantitative polymerase chain reaction (PCR) and viral protein quantification (HIV p24) are highly sensitive, even though these measures may overestimate the size of the latent reservoir to some degree [18]. This is due to the fact that most of the integrated virus in the latent reservoir is replication defective and will not contribute to viral rebound once ART is stopped [19]. Thus, assays such as the quantitative viral outgrowth assay (qVOA) are used to more specifically measure the replication-competent reservoir. However, qVOA is time intensive and requires large amounts of biological specimen. In addition, qVOA may actually underestimate the size of the clinically relevant reservoir due to sampling limitations of latently infected reservoir cells from the most critical body compartments and the inability to reactivate all intact virus-carrying cells in vitro [20]. As an alternative to qVOA, different assays are now available to determine the inducible HIV reservoir and the viral transcriptional activity, including TILDA (Tat/Rev-induced limiting dilution assay) [16], IPDA (intact pro-viral DNA assay) and Quadruplex qPCR (Q4PCR, [18, 21, 22]. In addition, several other innovative strategies, both laboratory- and image- (PAT-based imaging/nuclear medicine) based approaches are being developed to determine the actual size and tissue locations of the latent reservoir at different stages of HIV infection. These efforts may also help to resolve some of the controversy that exists concerning the specific markers of latently HIV-1-infected cells. For instance, the cell surface protein CD32a has been postulated to specifically define a cell population enriched in latently infected cells [23]; however, a series of subsequent publications have questioned this finding [24].
Together, given the uncertainties surrounding the relevant characteristics of the latent viral reservoir, especially in terms of exact location, size, genetic composition as well as its mechanisms for maintenance, it may be difficult to develop targeted interventions that can remove the latent HIV reservoir at once and completely [25]. As eluded to above, to date, only highly invasive strategies such as bone marrow transplants from CCR5-mutated donors have been successful in removing the entire reservoir. Thus, the persistence of parts of the viral reservoir remains one of the most significant hurdles in developing a cure for HIV infection.
3 Models of “Functional” Cure
As eluded to above, HIV cure can be an ambiguous term that is often used to refer either to a “sterilising cure” meaning the eradication of the virus from the infected individual or, alternatively, to the induction of a state of “functional cure” or ‘ART-free remission’ where pro-viral HIV DNA is still detectable in blood cells but plasma viraemia remains undetectable or at very low levels in the absence of ART. In contrast to eradication, in functional cure the virus is not being eliminated from the body, a viral reservoir persists and there remains a risk of late breakthrough of long-term controlled infection [26]. Persistent control of viraemia may be highly depend on effective antiviral immunity, and therapeutic vaccines may play a critical role in the induction of such an immune-based “functional” cure. However, therapeutic vaccination may face the critical challenge that the host immune system is at least partly dysfunctional due to HIV infection, which can drive inflammation and exhaustion of the innate and adaptive immune system even with ART suppressed viraemia. These detrimental effects are even more pronounced in the absence or delayed initiation of antiretroviral treatment, where the persistent antigen stimulation causes extended exhaustion of the virus-specific T cell response and/or can lead to a progressive escape of virus from T-cell-mediated immune surveillance. Still, studies of the HIV-specific cytotoxic T cell response in natural infection have shown that virus-specific CD8 T-cell responses critically contribute to virus control and their expansion and re-education by therapeutic vaccination may offer an opportunity to achieve sustained functional cure. This may parallel the situation in so-called long-term non-progressors (LTNP) or “HIV elite controllers” who represent < 0.5–1% of all people living with HIV but in whom clear evidence exists that their HIV infection with an intact, replication-competent virus can be controlled by adaptive immune mechanisms. The characterisation of the immune parameters and the fine specificity of T-cell responses mediating this control have been the subject of many studies and have given rise to different immunogen designs that are currently being employed in different therapeutic vaccine strategies [27, 28].
A similar but mechanistically possible different state of viral control is that of post-treatment controllers (PTC). These PTCs are HIV-infected individuals who are able to maintain suppressed undetectable or low-level viraemia after ART is stopped but without having received any prior immune therapeutic treatment. The incidence of PTC varies widely, depending on reports and the definition of sustained control. In the first report of a group of PTCs (n = 14) in a retrospective cohort [29], PTCs were estimated to be near 15% of HIV individuals who had received ART early after acquisition. Since then, several studies have reported a large range of PTC incidence. The most in-depth meta-analysis of PTC was conducted in the CHAMP (Control of HIV after antiretroviral Medication Pause) study, which included more than 700 HIV-infected individuals who discontinued ART in 14 selected interruption trials [30]. The investigators reported PTC rates of up to 13% in early treated individuals and less than 5% in individuals who initiated ART during chronic stages of infection. Importantly, the CHAMP study has identified different profiles of viral kinetics leading to PTC, including 31% of PTCs who presented viral rebounds with peak viraemia up to 10.000 copies/mL before controlling viral replication spontaneously (post-rebound controllers). These contrast with the PTCs that are able to maintain an undetectable/low-level plasma viral load for months/years after ATI (non-rebounders). Interestingly, PTCs are not enriched for individuals who express certain beneficial HLA-I alleles classically associated with HIV control (HLA B*57/HLA B*27 and others). Also, PTCs do not show particularly high magnitudes of interferon-gamma (IFN-γ) secreting, HIV-specific CD8 T-cell responses but seem to have preserved robust HIV-specific CD4 T-cell immunity [31]. These observations are of critical importance for the study of immune interventions aiming at functional cure as the rate of spontaneous control (i.e. PTC) and viral dynamics during treatment interruption needs to be considered when measuring effectiveness of functional cure strategies in clinical trials.
4 Measuring Viral Control During Analytical Treatment Interruptions
A major challenge with ethical, medical and public health dimensions arises in the clinical testing of functional cure therapies when it comes to how to best measure the effectiveness of such interventions. Since there are currently no robust virologic or immunologic correlates that can predict viral rebound once ART is stopped, analytical treatment interruption (ATI) is the only hard endpoint by which to assess the effectiveness of functional cure strategies [32]. Many of the current studies and clinical trials for HIV cure (https://www.treatmentactiongroup.org/cure/trials/) include analytical treatment interruptions (ATI), but ATI is not recommended in routine clinical practice and its implementation is limited to the scope of HIV research and through close clinical monitoring. In addition, potential adverse effects of treatment interruption and viral rebound need to be monitored. For instance, the Strategies for Management of Antiretroviral Therapy (SMART) study [33] showed that prolonged treatment interruptions—especially on those with low nadir CD4 T-cell counts, increases the risk of opportunistic infections or death, highlighting the potential risks of unsupervised treatment interruptions. In addition, presentation of acute retroviral syndrome or viral rebound in the CNS are other potential complications of extensive ATI that require close clinical monitoring [34]. On the other hand, short-term ATI as a test to measure outcome of therapeutic vaccination have proven to be relatively safe in terms of development of ART resistance mutations, reservoir re-seeding, and preservation of HIV-specific immune response, at least when ART was restarted within 6 months [19]. In studies with a longer antiretroviral therapy interruption, up to 48 weeks, an increase in reservoir levels measured as integrated HIV-1-DNA was observed, even though total HIV-1-DNA returned to the pre-ATI levels within less than six months thereafter [35].
Recent efforts [36] have tried to establish a consensus on monitoring intervals during ATIs and on virologic and immunological criteria for ART resumption. The current differences are not only due to preferences by individual investigators and clinicians, but are also impacted by the different positions taken by regulatory agencies in different countries, complicating the interpretation of data from different studies. Of critical importance to bear in mind is that stringent viral load thresholds for ART re-start in early-treated individuals may impact the outcome of the ATI and may miss potential post-intervention controllers [37]. In contrast, other studies that have allowed for longer intervals off treatment and for higher viral loads during ATI showed a somewhat better efficacy [35, 38]. By inference from non-human primate (NHP) models, where higher levels of control have been observed and where animals remain off treatment for time intervals that may not be considered appropriate in humans, participants in therapeutic vaccine/functional cure studies should possibly remain off ART long enough to observe the kinetics of the viral rebound until the ATI set-point is achieved. This will certainly not be achieved if the participants are put back on antiretroviral treatment as soon as viraemia becomes detectable. Considering that viral rebound and reservoir dynamics after ART cessation show a median rebound time of 2 to 4 weeks, latest recommendations suggest a cut-off for ART re-initiation after detecting > 1000 copies/mL for 4 consecutive weeks and up to 100,000 cop/mL in a single determination [36]. Independently from viral rebound kinetics though, clinical parameters also need to be considered, especially CD4 counts for which a drop to < 350 cells per µL or CD4% < 15%, dependent on the CD4 entry criteria, would prompt re-initiation of ART [36]. However, without better defined rebound kinetics, it is evidently difficult to establish the timing of the viral rebound after treatment interruption in the context of therapeutic vaccination. Of note, the immunological, virologic and host mechanisms influencing different control profiles (for instance PTC, elite controllers, vaccine controllers) are likely to be different from each other, although to date these differences have not yet been defined. In addition, the absence of markers of virus control and the recent description of profiles such as post-rebound controllers, both in the animal model [39] and in early treated HIV infection, requires ATI in clinical trials to be designed with placebo control groups if a sample size is sufficient to discern an effect of the intervention. Finally, a major challenge for clinical trials that include an ATI arises from how to manage the risk of HIV transmission to sexual partners, apart from the emotional stress that could manifest itself in people with previously undetectable viral load for a long time [40, 41]. Added to this is how to manage the risk of HIV superinfection in individuals off ART [42, 43]. HIV transmission prevention counselling and the use of pre-exposure prophylaxis (PrEP) in participants’ sexual partners can actively contribute to preventing new HIV infections and reducing the associated stress during the ATI period. Together, these considerations highlight that much work for the trial sponsors and involved physicians is required and only well-informed, psychologically stable and highly motivated participants should be included in interruption trials if one wants to minimise the loss to follow up and some of the outlined complications.
As described above, it is well established that ART does not completely remove HIV from the infected body due to the presence of the latent reservoir, homeostatic proliferation of latently infected cells and virus harbouring in anatomical reservoirs not accessible to ART and/or immune surveillance. As a consequence, when ART is interrupted, the virus rebounds typically within 4 weeks [44]. Since time to rebound has been associated with the size of the reservoir, several strategies to impact the size of the reservoir have been explored. In particular, the approach of intensified ART has widely been researched. However ART treatment intensification in long-term suppressed patients, for example with the integrase inhibitor raltegravir, showed no relevant differences between the intensified and non-intensified regimen when measuring reservoir levels using ultra-sensitive plasma viraemia and integrated and total HIV-DNA determination [45]. Similarly, upon intensifying an integrase-inhibitor-based regimen with maraviroc, no significant effect was observed in decreasing the reservoir size in early HIV infection [46]. On the other hand, in a large, early infection Thai cohort the “MegaHAART” strategy that combined tenofovir/emtricitabine/efavirenz/raltegravir/maraviroc showed better outcomes in terms of immune function and decreasing reservoir size compared to individuals who received regular ART (tenofovir/emtricitabine/efavirenz)” [8].
Similarly, early treated individuals showed a more profound decrease in cell-associated HIV DNA compared to patients treated at chronic stages of HIV infection [47, 48]. However, even in these favourable “reservoir conditions” achieved by very early treatment, a recent clinical trial showed that individuals initiating treatment in Fiebig stage I still rebounded quickly after ART cessation [37]. As hypothesised by the authors, the immune responses generated during Fiebig I—which are low compared to responses progressively induced in later stages—might be unable to affect viral rebound but could contribute to reduced viraemia. Of importance to note are the conservative ART resumption criteria which were used in the RV411 study and which may have blunted the possibility to detect post-treatment controllers after an initial viral rebound, as observed in other studies in which PTC have been described [30, 49]. Still, early treatment in the first six months after HIV acquisition may help to preserve the functionality of HIV-specific T-cell-immune response, limit the evolution of the virus and the number of immune escape mutations, reduce immune activation/exhaustion marker levels, and enhance immune recovery in comparison with non-early treatment [50, 51]. Thus, even though early treatment per-se may not prevent rapid HIV rebound once ART is stopped, the lower reservoir levels achieved and the improved immunologic and virologic parameters may provide critical advantages to early-treated individuals making them ideal candidates for early-stage trials researching a functional cure [37, 50]. This view is further supported by indirect evidence from individuals recruited from a pre-exposure prophylaxis program (PrEP) who started prophylactic tenofovir/emtricitabine an estimated 10 and 12 days after infection (detectable plasma HIV-1 RNA, antibody negative-Fiebig stage I). During the following 2 years, only low-level HIV RNA was intermittently detected from different CD4+ T cell subsets from one of the participants, despite wide sampling from different tissues (GALT, lymph nodes, bone marrow, CSF, CD4+ T-cell subsets and plasma). However, and despite the absence of detectable HIV, viral rebound was detected 225 days after a treatment interruption [52]. Likewise, in perinatally infected babies, small HIV reservoir sizes have been directly related to early ART initiation, promoting a long-term remission after discontinuation of treatment. Examples such as the “Mississippi baby” [53], the “French girl” [54] or the recently reported “South African boy” [55] show how early treatment may limit the size of the viral reservoir and provide these children with advantages in terms of time to rebound if ART should become interrupted.
Aside from early treatment initiation, the time on ART before treatment interruption may also be critical, as the reservoir shows a gradual decline under prolonged period of ART. This is illustrated by the SPARTAC study, which included participants with primary HIV infection who were randomised to either a long (48-week) or short (12-week) course of ART or no ART at all [56]. In this study, control was defined as plasma viral load < 400 copies/mL for > 16 weeks. After ATI, the 48-week ART arm showed a higher frequency of post-treatment controllers (18.6%) in contrast to the 12-week ART arm (11.3%) and to the no ART arm (7.9%) [57].
5 Kick-and-Kill and Block-and-Lock Strategies
Described by Siliciano and Deeks in 2012, “Kick and kill” strategies are combined interventions that are based on the reactivation of latently infected cells through latency reversing agents (LRA) and an immunotherapeutic intervention (such as a therapeutic T-cell vaccine), that lead to the elimination of cells with reactivated virus [58, 59]. Since neither component when tested alone may show clinical benefit, combination strategies may need to be tested. In addition, many proposed LRAs may have considerable safety risks and/or are only modestly effective in reactivating latent cells in vivo. In fact, as different mechanisms are involved in the maintenance of latency and various cell compartments contribute to the total viral reservoir, a single LRA may not be sufficiently effective and the complete reactivation of the entire latent viral reservoir may be a major hurdle for effective curative strategies [25].
Different classes of LRAs have been and are currently being developed and tested clinically, including epigenetic modifiers, protein kinase C agonists, NF-κB agonists, Pi3K/Akt pathway activators, toll like receptor (TLR) agonists, immune checkpoint blockers and other small molecules with different mechanisms and potential to disrupt viral latency [60]. Of these approaches, the use of Histone Deacetylase Inhibitors (HDACi) has been widely explored. During viral latency, histones are deacetylated, leading to highly compacted chromatin, which hinders (viral) gene expression. Inhibition of these histone deacetylases thus enables the unpacking of chromatin and transcription of viral genes, rendering the infected cells susceptible to immune recognition by the cellular arm of the immune response. Initially developed in the field of oncological therapies, the HDACis vorinostat, panobinostat and romidepsin have been shown to reactivate HIV in vitro and in vivo [61]. The first in-vivo proof-of-concept with vorinostat [suberoylanilide hydroxamic acid (SAHA)] in chronically HIV-infected individuals on suppressive ART, showed an increase in HIV gag gene transcription measured by HIV RNA expression in resting CD4 cells [62]. Similarly, panobinostat, a non-selective HDACi (pan-HDACi), demonstrated in vitro and in vivo activity in a clinical trial with 15 aviraemic patients, increasing cell-associated HIV-RNA and allowing for the detection of HIV RNA in the peripheral blood [63]. Romidepsin is the HDACi that has shown the highest activity on histone acetylation and HIV replication in several in vitro studies. However, as for panobinostat or vorinostat, no changes in reservoir size measured by total HIV-DNA, Tat/rev Induced Limiting Dilution Assay (TILDA) or infectious units per million (IUMP) were observed [64]. Still, HDACis have been tested in interventions that include a treatment interruption period to assess in vivo efficacy to prevent viral rebound. Despite some promising results from the recent open-label single arm BCN02 study in early-treated HIV-1-infected individuals using MVA.HIVconsv vaccine [65], these molecules used as single LRA are likely not sufficiently potent to reactivate a substantial proportion of the latent reservoir and reduce the overall reservoir size. In addition, in vitro studies and data emerging from the BCN02 trial strongly suggest that romidepsin and possible other HDACi as well, may impair host immunity [66, 67], further limiting the use of this class of LRAs in HIV cure strategies.
Aside from the above-mentioned interventions that aim to reverse viral latency and activate viral gene expression, there are strategies that aim to achieve the opposite. These “Block-and-Lock” strategies aim to lock HIV into a non- or hard-to-reverse latent form and thereby reduce the chance of viral reactivation and replication. To date, multiple targets and drugs have been postulated to serve as such latency-promoting agents (LPA). In particular, molecules that inhibit classical transcriptional factors such as NF-κB, NFAT (CsA), P-TEFb complex, mTOR complex and Transcription Factor II H (TFIIH) as well as other drugs such as levosimendan [68], spironolactone [69] and complex-targeting curaxin CBL0100 [70] have been tested outside of HIV infection but may well serve as future agents that could be explored for their transcriptional blocking strategy [71, 72].
6 Combination Strategies Using T-Cell Vaccines, Passive Nab Immunisation and Immune Check Point Inhibitors
Therapeutic vaccines may prove critical in “kick and kill” and functional cure strategies as they may be needed to remove reactivated virally infected cells as soon as they become visible to the immune system. By inference from natural HIV infection and studies in HIV controllers who do not take ART, increasing the breadth and the magnitude of the cellular immune responses towards selected and the most vulnerable targets of the virus appears to be a central pre-requisite for successful cure approaches [73]. Different strategies to induce such responses have been developed, and immunogen sequence design and vaccine vector choices can differ significantly. In addition, most potent vaccination strategies may need to include elaborate prime-boost regimen, adding further complexity and costs to the production and clinical testing of promising strategies [74].
Aside from vectored vaccines, cell-based approaches for immune restoration have also been explored in HIV cure strategies. Of special interest in this context have been dendritic cells, which as professional antigen present cells (APC) play an important role the induction of both the humoral as well as cellular adaptive immunity. Different studies, among them the DCV-2 trials [75], provided proof of concept that using autologous monocyte-derived dendritic cells pulsed with inactivated virus can induce broad virus-specific T-cell immune responses with the potential to impact viral setpoint during an ART interruption phase [38]. Similarly, the AGS-004 Phase IIb trial in early-treated HIV-infected individuals employed a regimen of autologous DC electroporated with RNA encoding autologous viral antigens and showed an increase in memory T-cell responses, which however, was not able to delay viral rebound during ATI [76].
Of note, immunotherapies that are not based on active vaccination strategies, have also been tested in the HIV cure field. In particular, immune therapeutic interventions using broadly neutralising antibodies (bNAb) have shown very encouraging results. Such bNAbs can, aside from direct neutralisation of the virus, mediate various effector functions including ADCC (antibody-dependent cellular toxicity), ADCP (antibody-dependent cellular phagocytosis), ADCDC (antibody-dependent complement-dependent cytotoxicity) or ADCT (antibody-dependent trogocytosis), which may contribute to variable levels of HIV control [77,78,79]. These effects may be further increased, and viral escape largely prevented by the use of combination of various bNAbs. Indeed, the successful combinations of three bNAbs in pre-clinical phases [80, 81] prompted the strategy to be tested in HIV-infected individuals, which has shown promise in a recent a Phase I clinical trial that combined the bNAb 3BNC117 and 10–1074 (NCT03571204). In this trial, effective suppression of viral rebound for a median of 21 weeks was observed, further raising hopes that such interventions alone or in combination with T-cell vaccines could provide significantly prolonged virus control [82]. The clinical benefit of such bNAb-based interventions may be further increased by targeted antibody engineering that allows the potentiation of specific effector functions (for instance through modifications in the Fc portion of the Ab), and the creation of multi-specific antibodies, which might enhance the possible vaccinal-effect of antibodies. This could drive T-cell immunity [83, 84] and possibly, most importantly, increase the in vivo half-life of these Abs [85]. Together with increased half-lives, the “vaccinal effect”, whereby the formation of immune complexes of reactivated virus and nAb is believed to boost virus-specific T-cell immunity and effective CTL responses [86], could provide critical advances to combination approaches. Additional ways to harness the full capacities of the humoral immune response and bNAb for HIV control is their combination with LRAs and immune modulators. In this regard, a recent study by Borducchi et al. [39] demonstrated that the combination of the TLR7 agonist GS9620 (vesatolimod) together with the bNAb PGT121 was able to delay SIV rebound for more than 6 months. Mechanistically, GS-9620 may have activated parts of the latent viral reservoir, which in the presence of PGT121 led to virus neutralisation. Other TLR ligands, in particular TLR9 agonist (MGN1703, lefitolimod) have been tested clinically and may show some effect on viral reservoir as well as the virus-specific T-cell response [87]. However, the effect on reservoir reactivation needs to be assessed and interpreted critically, as none of the existing data suggest a broad reactivation of latent virus in vivo.
Finally, alternative strategies to potentiate T-cell vaccine have been explored, including the use of immune check-point inhibitors (ICB). The rational for these strategies is based on the observation that HIV-specific CD8 T cells express high levels of PD1 [88] and that anti-PD1 mAb and other immune checkpoint inhibitors (CTLA-4, LAG-3, TIM-3, TIGIT y 2B4) have shown promise in the cancer field to restore anti-cancer immunity. These approaches are currently being explored in the HIV field, in some cases in combination with therapeutic vaccination and a LRA. In addition, PD1 is believed to play an important role in the establishment and maintenance of latently infected cells [89] and PD1-blocking treatment has been proposed to increase HIV DNA transcription [90]. Since monoclonal antibodies blocking PD-1 (nivolumab and pembrolizumab) and CTL4 (ipilimumab) are now being used in HIV-infected individuals as treatments for malignancies, their potential as HIV viral reservoir activators can be assessed. Of interest, recent results in a French study showed a significant decrease of the viral reservoir during the treatment of a non-small-cell lung cancer with nivolumab in an individual patient [91]. However, this effect was not observed in a subsequent small sample of patients (n = 3) treated with anti PD-1 in a similar malignancy [92]. Ongoing studies (among them NCT02408861, NCT02595866) that are enrolling a larger number of patients receiving different anti-PD1 treatments for the treatment of neoplasia may help to better assess the potential of these agents in the cure area.
7 Conclusions
In conclusion, much progress has been made over the last few years in designing and testing different strategies to achieve a functional cure and eradication of HIV. To date, no scalable therapeutic interventions exist that provide lasting suppression of viral replication in the absence of antiretroviral treatment. Therapeutic vaccines, the passive administration of bNAb, latency-reversing agents, immune adjuvant as well as immune checkpoint modulators have all been tested individually or in combination in NHP models and in human clinical trials. Since each individual approach has its specific shortcomings, the clinical effectiveness of a future approach will likely only be established when all the required components are combined in the right order, schedule and dose, and in the most suitable patent population, making the clinical development of such strategies a formidable challenge.
Change history
03 June 2020
The first author currently listed as Lucia Bailon Alvarez should be Lucia Bailon.
References
UNAIDS. Data 2019. https://www.unaids.org/en/resources/documents/2019/2019-UNAIDS-data. 2019.
Hütter G, Nowak D, Mossner M, Ganepola S, Müßig A, Allers K, Schneider T, Hofmann J, Kütcherer C, Blau O, Hofmann IWK. Long-term control of HIV by stem cell transplantation. N Engl J Med. 2009;360:692–8.
Gupta RK, Peppa D, Hill AL, Gálvez C, Salgado M, Pace M, et al. Evidence for HIV-1 cure after CCR5Δ32/Δ32 allogeneic haemopoietic stem-cell transplantation 30 months post analytical treatment interruption: a case report. Lancet HIV. 2020;1(20):1–8.
Dash PK, Kaminski R, Bella R, Su H, Mathews S, Ahooyi TM, et al. Sequential LASER ART and CRISPR treatments eliminate HIV-1 in a subset of infected humanized mice. Nat Commun. 2019;10(1):1–20.
Rasmussen TA, Søgaard OS. Clinical interventions in HIV cure research. Adv Exp Med Biol. 2018;1075:285–318.
Castro-Gonzalez S, Colomer-Lluch M, Serra-Moreno R. Barriers for HIV cure: the latent reservoir. AIDS Res Hum Retroviruses. 2018;34(9):739–59.
Whitney JB, Hill AL, Sanisetty S, Penaloza-macmaster P, Liu J, Shetty M, et al. Rapid seeding of the viral reservoir prior to SIV viremia in rhesus monkeys. Nature. 2015;512(7512):74–7.
Ananworanich J, Schuetz A, Vandergeeten C, Sereti I, Souza Mark D, Rerknimitr R, et al. Impact of multi-targeted antiretroviral treatment on gut t cell depletion and HIV reservoir seeding during acute hiv infection. PLoS ONE. 2012;7:3.
Ho Y-C, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DIS, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155(3):540–51.
Banga R, Procopio FA, Noto A, Pollakis G, Cavassini M, Ohmiti K, et al. PD-1 + and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat Med. 2016;22(7):754–61.
Katlama C, Deeks SG, Autran B, Martinez-Picado J, Van J, Rouzioux C, et al. Barriers to a cure: new concepts in targeting and eradicating HIV-1 reservoirs. Lancet. 2014;381:9883.
Martinez-Picado J, Deeks SG. Persistent HIV-1 replication during antiretroviral therapy. Curr Opin HIV AIDS. 2016;11(4):417–23.
Stevenson M. HIV persistence in macrophages. Nat Med. 2017;23(5):538–9.
Machado Andrade V, Mavian C, Babic D, Alvarado T, Sharkey M, Barrios L, et al. A minor population of macrophage-tropic HIV-1 variants is identified in recrudescing viremia following analytic treatment interruption. Proc Natl Acad Sci U S A. 2020;117(18):9981–90. https://doi.org/10.1073/pnas.1917034117.
Wong ME, Jaworowski A, Hearps AC. The HIV reservoir in monocytes and macrophages. Front Immunol. 2019;2019:10.
Deleage C, Baris T, Estes JD. Imaging Lymphoid tissues in nonhuman primates to understand SIV pathogenesis and persistence. Curr Opin Virol. 2017;19:77–84.
Wang Z, Simonetti FR, Siliciano RF, Laird GM. Measuring replication competent HIV-1: advances and challenges in defining the latent reservoir. Retrovirology. 2018;15(1):1–9.
Henrich TJ, Deeks SG, Pillai SK. Measuring the size of the latent human immunodeficiency virus reservoir: the present and future of evaluating eradication strategies. J Infect Dis. 2017;215(Suppl 3):S134–S141141.
Clarridge KE, Blazkova J, Einkauf K, Petrone M, Refsland EW, Justement JS, et al. Effect of analytical treatment interruption and reinitiation of antiretroviral therapy on HIV reservoirs and immunologic parameters in infected individuals. PLoS Pathog. 2018;14(1):1–16.
Olvera A, Martinez JP, Casadellà M, Llano A, Rosás M, Mothe B, et al. Benzyl-2-acetamido-2-deoxy-α-d-galactopyranoside increases human immunodeficiency virus replication and viral outgrowth efficacy in vitro. Front Immunol. 2018;8:2010.
Bruner KM, Wang Z, Simonetti FR, Bender AM, Kwon KJ, Sengupta S, et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature. 2019;566(7742):120–5. https://doi.org/10.1038/s41586-019-0898-8.
Gaebler C, Lorenzi JCC, Oliveira TY, Nogueira L, Ramos V, Lu CL, et al. Combination of quadruplex qPCR and next-generation sequencing for qualitative and quantitative analysis of the HIV-1 latent reservoir. J Exp Med. 2019;216(10):2253–64.
Descours B, Petitjean G, López-Zaragoza JL, Bruel T, Raffel R, Psomas C, et al. CD32a is a marker of a CD4 T-cell HIV reservoir harbouring replication-competent proviruses. Nature. 2017;543(7646):564–7.
Badia R, Ballana E, Castellví M, García-Vidal E, Pujantell M, Clotet B, et al. CD32 expression is associated to T-cell activation and is not a marker of the HIV-1 reservoir. Nat Commun. 2018;9(1):1–10.
Grau-Expósito J, Luque-Ballesteros L, Navarro J, Curran A, Burgos J, Ribera E, et al. Latency reversal agents affect differently the latent reservoir present in distinct CD4+ T subpopulations. PLOS Pathog. 2019;15(8):e1007991.
Rosás-Umbert M, Llano A, Bellido R, Olvera A, Ruiz-Riol M, Rocafort M, et al. Mechanisms of abrupt loss of virus control in a cohort of previous HIV controllers. J Virol. 2019;4:93.
Mothe B, Llano A, Ibarrondo J, Daniels M, Miranda C, Zamarreño J, et al. Definition of the viral targets of protective HIV-1-specific T cell responses. J Transl Med. 2011;9:208.
Lopez-Galindez C, Pernas M, Casado C, Olivares I, Lorenzo-Redondo R. Elite controllers and lessons learned for HIV-1 cure. Curr Opin Virol. 2019;38:31–6.
Sáez-Cirión A, Bacchus C, Hocqueloux L, Avettand-Fenoel V, Girault I, Lecuroux C, et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI study. PLoS Pathog. 2013;9(3):e1003211.
Namazi G, Fajnzylber JM, Aga E, Bosch R, Edward P, Sharaf R, et al. The control of HIV after antiretroviral medication pause (CHAMP) study: post-treatment controllers identified from 14 clinical studies. J Infect Dis. 2018;218(12):1954–63.
Etemad B, Esmaeilzadeh E, Li JZ. Learning from the exceptions: HIV remission in post-treatment controllers. Front Immunol. 2019;10(July):1749.
Margolis DM, Deeks SG. How unavoidable are analytical treatment interruptions in HIV cure-related studies? J Infect Dis. 2019;220(Suppl 1):24–6.
Gordin F, Med- WVA, Abrams D, Francisco S, Babiker A, Re M, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med. 2006;355(22):2283–96.
Routy J, Boulassel M, Nicolette C, Jacobson J. Assessing risk of a short-term antiretroviral therapy discontinuation as a read-out of viral control in immune-based therapy. J Med Virol. 2012;84:885–9.
Montserrat M, Plana M, Guardo AC, Andrés C, Climent N, Gallart T, et al. Impact of long-term antiretroviral therapy interruption and resumption on viral reservoir in HIV-1 infected patients. Aids. 2017;31(13):1895–7.
Julg B, Dee L, Ananworanich J, Barouch DH, Bar K, Caskey M, et al. Recommendations for analytical antiretroviral treatment interruptions in HIV research trials—report of a consensus meeting. Lancet HIV. 2019;6(4):e259–e268268.
Colby DJ, Trautmann L, Pinyakorn S, Leyre L, Pagliuzza A, Kroon E, et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nat Med. 2018;24(7):923–6.
García F, Climent N, Guardo AC, Gil C, León A, Autran B, et al. A dendritic cell-based vaccine elicits T cell responses associated with control of HIV-1 replication. Sci Transl Med. 2013;5:166.
Borducchi EN, Liu J, Nkolola JP, Cadena AM, Yu WH, Fischinger S, et al. Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature. 2018;563(7731):360–4.
Garner SA, Rennie S, Ananworanich J, Dube K, Margolis DM, Sugarman J, et al. Interrupting antiretroviral treatment in HIV cure research: scientific and ethical considerations. J Virus Erad. 2017;3(2):82–4.
Eyal N. How to address the risk of HIV transmission in remission studies with treatment interruption: the low- hanging fruit approach. J Infect Dis. 2019;220(Suppl 1):7–11.
Rachinger A, Navis M, van Assen S, Groeneveld PHP, Schuitemaker H. Recovery of viremic control after superinfection with pathogenic HIV type 1 in a long-term elite controller of HIV type 1 infection. Clin Infect Dis. 2008;47(11):e86–e8989.
Brener J, Gall A, Hurst J, Batorsky R, Lavandier N, Chen F, et al. Rapid HIV disease progression following superinfection in an HLA-B*27:05/B*57: 01-positive transmission recipient. Retrovirology. 2018;15(1):1–13.
Wen Y, Bar KJ, Li JZ. Lessons learned from HIV antiretroviral treatment interruption trials. Curr Opin HIV AIDS. 2018;13(5):416–21.
Llibre JM, Buzón MJ, Massanella M, Esteve A, Dahl V, Puertas MC, et al. Treatment intensification with raltegravir in subjects with sustained HIV-1 viraemia suppression: a randomized 48-week study. Antivir Ther. 2012;17(2):355–64.
Puertas MC, Massanella M, Llibre JM, Ballestero M, Buzon MJ, Ouchi D, et al. Intensification of a raltegravir-based regimen with maraviroc in early HIV-1 infection. Aids. 2014;28(3):325–34.
Buzon MJ, Martin-Gayo E, Pereyra F, Ouyang Z, Sun H, Li JZ, et al. Long-Term antiretroviral treatment initiated at primary HIV-1 infection affects the size, composition, and decay kinetics of the reservoir of HIV-1-infected CD4 T cells. J Virol. 2014;88(17):10056–65.
Gianella S, von Wyl V, Fischer M, Niederoest B, Battegay M, Bernasconi E, et al. Effect of early antiretroviral therapy during primary HIV-1 infection on cell-associated HIV-1 DNA and plasma HIV-1 RNA. Antivir Ther. 2011;16(4):535–45.
Sneller MC, Justement JS, Gittens KR, Petrone ME, Clarridge KE, Proschan MA, et al. A randomized controlled safety/efficacy trial of therapeutic vaccination in HIV-infected individuals who initiated antiretroviral therapy early in infection. Sci Transl Med. 2017;9:419.
Ananworanich J, Chomont N, Eller LA, Kroon E, Tovanabutra S, Bose M, et al. HIV DNA set point is rapidly established in acute HIV infection and dramatically reduced by early ART. EBioMedicine. 2016;11:68–72.
Jain V, Hartogensis W, Bacchetti P, Hunt PW, Hatano H, Sinclair E, et al. Antiretroviral therapy initiated within 6 months of HIV infection is associated with lower T-cell activation and smaller HIV reservoir size. J Infect Dis. 2013;208(8):1202–11.
Henrich TJ, Hatano H, Bacon O, Hogan LE, Rutishauser R, Hill A, et al. HIV-1 persistence following extremely early initiation of antiretroviral therapy (ART) during acute HIV-1 infection: an observational study. PLoS Med. 2017;14(11):1–22.
Persaud D, Gay H, Ziemniak C, Chen YH, Piatak M, Chun T-W, et al. Absence of detectable HIV-1 viremia after treatment cessation in an infant. N Engl J Med. 2013;19(7):1828–63.
Frange P, Faye A, Avettand-Fenoël V, Bellaton E, Descamps D, Angin M, et al. HIV-1 virological remission lasting more than 12 years after interruption of early antiretroviral therapy in a perinatally infected teenager enrolled in the French ANRS EPF-CO10 paediatric cohort: a case report. Lancet HIV. 2016;3(1):e49–54.
Violari A, Cotton MF, Kuhn L, Schramm DB, Paximadis M, Loubser S, et al. A child with perinatal HIV infection and long-term sustained virological control following antiretroviral treatment cessation. Nat Commun. 2019;10(1):1–11.
Fidler S, Porter K, Ewings F, Frater J, Ramjee G, Cooper D, et al. Short-course antiretroviral therapy in primary HIV infection. N Engl J Med. 2013;368(3):207–17.
Martin GE, Gossez M, Williams JP, Stöhr W, Meyerowitz J, Leitman EM, et al. Post-treatment control or treated controllers? Viral remission in treated and untreated primary HIV infection. AIDS. 2017;31(4):477–84.
Shan L, Deng K, Shroff NS, Durand C, Rabi SA, Zhang H, et al. Elimination of Latent Viral Reservoir After Virus Reactivation. Immunity. 2012;36(3):491–501.
Deeks SG. Shock and kill. Abilities Nature. 2012;487:439–40.
Kim Y, Anderson JL, Lewin SR. Getting the “Kill” into “Shock and Kill”: strategies to eliminate latent HIV. Cell Host Microbe. 2018;23(1):14–26.
Bose P, Dai Y, Grant S. Histone deacetylase inhibitor (HDACI) mechanisms of action: emerging insights. Pharmacol Ther. 2014;143(3):323–36.
Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487(7408):482–5.
Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, et al. Panobinostat, a histone deacetylase inhibitor, for latent virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV. 2014;1(1):e13–21.
Søgaard OS, Graversen ME, Leth S, Olesen R, Brinkmann CR, Nissen SK, et al. The depsipeptide romidepsin reverses HIV-1 latency in vivo. PLoS Pathog. 2015;11(9):1–22.
Mothe B, Rosas-Umbert M, Coll P, Manzardo C, Puertas MC, Moron-Lopez S, et al. HIVconsv vaccines and romidepsin in early-treated HIV-1- infected individuals: Safety, immunogenicity and effect on the viral reservoir (study BCN02). Front Immunol. 2020;11:823. https://doi.org/10.3389/fimmu.2020.00823.
Jones RB, O’Connor R, Mueller S, Foley M, Szeto GL, Karel D, et al. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic t-lymphocytes. PLoS Pathog. 2014;10:8.
Rosás-Umbert M, Ruiz-Riol M, Fernández MA, Marszalek M, Coll P, Manzardo C, et al. In vivo effects of romidepsin on T-cell activation, apoptosis and function in the BCN02 HIV-1 Kick & Kill clinical Trial. Front Immunol. 2020;11(March):1–11.
Hayashi T, Jean M, Huang H, Simpson S, Santoso NG, Zhu J. Screening of an FDA-approved compound library identifies levosimendan as a novel anti-HIV-1 agent that inhibits viral transcription. Antiviral Res. 2017;146:76–85.
Lacombe B, Morel M, Margottin-Goguet F, Ramirez BC. Specific inhibition of HIV infection by the action of spironolactone in T cells. J Virol. 2016;90(23):10972–80.
Jean MJ, Hayashi T, Huang H, Brennan J, Simpson S, Purmal A, et al. Curaxin CBL0100 blocks HIV-1 replication and reactivation through inhibition of viral transcriptional elongation. Front Microbiol. 2017;8:1–13.
Jean MJ, Fiches G, Hayashi T, Zhu J. Current strategies for elimination of HIV-1 latent reservoirs using chemical compounds targeting host and viral factors. AIDS Res Hum Retroviruses. 2019;35(1):1–24.
Darcis G, Van Driessche B, Van Lint C. HIV latency: should we shock or lock? Trends Immunol. 2017;38(3):217–28.
Mylvaganam GH, Silvestri G, Amara RR. HIV therapeutic vaccines: moving towards a functional cure. Curr Opin Immunol. 2015;35:1–8.
Mothe B, Brander C. HIV T-cell vaccines. Adv Exp Med Biol. 2018;2018:31–51.
García F, Climent N, Assoumou L, Gil C, González N, Alcamí J, et al. A therapeutic dendritic cell-based vaccine for HIV-1 infection. J Infect Dis. 2011;203(4):473–8.
Gay CL, Debenedette MA, Tcherepanova IY, Gamble A, Lewis WE, Cope AB, et al. Immunogenicity of AGS-004 dendritic cell therapy in patients treated during acute HIV Infection. AIDS Res Hum Retroviruses. 2018;34(1):111–22.
Caskey M, Klein F, Lorenzi JCC, Seaman MS, West AP, Buckley N, et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature. 2015;522(7557):487–91.
Lynch RM, Boritz E, Coates EE, DeZure A, Madden P, Costner P, et al. Virologic effects of broadly neutralizing antibody VRC01 administration during chronic HIV-1 infection. Sci Transl Med. 2015;7(319):1–15.
Bar KJ, Sneller MC, Harrison LJ, Justement JS, Overton ET, Petrone ME, et al. Effect of HIV antibody VRC01 on viral rebound after treatment interruption. N Engl J Med. 2016;375(21):2037–50.
Klein F, Halper-Stromberg A, Horwitz JA, Gruell H, Scheid JF, Bournazos S, et al. HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature. 2012;492(7427):118–22.
Barouch DH, Whitney JB, Moldt B, Klein F, Oliveira TY, Liu J, et al. Therapeutic efficacy of potent neutralizing HIV-1-specific monoclonal antibodies in SHIV-infected rhesus monkeys. Nature. 2013;503(7475):224–8.
Mendoza P, Gruell H, Nogueira L, Pai JA, Butler AL, Millard K, et al. Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature. 2018;561(7724):479–84.
Martinez-Navio JM, Fuchs SP, Pantry SN, Lauer WA, Duggan NN, Keele BF, et al. Adeno-associated virus delivery of anti-HIV monoclonal antibodies can drive long-term virologic suppression. Immunity. 2019;50(3):567–575.e5.
Xu L, Pegu A, Rao E, Doria-Rose N, Beninga J, McKee K, et al. Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science. 2017;358(6359):85–90.
Grobben M, Stuart RAL, van Gils MJ. The potential of engineered antibodies for HIV-1 therapy and cure. Curr Opin Virol. 2019;38(1):70–80.
Nishimura Y, Gautam R, Chun TW, Sadjadpour R, Foulds KE, Shingai M, et al. Early antibody therapy can induce long-lasting immunity to SHIV. Nature. 2017;543(7646):559–63.
Offersen R, Nissen SK, Rasmussen TA, Østergaard L, Denton PW, Søgaard OS, et al. A novel toll-like receptor 9 agonist, MGN1703, enhances HIV-1 transcription and NK cell-mediated inhibition of HIV-1-infected autologous CD4 + T cells. J Virol. 2016;90(9):4441–53.
Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443(7109):350–4.
Evans VA, Van Der Sluis RM, Solomon A, Dantanarayana A, McNeil C, Garsia R, et al. Programmed cell death-1 contributes to the establishment and maintenance of HIV-1 latency. Aids. 2018;32(11):1491–7.
Boyer Z, Palmer S. Targeting immune checkpoint molecules to eliminate latent HIV. Front Immunol. 2018;9:8–10.
Guihot A, Marcelin A, Massiani M, Samri A, Soulie C, Autran B, et al. Drastic decrease of the HIV reservoir in a patient treated with nivolumab for lung cancer. Ann Oncol. 2018;29(2):517–8.
Scully EP, Rutishauser RL, Simoneau CR, Delagrèverie H, Euler Z, Thanh C, et al. Inconsistent HIV reservoir dynamics and immune responses following anti-PD-1 therapy in cancer patients with HIV infection. Ann Oncol. 2018;29(10):2141–2.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The present work was funded in parts by the European Union’s Horizon 2020 research and innovation programme under grant agreement 681137-EAVI2020 and by NIH Grant P01-AI131568. BM is a consultant for AELIX Therapeutics, S.L., outside the submitted work. CB is CSO and shareholder. AELIX Therapeutics, S.L., outside the submitted work. LBe is former CMO of AELIX Therapeutics, S.L. LBa declares no conflicts of interest.
Additional information
The original article has been updated: Due to first author name update.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bailon, L., Mothe, B., Berman, L. et al. Novel Approaches Towards a Functional Cure of HIV/AIDS. Drugs 80, 859–868 (2020). https://doi.org/10.1007/s40265-020-01322-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-020-01322-y