
Abstract
Target-specific oral anticoagulants have become increasingly available as alternatives to traditional agents for the management of a number of thromboembolic disorders. To date, the direct Factor Xa inhibitor rivaroxaban is the most widely approved of the new agents. The dosing of rivaroxaban varies and adheres to specific schedules in each of the clinical settings in which it has been investigated. These regimens were devised based on the results of phase II dose-finding studies and/or pharmacokinetic modeling, and were demonstrated to be successful in randomized, phase III studies. In most cases, the pharmacodynamic profile of rivaroxaban permits once-daily dosing. A once-daily dose is indicated for the prevention of venous thromboembolism (VTE) in patients undergoing hip or knee replacement surgery, the long-term prevention of stroke in patients with non-valvular atrial fibrillation, and the long-term secondary prevention of recurrent VTE. Twice-daily dosing is required in the acute phase of treatment in patients with VTE and in the combination of rivaroxaban with standard single or dual antiplatelet therapy for secondary prevention after acute coronary syndrome events. This article reviews the empirical and clinical rationale supporting the dose regimens of rivaroxaban in each clinical setting.
Similar content being viewed by others
1 Introduction
Hospital-based and outpatient-focused physicians are increasingly prescribing target-specific oral anticoagulants (TSOACs) as an alternative to traditional heparins and/or vitamin K antagonists (VKAs) for the prophylaxis and treatment of thromboembolic disorders. Rivaroxaban, dabigatran etexilate, and apixaban are all approved in Europe and the United States for the prevention of stroke and systemic embolism in patients with non-valvular atrial fibrillation (AF) who are at risk of stroke [1–6]. All three agents are also licensed in Europe for the prevention of venous thromboembolism (VTE) after elective hip or knee replacement surgery [1, 3, 5], and rivaroxaban and apixaban are approved for this indication in the United States [2]. In addition, TSOACs are available in the United States (dabigatran and rivaroxaban) and Europe (rivaroxaban only) for the treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE), as well as for the secondary prevention of VTE recurrence (Table 1) [1, 2]. In Europe, rivaroxaban is approved for the prevention of atherothrombotic events in patients with recent acute coronary syndrome (ACS) and elevated cardiac biomarkers, in combination with standard antiplatelet therapy (Table 1) [1].
The maintenance of vascular integrity amidst vessel wall injury requires a complex interplay between the vascular endothelium, platelets, and circulating proteins that either promote (procoagulant) or limit (anticoagulant) the formation of thrombosis. The successive activation of procoagulant factors leading to thrombin generation is recognized as the coagulation cascade, and results in the conversion of soluble fibrinogen to insoluble fibrin, and the formation of a stable thrombus (Fig. 1). Inhibition of the coagulation cascade represents an important target in the management of various thromboembolic disorders. Traditional anticoagulants, such as unfractionated or low-molecular-weight heparin (LMWH), target multiple points in the coagulation cascade in concert with a cofactor. VKAs, previously the only option for long-term oral anticoagulation, do not directly target coagulation factors, but lead to the synthesis of dysfunctional clotting Factors II, VII, IX, and X [7]. TSOACs inhibit specific coagulation factors: Factor Xa in the case of rivaroxaban apixaban, and edoxaban and Factor IIa (thrombin) for dabigatran (without the need for a cofactor; Fig. 1) [8]. Rivaroxaban, apixaban, and edoxaban directly and selectively bind to the active site of Factor Xa, regardless of whether it is freely circulating, bound to the prothrombinase complex, or found within an existing thrombus [9, 10]. Differences in the mechanism of action of TSOACs compared with VKAs have important implications for the management of these agents.

Coagulation factors targeted by the target-specific oral anticoagulants
Rivaroxaban and the other TSOACs, apixaban, edoxaban, and dabigatran, overcome many of the perceived limitations of traditional anticoagulants. Unlike heparins, they are taken orally, but have a comparably rapid onset of action. Compared with warfarin and other VKAs, the TSOACs have predictable pharmacokinetics and pharmacodynamics that allow for fixed dosing (in the patient populations studied to date), do not require routine coagulation monitoring, and have fewer drug and food interactions [8]. Despite these advantages, the long-term use of TSOACs presents different challenges, including drug accumulation in patients with renal dysfunction (varies by drug), lack of an antidote or standardized reversal strategy when life-threatening bleeding is encountered, and difficulties in quantifying the degree of anticoagulation with commonly available assays in emergency situations such as urgent surgery (specific anti-Factor Xa assays, although preferred, may not be routinely available or practical in acute situations) [8]. As mentioned previously, predictable dose–concentration response with TSOACs allows for standard fixed dosing for various populations. Dosing for TSOACs is based on phase II and III clinical trial data. This represents a paradigm shift from the dosing of traditional anticoagulants that are typically titrated to specific goals via coagulation assay monitoring.
Dosing in the various clinical settings for the TSOACs takes into account many factors, including the specific pharmacokinetics of each agent, the pharmacodynamic effects observed (which may have a different timeline than the clearance or half-life of the specific agent), decisions on the acceptable peak-to-trough ratios observed with once-daily or twice-daily dosing schemes, as well as the clinical outcomes of different regimens tested in phase II and III clinical trials. Specific dosing regimens for rivaroxaban are approved for each clinical setting in which it is used. The dosing regimens have been developed based on preclinical data and clinical studies to take into account the balance of antithrombotic efficacy and bleeding risk. In many clinical scenarios, pharmacokinetic and pharmacodynamic data support the use of once-daily rivaroxaban doses because its antithrombotic effect persists for at least 24 hours for doses above 5 mg [11]. In this review, the empirical and clinical rationale supporting the dose regimens of rivaroxaban in each clinical setting will be discussed, with consideration of both the general populations and relevant special patient groups.
2 Pharmacokinetic and Pharmacodynamic Characteristics of Rivaroxaban in Healthy Subjects and Specific Populations
2.1 Healthy Subjects
In phase I studies in healthy volunteers, rivaroxaban was rapidly absorbed and demonstrated a high oral bioavailability (Table 2) [11]. Maximum concentrations occurred within a few hours of dosing and the half-life, which varied by age, ranged between 5 and 9 hours in healthy young subjects and from 11 to 13 hours in healthy elderly subjects [11–13]. Rivaroxaban was the principal circulating drug and no pharmacologically active metabolites were detected in plasma [14]. Approximately two-thirds of a dose was found to undergo metabolic degradation to inactive metabolites, of which one third was eliminated renally and the other third was eliminated via the hepatobiliary route; the final third was excreted unchanged by renal filtration and renal secretion (Table 2) [14, 15]. Rivaroxaban was well tolerated and had predictable pharmacokinetics at different single and multiple doses in healthy volunteers [11, 12, 16]. The pharmacokinetics of rivaroxaban were not affected to a clinically relevant degree by age [13, 17], gender [13, 17–19], or body weight [18]. Pharmacokinetics were also similar between Caucasian and Chinese subjects [19, 20]. Inhibition of Factor Xa activity was found to be dose-dependent and correlated closely with rivaroxaban plasma levels; the maximum effect occurred after approximately 3 hours and inhibition continued for at least 12 hours with 10-, 20-, and 30-mg twice-daily doses [12], and for more than 24 hours with a 30-mg dose [21]. The effect on coagulation assays also depended on dose [12]. The inhibition constant of rivaroxaban (0.4 nM) [10] is equivalent to a plasma concentration of unbound rivaroxaban of 0.17 μg/L, which is typically exceeded even at trough concentrations after 10- and 20-mg once-daily doses [10, 11, 21]. Through population pharmacokinetic analysis the expected peak and trough plasma concentrations for rivaroxaban in the treatment of VTE are 270 μg/L (189–491 μg/L) and 26 μg/L (6–87 μg/L), respectively. For atrial fibrillation the concurrent values are 249 μg/L (184–343 μg/mL) and 44 μg/L (12–137 μg/L) [22]. Based on these data along with the typical half-life in healthy and elderly patients of 11–13 hours (i.e., those in whom the drug persists longest) [1, 2], anticoagulant effects would be expected to persist for longer than 24 hours after these doses.
2.2 Renal and Hepatic Impairment
Exposure to rivaroxaban (area under the concentration–time curve [AUC]) was increased by 44, 52, and 64 % in patients with mild (creatinine clearance [CrCl] 50–79 mL/min), moderate (CrCl 30–49 mL/min), and severe (CrCl <30 mL/min) renal impairment, respectively. However, the peak concentration was less variable and the overall effect, even for those with severe renal impairment, was considered by the authors to be of moderate clinical relevance only (Table 2) [23]. Nevertheless, clinical use of rivaroxaban must take into account renal function, and caution should be exercised in patients with moderate (CrCl 30–49 mL/min) renal impairment. In Europe, rivaroxaban may be used with caution in patients with severe renal impairment (CrCl 15–29 mL/min); however, in the United States rivaroxaban is not advised in these patients, except in those with AF. In all cases, rivaroxaban should be avoided in patients with a CrCl <15 mL/min [1, 2].
Mild (Child–Pugh A) hepatic impairment did not cause a clinically relevant alteration of rivaroxaban pharmacokinetics (15 % increase in AUC compared with healthy subjects), but moderate impairment led to a marked increase in exposure (Table 2) and pharmacodynamic effects (159 % increase in the effect–time curve AUC and significant prolongation of prothrombin time) [24]. As such, rivaroxaban therapy is not recommended in patients with moderate to severe hepatic impairment, including hepatic disease associated with coagulopathy and clinically relevant bleeding risk, and in cirrhotic patients (Child–Pugh B and C) [1, 2].
2.3 Food Effects and Drug–Drug Interactions
Oral bioavailability was high (80–100 %) for a 10-mg dose regardless of food intake, but at higher doses of 15 or 20 mg in the absence of food, bioavailability and absorption rate were less than dose proportional [16, 25]. Without food, the bioavailability of a 20-mg dose of rivaroxaban was 66 % and the AUC was 1,447 μg·h/L. When this dose was given with food, the bioavailability was nearly 100 % with a 39 % increase in the AUC (2,048 μg·h/L) (Table 2) [26]. Therefore, while taking rivaroxaban with food is necessary for optimal absorption, if food could not be administered for a single day due to the patient being NPO (nil by mouth), there is still about two thirds of the dose absorbed. Clinicians need to weigh the potential clinical impact of this change in absorption. The decreased absorption rate appears to be due to the limited aqueous solubility of rivaroxaban at higher doses, leading to a lower absolute oral bioavailability for the 20-mg tablet under fasting conditions compared with fed conditions [26]. Rivaroxaban absorption was unaffected by changes in gastric pH due to ranitidine or antacid use [25], and pharmacokinetics were unaltered by digoxin or atorvastatin [27]. Concomitant administration of other anticoagulants, non-steroidal anti-inflammatory drugs, or antiplatelet agents may increase bleeding risks, and potential interactions of rivaroxaban with these agents have been investigated in phase I studies. Co-administration with enoxaparin did not affect pharmacokinetics but led to an additive effect on anti-Factor Xa inhibition [28]. Co-administration of naproxen with rivaroxaban did not prolong bleeding time to a clinically relevant extent, but there was evidence of a more pronounced response in some individuals [29]. When co-administered with antiplatelet agents, the bioavailability and pharmacokinetics of a 15-mg dose of rivaroxaban were not affected by steady-state clopidogrel, although bleeding time increased to a clinically relevant extent [30]. No clinically relevant pharmacokinetic or pharmacodynamic effects occurred with the combination of rivaroxaban and acetylsalicylic acid (ASA; 500 mg loading dose then 100 mg the next day plus rivaroxaban 15 mg) [31]. Rivaroxaban is eliminated by multiple pathways, which include, among others, metabolic degradation by cytochrome P450 3A4 (CYP3A4) and it is a substrate for P-glycoprotein (P-gp)-dependent pathways [15, 32], and a relevant increase in rivaroxaban exposure was seen with co-administration of strong inhibitors of both CYP3A4 and P-gp (e.g., ketoconazole, ritonavir; Table 2) [1, 2, 15]. Because of the multiple routes of elimination, drugs that inhibit only one of these pathways and those that moderately inhibit both pathways did not have a clinically relevant effect on the pharmacokinetics of rivaroxaban. However, care must be taken with concomitant administration of moderate inhibitors in the setting of patients with chronic kidney disease (CrCl 30–49 mL/min) because the risk of bleeding may be increased [1, 2]. CYP3A4 inducers (e.g., rifampicin) have the potential to reduce rivaroxaban plasma concentrations to a significant degree, and concomitant therapy is not recommended because of the risk of reduced efficacy [1, 2].
3 Prevention of Venous Thromboembolism in Patients Undergoing Elective Hip or Knee Replacement Surgery
Patients undergoing major orthopedic surgery to replace hip or knee joints have an incidence of VTE of approximately 40–60 % if no thromboprophylaxis is given [33]. As per international guidelines, acceptable options to reduce the risk of VTE include parenteral anticoagulation with unfractionated heparin, LMWH, or fondaparinux, or oral antithrombotic therapy with ASA, warfarin, or a TSOAC. Depending on the agent selected, therapy is started around the time of surgery and continued for 2–6 weeks [34]. Although a lower-grade recommendation, LMWH therapy is still considered the preferred option given the amount of evidence available [34]. However, in patients given parenteral agents, high rates of non-adherence to the recommended duration of anticoagulation have been reported, owing partly to the burden of self-injection [35]. Rivaroxaban has a similarly rapid onset of action to the commonly used LMWH enoxaparin [28], which is important in this setting, and could be more convenient for patients (particularly after discharge) because of its once-daily administration and oral rather than injectable formulation.
Rivaroxaban was initially evaluated in patients undergoing total hip and knee replacement surgery in a series of four phase II dose-finding studies. Rivaroxaban doses ranging from 2.5 to 30 mg twice daily or from 5 to 40 mg once daily, starting 6–8 hours after surgery, were compared with standard enoxaparin prophylactic doses (30 mg twice daily [bid] or 40 mg once daily [od]). Bilateral venography was performed between 5 and 9 days after surgery to verify the incidence of VTE, and bleeding outcomes were recorded [36–39]. Empirical analysis of the results of the four studies taken together suggested that rivaroxaban doses, in the range of 5–20 mg daily, had a comparable efficacy and safety profile to enoxaparin. As expected, lower daily doses were associated with a greater incidence of VTE, whereas higher doses led to an increased incidence of major bleeding. Overall, a 10 mg once-daily dose of rivaroxaban appeared to offer the potential for superior VTE prevention compared with enoxaparin 40 mg once daily and a similar incidence of major bleeding (Fig. 2) [36–39]. Further support to continue the 10-mg once-daily dose into phase III clinical trials came from previous preclinical work demonstrating that reasonable anti-Factor Xa activity was still present at 24 hours, indicating that patients would receive adequate levels of anticoagulation throughout the dosing interval (Fig. 3) [11].

Composite efficacy and safety profile of different rivaroxaban doses compared with enoxaparin for the prevention of VTE after hip and knee replacement surgery in four phase II dose-finding studies [36–39]. Doses for which rivaroxaban bars remained below the line performed better overall than enoxaparin 40 mg od. The composite outcome depicted here was not a predefined endpoint of these trials. bid twice daily, od once daily, VTE venous thromboembolism

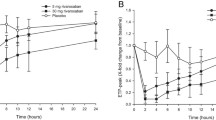
Median percentage change from baseline in Factor Xa inhibition after administration of rivaroxaban (with permission [11])
Data from these trials were used to construct two population pharmacokinetic and pharmacodynamic models, one of which compared once- and twice-daily doses in patients undergoing hip replacement surgery [40], and the other also included patients undergoing knee replacement surgery [41]. The models were used to simulate the effect of different dosing regimens, variations in population demographic factors, and the effect of co-medications. In the first model, the peak plasma concentration with a 10-mg once-daily dose was similar to that in healthy volunteers (median 125 vs 141 ng/mL) [11, 40], and trough concentrations between doses (~9 μg/L) were not significantly different from those with 5-mg twice-daily dosing, further supporting the use of a once-daily dose [40]. The average effect of variations in body weight, age, renal function, and other factors remained within the variability of the overall population, indicating that rivaroxaban could be given at a fixed dose to a variety of patients undergoing hip replacement surgery [40]. Even when extreme scenarios were simulated (age 90 years, moderate-to-severe renal impairment [CrCl 30 mL/min], low body weight [40 kg], and combined age of 90 years and body weight of 40 kg), the plasma concentration–time profiles of rivaroxaban remained within the predicted 90 % confidence intervals for the average population (Fig. 4). In the second model, in which patients undergoing knee replacement surgery were also included, the only relevant difference in pharmacokinetics between hip and knee replacement surgery patients was a 26 % lower drug clearance, which led to an approximately 30 % greater exposure to rivaroxaban [41].

Simulations of mean rivaroxaban plasma concentrations after a 10-mg once-daily dose in patients who have undergone hip replacement surgery (with permission [40]). Patients who are elderly, have moderate-to-severe renal impairment, have low body weight, or are elderly with low body weight, have predicted average plasma concentrations that fall within the boundaries for the overall population (90 % confidence intervals). CrCl creatinine clearance
The 10-mg once-daily dose was taken forward to four phase III clinical studies in the RECORD (REgulation of Coagulation in ORthopedic Surgery to Prevent DVT and PE) program, in which more than 12,500 patients were randomized to receive rivaroxaban 10 mg once daily started 6–8 hours after surgery, or enoxaparin started before (40 mg once daily) or after (30 mg twice daily) surgery [42]. In patients undergoing total hip replacement surgery, rivaroxaban given for 5 weeks was compared with enoxaparin 40 mg once daily given for 5 weeks (RECORD1 [43]) or 2 weeks (RECORD2 [44]). In patients undergoing total knee replacement surgery, rivaroxaban was compared with enoxaparin 40 mg once daily (RECORD3 [45]) and enoxaparin 30 mg twice daily (RECORD4 [46]), with all regimens given for 2 weeks. A variety of comparisons were thus possible, including the effect of longer compared with shorter duration thromboprophylaxis (RECORD2), and the efficacy and safety of rivaroxaban against the enoxaparin regimens most commonly used in Europe (RECORD1–3) and North America (RECORD4). In all trials, rivaroxaban was superior to enoxaparin for the prevention of VTE and there was no significant increase in major bleeding. In a pooled analysis of all the trials comparing rivaroxaban with enoxaparin 40 mg once daily (RECORD1–3; n = 9,581), rivaroxaban significantly reduced the incidence of VTE plus all-cause mortality (Table 3). The incidence of major bleeding was similar between the treatments at these time points [47]. When the trial of rivaroxaban compared with enoxaparin 30 mg twice daily (RECORD4; n = 12,729) was included in a pooled analysis of all four RECORD trials, rivaroxaban was associated with a significantly lower incidence of symptomatic VTE plus all-cause mortality than enoxaparin on treatment (Table 3), with no significant differences between treatments in terms of major bleeding, major plus non-major clinically relevant bleeding, or any bleeding [42]. Confirming the earlier studies, fixed-dose, oral rivaroxaban 10 mg once daily was found to have improved efficacy and similar safety to injectable enoxaparin across patient subgroups in this indication.
4 Treatment of Deep Vein Thrombosis and Pulmonary Embolism, and Prevention of Recurrent Venous Thromboembolism
Venous thromboembolism, comprising DVT and PE, is an established public health concern and is estimated to be responsible for almost half a million deaths in the European Union and approximately 300,000 fatalities in the United States annually [48, 49]. The standard treatment approach for all patients with VTE (specifically proximal DVT and PE, management of calf vein DVT is less established and varies by global region) includes therapeutic anticoagulation, and for patients not considered at high risk of immediate death, treatment usually starts with a parenteral anticoagulant such as a LMWH. An oral VKA is then added in parallel and the parenteral drug is discontinued after a minimum of 5 days of concomitant therapy and once the international normalized ratio (INR) is stable within the target range of 2.0–3.0 [50, 51]. All patients should receive a minimum of 3 months of therapeutic anticoagulation for the management of proximal DVT or PE, and many patients are candidates for longer durations of therapy to prevent recurrent events [50, 51]. This dual-drug paradigm (parenteral agent plus VKA) has been demonstrated to be very effective, but requires careful management, with patients subject to regular coagulation monitoring for the duration of their VKA treatment to keep the INR within the target range. By contrast, oral rivaroxaban can be used alone from the outset of treatment without the need for routine coagulation monitoring.
Two phase II studies were conducted to test the proof of concept of rivaroxaban as a single drug approach to treat VTE as compared with the standard approach of injectable anticoagulant plus vitamin K antagonist (VKA). Data from both trials were used to help determine an appropriate rivaroxaban dosing schedule for VTE treatment in phase III testing. The first trial evaluated single daily doses of rivaroxaban 20, 30, or 40 mg on asymptomatic deterioration in thrombotic burden or symptomatic VTE compared with LMWH plus VKA [52]. The companion trial evaluated dosing regimens of 10, 20, or 30 mg twice daily as well as 40 mg once daily compared with standard LMWH plus VKA on improvement in thrombotic burden at 21 days without recurrent symptomatic VTE or VTE-related death [53]. In the latter trial, no significant difference was observed in the dose–response relationship between twice-daily rivaroxaban doses and the primary endpoint. However, it was noted that patients receiving the 40-mg once-daily dose had fewer patients with improvement in thrombus burden at 21 days (43.8 %) compared with twice-daily regimens (53–59.2 %) (Fig. 5a). The 10- and 20-mg twice-daily rivaroxaban regimens had fewer cases of major bleeding than the 30-mg twice-daily dose.

Composite efficacy and safety profile of different rivaroxaban doses compared with enoxaparin/VKA for the treatment of deep vein thrombosis in two phase II dose-finding studies. a 21-day thrombus regression and major bleeding; b 3-month thrombus burden and clinically relevant bleeding [52, 53]. Rivaroxaban bars that remained below the line performed better overall than enoxaparin/VKA. Clinically relevant bleeding was the composite of major and non-major clinically relevant bleeding. The composite outcome depicted here was not a pre-defined endpoint of these trials. bid twice daily, od once daily, VKA vitamin K antagonist
In the trial evaluating once-daily doses over the course of 3 months, the 20-, 30-, and 40-mg once-daily rivaroxaban regimens compared favorably with standard therapy in terms of reducing thrombus burden, with a similar incidence of major bleeding (Fig. 5b). Based on the outcomes of these two studies, the lowest effective daily dose for rivaroxaban in the treatment of VTE was identified as 20 mg daily. In addition, phase II results also raised the possibility that an initial twice-daily regimen may provide better clot resolution during the first 21 days of therapy. Utilization of a higher dose during initial therapy is also supported by other VTE trials that have shown that the highest risk of recurrent VTE occurs in the acute phase (initial 3–4 weeks) of treatment [54, 55]. Taking these considerations together, the EINSTEIN investigators chose a 15-mg twice-daily dose for 21 days, followed by transition to a 20-mg once-daily dose for the duration of therapy [56–58]. The EINSTEIN investigators did not discuss their selection of the 15 mg twice-daily dose in their methods paper as it was not studied in phase II testing [58]. However, we might speculate they simply picked a dose between the 10- and 20-mg twice-daily dosing regimens that offered efficacy with the lowest risk of bleeding.
The proposed two-step dose regimen for rivaroxaban of 15 mg twice daily for 3 weeks followed by 20 mg once daily was evaluated using pharmacokinetic data from 870 patients with acute DVT in the phase II studies to create a population model and provide more information on the transition between once- and twice-daily dosing as required by the rivaroxaban VTE treatment regimen [56]. The model showed that age and renal function had a moderate effect on the pharmacokinetics of rivaroxaban, but variations remained minimal from the overall population. As such, no dose adjustment for patient populations who might be at risk for drug accumulation (renal dysfunction, extremes in body weight, age, etc.) was utilized in phase III testing for VTE. Simulations predicted a consistent exposure (i.e., no substantial changes in maximum concentration) to rivaroxaban during the transition from 15-mg twice-daily to 20-mg once-daily dosing (Fig. 6). Although the rivaroxaban AUC increased in a dose-dependent fashion with both once- and twice-daily dosing, the AUC results obtained for both regimens were similar. When comparing the same total daily doses, the peak plasma concentration was approximately 20 % higher and the trough concentration was approximately 60 % lower with the use of once-daily compared with twice-daily dosing. Because the 5th–95th percentile ranges for these parameters had substantial overlap, once-daily dosing should not expose patients to a greater risk of bleeding at the peak plasma concentration, or thrombus growth at the trough plasma concentration) compared with twice-daily dosing [56]. The model also demonstrated that significant plasma concentrations of rivaroxaban, expected to provide an adequate anticoagulant effect, would be present at the end of the 24-hour dosing interval during long-term therapy with a once-daily dose [56].

Simulated venous thromboembolism treatment dosing regimen of rivaroxaban 15 mg bid for 3 weeks, followed by 20 mg od (with permission [56]). Rivaroxaban exposure remains consistent during the transition, indicating that antithrombotic activity should be maintained. bid twice daily, od once daily
Two phase III studies of rivaroxaban for acute VTE treatment were conducted [57, 59]. EINSTEIN DVT recruited patients with symptomatic, confirmed DVT without PE [57], whereas EINSTEIN PE included patients with PE with or without concurrent DVT [59]. PE was studied separately because it has a distinct clinical course to DVT and is also less common; therefore, trials involving a general VTE population may not recruit a sufficient proportion of patients with PE to confirm outcomes in this group. Both trials followed the same design, with patients randomized to receive 3, 6, or 12 months of treatment with rivaroxaban 15 mg twice daily for 3 weeks followed by 20 mg once daily, or enoxaparin 1 mg/kg twice daily overlapping with and transitioning to a VKA (warfarin or acenocoumarol) with the dose adjusted to maintain an INR of 2.0–3.0 [57, 59].
In EINSTEIN PE, the first 400 patients included in the study were evaluated separately to confirm that the dose regimen, which had been selected based on outcomes in patients with symptomatic DVT without symptomatic PE, was efficacious and safe for patients with PE. Based on the incidence of the composite of symptomatic recurrent VTE and asymptomatic deterioration on repeat lung imaging at 3 weeks, the regimen was deemed to be appropriate [59]. In both EINSTEIN DVT and EINSTEIN PE, rivaroxaban was non-inferior to enoxaparin/VKA for the prevention of recurrent, symptomatic VTE (Table 3). There was also no difference between the regimens in the incidence of major plus non-major clinically relevant bleeding [57, 59]. However, in EINSTEIN PE, rivaroxaban provided a 51 % relative risk reduction in the incidence of major bleeding (Table 3). Outcomes were consistent in subpopulations of patients such as the elderly, those with renal impairment, and patients with high or low body weight, suggesting that the rivaroxaban regimen would be appropriate without the need for adjustment in this setting. It is important to note that patients with an estimated CrCl <30 mL/min were excluded from the EINSTEIN trials, and that patients receiving thrombolytic therapy were also excluded from the EINSTEIN PE trial [57, 59]. An extension study of long-term rivaroxaban treatment to prevent recurrent VTE, EINSTEIN EXT, was also conducted. Patients who had been successfully treated for an initial VTE, but for whom the decision to continue or stop anticoagulation was uncertain, received either rivaroxaban 20 mg once daily or placebo for a further 6 or 12 months [57]. Rivaroxaban was superior to placebo for the prevention of recurrent VTE, without a significant increase in major bleeding (Table 3) [57].
5 Prevention of Stroke and Systemic Embolism in Patients with Non-Valvular Atrial Fibrillation
AF is the most common dysrhythmia that occurs in clinical practice and is responsible for approximately 500,000 hospitalizations in the United States alone [60]. Conservative estimates suggest that 2.7 million patients in the United States and 4.5 million in the European Union have AF [60, 61]. These numbers are expected to at least double in the next 30 years because of the aging population [62]. Patients with AF have an approximately five-fold increase in the risk of stroke compared with the general population [63]. Patients with AF are routinely prescribed oral anticoagulant therapy such as warfarin or another VKA. The limitations of VKA therapy have been well described, and the use of an oral anticoagulant that does not require dose adjustment (fixed dose) or lifetime therapeutic monitoring would be a significant advantage in the management of these patients. Whereas dabigatran and apixaban are both administered twice daily for the reduction of risk of stroke in patients with AF, rivaroxaban is taken once daily. This adds to the simplicity of the regimen and may improve adherence [64].
The once-daily dosing of rivaroxaban that has been investigated to reduce the risk of stroke in patients with AF was developed from a population pharmacokinetic model [56]. This model used data on rivaroxaban pharmacokinetics from the two previously described phase II studies of patients treated for DVT [52, 53]. The purpose of this modeling study was to simulate the pharmacokinetics of rivaroxaban in a virtual patient population with AF, and thus DVT treatment population data were modified to reflect the demographic characteristics of a patient population with AF [56]. The demographic data used to represent this typical AF population came from the SPORTIF (Stroke Prevention Using Oral Thrombin Inhibitor in Atrial Fibrillation) III and V trials of patients with AF [65, 66]. Simulations were conducted to determine which demographic factors influenced the rivaroxaban exposure in AF patients [56]. The results of these simulations determined that moderate renal impairment (CrCl 30–49 mL/min), and, to a lesser extent, age ≥75 years, led to a slight increase in rivaroxaban exposure. The simulated plasma concentration–time profile for patients with normal renal function receiving rivaroxaban 20 mg once daily was similar for simulated AF and DVT patients. Additionally, the increase in rivaroxaban exposure because of decreased renal function in this simulation was corrected by dose adjustment to 15 mg once daily (Fig. 7) [56]. Therefore, for phase III clinical testing, the 20-mg once-daily dose was chosen for patients with normal renal function, and the 15-mg once-daily dose was chosen for patients with moderate renal insufficiency.

Simulated rivaroxaban plasma concentration–time profiles for a virtual population of patients with atrial fibrillation (with permission [56]). For patients with mildly impaired or normal CrCl (>50 mL/min), exposure is the same with a 20 mg od dose as for patients with moderate renal impairment (≤50 mL/min) with a 15 mg od dose. CrCl creatinine clearance, od once daily
The suitability of the aforementioned dose was confirmed in the randomized, double-blind, double-dummy, phase III trial, ROCKET AF (Rivaroxaban Once-daily Oral Direct Factor Xa Inhibition Compared with Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation) [67]. A total of 14,264 patients with non-valvular AF at moderate to high risk of stroke were randomized to rivaroxaban, dosed as above, or dose-adjusted warfarin (managed to an INR of 2.0–3.0). In the intention-to-treat analysis, the primary endpoint of stroke or systemic embolism occurred at a rate of 2.1 % per year in patients receiving rivaroxaban and 2.4 % per year in patients receiving warfarin. This 12.5 % relative risk reduction with rivaroxaban met the criteria for non-inferiority (p < 0.001), but not superiority (p = 0.12). The primary analysis of the trial was designed to be the per-protocol, as-treated population. In this group, a benefit of rivaroxaban was demonstrated compared with warfarin (hazard ratio 0.79; 95 % CI 0.66–0.96; Table 3). Among patients in the on-treatment safety population, rivaroxaban was superior to warfarin (p = 0.01). Rates of clinically relevant bleeding and major bleeding were similar between the treatment arms (Table 3). However, rivaroxaban use led to a significant decrease in intracranial hemorrhage (p = 0.02) and fatal bleeding (p = 0.003), although the incidence of gastrointestinal bleeding was higher in the rivaroxaban arm (p < 0.001) [67]. Pharmacokinetic analysis of 161 patients in ROCKET AF supported the predictions of the modeling work, and confirmed that parameters for patients with moderate renal impairment (who were given rivaroxaban 15 mg once daily) were similar to those with normal renal function who received 20 mg twice daily [68]. Therefore, ROCKET AF demonstrated that rivaroxaban provides comparable efficacy to warfarin, with less intracranial and fatal bleeding at a cost of more gastrointestinal bleeding.
6 Prevention of Cardiovascular Events in Patients with Recent Acute Coronary Syndrome
An ACS event is a common complication of coronary heart disease and is associated with more than 1.2 million hospitalizations in the United States and close to 3 million hospitalizations worldwide annually [60, 69]. ACS comprises unstable angina (UA), non-ST-segment elevation myocardial infarction (NSTEMI), and ST-elevation myocardial infarction (STEMI), with UA and NSTEMI commonly grouped together as non-ST-segment elevation ACS (NSTE ACS). Standard chronic management of patients who have had an ACS event includes treatment with dual antiplatelet therapy consisting of ASA and a P2Y12 inhibitor. Despite this aggressive antiplatelet strategy, the rate of recurrent cardiovascular events remains high (~10 %) [70, 71]. There is evidence of sustained coagulation activation after an ACS event [72, 73], and previous studies have shown that the addition of an oral anticoagulant (e.g., warfarin) to antiplatelet therapy can further reduce recurrent cardiovascular events but increases the risk of bleeding [74, 75].
The phase II dose-ranging study ATLAS ACS TIMI 46 (Anti-Xa Therapy to Lower Cardiovascular Events in Addition to Standard Therapy in Subjects With Acute Coronary Syndrome ACS 2–Thrombolysis In Myocardial Infarction 46: Rivaroxaban Versus Placebo in Patients with Acute Coronary Syndromes) was conducted in patients who had experienced a recent ACS event [76]. Patients were randomized to receive rivaroxaban at doses of 5, 10, or 20 mg once daily or the same total doses twice daily, or equivalent placebo, in addition to single or dual antiplatelet therapy (depending on the decision of the individual investigator). Rivaroxaban reduced the incidence of death, MI, or stroke, but increased the risk of clinically significant bleeding dose-dependently [76]. Overall, doses of 2.5 and 5 mg twice daily appeared to offer the best balance of efficacy and safety (Fig. 8), and these were selected for further investigation. These doses of rivaroxaban are much lower than those used in stroke prevention in atrial fibrillation and in the treatment of VTE. While coagulation plays a role in thrombosis in the arterial vascular, most of the process is dominated by platelets. Therefore, in combination with single or dual antiplatelet therapy, it is likely that less intense long-term anticoagulation would be optimal. Data from 2,290 patients with pharmacokinetic values from the ATLAS ACS TIMI 46 trial were entered into a population pharmacokinetic model. The data indicated that pharmacokinetic parameters, and the effects of factors such as age, renal function, and body weight, would be similar for patients with ACS receiving rivaroxaban 2.5 mg twice daily as for the other population models in patients receiving rivaroxaban for VTE prevention, VTE treatment, and for stroke prevention in patients with AF [77].

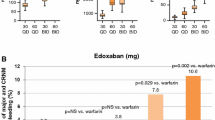
Composite efficacy and safety of different doses of rivaroxaban compared with placebo, both combined with standard antiplatelet therapy, for the prevention of recurrent events in patients with ACS in a phase II dose-finding study [76]. Clinically relevant bleeding was the composite of major and non-major clinically relevant bleeding. The composite outcome depicted here was not a pre-defined endpoint of this trial. ACS acute coronary syndrome, bid twice daily, od once daily
In the randomized phase III ATLAS ACS 2 TIMI 51 study, rivaroxaban 2.5 or 5 mg twice daily added to standard antiplatelet therapy (ASA ± a thienopyridine) was assessed against antiplatelets alone in patients with a recent ACS event [78]. The addition of rivaroxaban at either dose significantly reduced the incidence of death from cardiovascular causes, MI, or stroke (Table 3) but, unlike the higher dose, the 2.5-mg twice-daily dose led to significantly reduced rates of death from cardiovascular causes (p = 0.002) and any cause (p = 0.002). Rivaroxaban was associated with an increased incidence of major bleeding not related to coronary artery bypass grafting (1.8 % for the 2.5-mg dose and 2.4 % for the 5-mg bid dose vs 0.6 %; p < 0.001 for both rivaroxaban doses combined vs placebo) and intracranial bleeding (0.4 % for the 2.5-mg bid dose and 0.7 % for the 5-mg bid dose vs 0.2 %; p = 0.009 for both rivaroxaban doses combined vs placebo), but fatal bleeding was not increased (0.3 vs 0.2 %; p = 0.66). The lower dose resulted in a lower rate of fatal bleeding than the higher dose (0.1 vs 0.4 %; p = 0.04) [78]. Based on these data, the 2.5-mg twice-daily dose was approved in Europe as an adjunct to antiplatelet therapy, but only in patients with a recent ACS event and elevated cardiac biomarkers, suggesting a high likelihood of recurrence. Rivaroxaban is currently not approved in the United States for this indication, as the FDA has raised concerns with trial results and data management.
7 Implications for Daily Clinical Practice
The rivaroxaban clinical study program, including phase I pharmacokinetic and pharmacodynamic investigations, phase II dose-finding studies, and randomized phase III clinical trials, as well as pharmacokinetic modeling and simulation work, provide a body of evidence to support the doses that are now used in each approved clinical indication (Table 1). Despite a half-life that would suggest the need for twice-daily dosing (5–9 h in healthy young subjects, 11–13 h in healthy elderly patients), once-daily doses are appropriate in many situations based on positive results from phase II and III clinical trials for patients with AF, patients who have undergone hip or knee replacement surgery, and those receiving long-term treatment for the prevention of recurrent VTE. In addition, it is important to consider that similar to what has been observed for LMWHs, the pharmacodynamic effects for rivaroxaban exceed the pharmacokinetic half-life. As such, the duration of pharmacodynamic effect (anti-Xa activity) should be the main determinant of the daily dosing schedule. Nevertheless, it is important to consider clinical circumstances in which daily dosing is not the case, namely the initial 3 weeks of twice-daily rivaroxaban treatment for acute DVT or PE and for patients with ACS; (Table 1). The clinical pharmacology of rivaroxaban supports a once-daily dosing regimen for VTE prophylaxis after hip and knee replacement surgery (10 mg) and the prevention of recurrent VTE and stroke (20 mg) because of a long-lasting effect on thrombin generation and trough plasma drug concentrations sufficient to provide an effective antithrombotic effect for 24 hours or longer [10, 11, 21, 40, 56].
Routine dose reduction for renal impairment is recommended at this time only for patients with AF who are receiving rivaroxaban for stroke prevention and who have moderate (CrCl 30–49 mL/min) or severe (CrCl 15–29 mL/min) renal impairment. In these patients, a 15-mg once-daily dose is recommended instead of the normal 20-mg once-daily dose [1, 2]. This dose reduction was as effective as standard therapy when clinically tested in patients with renal impairment in ROCKET AF [67]. By contrast, patients receiving rivaroxaban for long-term secondary prevention of recurrent VTE, for whom the same 20-mg once-daily dose is recommended, should not routinely be given a reduced dose if they have renal impairment (CrCl 15–50 mL/min), as they did not receive such a dose in the phase III EINSTEIN trials [57, 59]. The rationale is that VTE patient populations are generally younger and have fewer co-morbidities than AF populations and are, therefore, less susceptible to drug accumulation. As previously stated, pharmacokinetic modeling for this patient population did not indicate a large variation in plasma concentrations accompanying changes in either age or renal function [56]. In addition, clinical trial results support this approach. In a combined pooled analysis of both the EINSTEIN DVT and PE trials, it was found that in patients with a calculated creatinine clearance <50 mL/min, rivaroxaban demonstrated a lower risk of bleeding as compared with patients receiving LMWH/warfarin with a similar effect on efficacy. Similar favorable results with safety were also seen in patients >75 years of age, or who weighed less than 60 kg. These results further support maintaining patients at a dose of 20 mg once daily down to a creatinine clearance of 30 mL/min [79]. Although these results stem from a secondary analysis of the EINSTEIN trials, it is important to note that the subpopulation for reduced renal function totaled 649 patients, a sample size that is far larger than exists for any other anticoagulant in this patient population. Regardless, the European Summary of Product Characteristics states that a 15-mg once-daily dose may be considered in patients receiving long-term rivaroxaban for prevention of secondary VTE if the risk of bleeding is high and outweighs the evaluated risk for recurrent VTE [1]. However, this recommendation is not included in the US Prescribing Information [2]. In general, with the exception of ROCKET AF, few of the dose reductions recommended for TSOACs have actually been tested in clinical trials. It is important that patients with a CrCl <15 min/mL should not receive rivaroxaban for any of the approved indications because of the risk of drug accumulation.
Rivaroxaban is appropriate for a broad population of patients because its pharmacokinetic and pharmacodynamic properties generally do not vary to a clinically relevant degree with variations in gender, age, body weight, ethnicity, and mild or moderate renal and mild hepatic impairment [39, 56, 77]. However, there are circumstances and patient groups in which these agents are either not appropriate or relatively untested. Critically ill patients with PE who have shock and/or hypotension and require urgent thrombolytic therapy or thromboectomy fall into this category, and there are no data on the use of TSOACs in this setting. There are also no data on the use of these drugs during pregnancy, in patients aged <18 years, or in patients with a CrCl <15 mL/min; therefore, use of rivaroxaban is not recommended in these patients [1, 2]. Use in patients with moderate or severe hepatic disease (Child–Pugh B or C) is also not recommended because of a clinically relevant increased risk of bleeding. In general, more data are needed in patients with moderate renal impairment and those with cancer, relatively few of whom have been included in the phase III trials of the TSOACs, and also in patients with severe renal impairment (CrCl 15–29 mL/min), who were excluded from phase III studies because of their high bleeding risk. Although risk of major bleeding appears to be comparable to other anticoagulants, it is important to note that a small number of patients on rivaroxaban will experience a bleeding event and further guidance is needed with regards to effective reversal strategies in the case where bleeding may be life-threatening.
Rivaroxaban is increasingly used by clinicians in a broad range of thromboembolic disorders. Use of the specific dose regimen for each indication is vital to optimize therapeutic benefit and minimize risk of bleeding.
References
Bayer Pharma AG. Xarelto® (rivaroxaban) Summary of Product Characteristics. 2014. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000944/human_med_001155.jsp. Accessed 28 May 2014.
Janssen Pharmaceuticals Inc. Xarelto® (rivaroxaban) Prescribing Information. 2014. http://www.xareltohcp.com/sites/default/files/pdf/xarelto_0.pdf#zoom=100. Accessed 28 May 2014.
Boehringer Ingelheim International GmbH. Pradaxa® (dabigatran etexilate) Summary of Product Characteristics. 2014. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000829/human_med_000981.jsp. Accessed 28 May 2014.
Boehringer Ingelheim Pharmaceuticals Inc. Pradaxa® (dabigatran etexilate) Prescribing Information. 2014. http://bidocs.boehringer-ingelheim.com/BIWebAccess/ViewServlet.ser?docBase=renetnt&folderPath=/Prescribing%20Information/PIs/Pradaxa/Pradaxa.pdf. Accessed 28 May 2014.
Bristol–Myers Squibb, Pfizer EEIG. Eliquis® (apixaban) Summary of Product Characteristics. 2014. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002148/human_med_001449.jsp&mid=WC0b01ac058001d124. Accessed 28 May 2014.
Bristol–Myers Squibb Company, Pfizer Inc. Eliquis® (apixaban) Prescribing Information. 2014. http://packageinserts.bms.com/pi/pi_eliquis.pdf. Accessed 28 May 2014.
Ageno W, Gallus AS, Wittkowsky A, Crowther M, Hylek EM, Palareti G. Oral anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141:e44S–88S.
Eikelboom JW, Weitz JI. New anticoagulants. Circulation. 2010;121:1523–32.
Wong PC, Pinto DJ, Zhang D. Preclinical discovery of apixaban, a direct and orally bioavailable factor Xa inhibitor. J Thromb Thrombolysis. 2011;31:478–92.
Perzborn E, Roehrig S, Straub A, Kubitza D, Misselwitz F. The discovery and development of rivaroxaban, an oral, direct Factor Xa inhibitor. Nat Rev Drug Discov. 2011;10:61–75.
Kubitza D, Becka M, Voith B, Zuehlsdorf M, Wensing G. Safety, pharmacodynamics, and pharmacokinetics of single doses of BAY 59-7939, an oral, direct Factor Xa inhibitor. Clin Pharmacol Ther. 2005;78:412–21.
Kubitza D, Becka M, Wensing G, Voith B, Zuehlsdorf M. Safety, pharmacodynamics, and pharmacokinetics of BAY 59-7939—an oral, direct Factor Xa inhibitor—after multiple dosing in healthy male subjects. Eur J Clin Pharmacol. 2005;61:873–80.
Kubitza D, Becka M, Roth A, Mueck W. The influence of age and gender on the pharmacokinetics and pharmacodynamics of rivaroxaban—an oral, direct Factor Xa inhibitor. J Clin Pharmacol. 2013;53:249–55.
Weinz C, Schwarz T, Kubitza D, Mueck W, Lang D. Metabolism and excretion of rivaroxaban, an oral, direct Factor Xa inhibitor, in rats, dogs and humans. Drug Metab Dispos. 2009;37:1056–64.
Mueck W, Kubitza D, Becka M. Co-administration of rivaroxaban with drugs that share its elimination pathways: pharmacokinetic effects in healthy subjects. Br J Clin Pharmacol. 2013;76:455–66.
Mueck W, Becka M, Kubitza D, Voith B, Zuehlsdorf M. Population model of the pharmacokinetics and pharmacodynamics of rivaroxaban—an oral, direct Factor Xa inhibitor—in healthy subjects. Int J Clin Pharmacol Ther. 2007;45:335–44.
Kubitza D, Becka M, Roth A, Mueck W. Dose-escalation study of the pharmacokinetics and pharmacodynamics of rivaroxaban in healthy elderly subjects. Curr Med Res Opin. 2008;24:2757–65.
Kubitza D, Becka M, Zuehlsdorf M, Mueck W. Body weight has limited influence on the safety, tolerability, pharmacokinetics, or pharmacodynamics of rivaroxaban (BAY 59-7939) in healthy subjects. J Clin Pharmacol. 2007;47:218–26.
Jiang J, Hu Y, Zhang J, Yang J, Mueck W, Kubitza D, Bauer RJ, Meng L, Hu P. Safety, pharmacokinetics and pharmacodynamics of single doses of rivaroxaban—an oral, direct Factor Xa inhibitor—in elderly Chinese subjects. Thromb Haemost. 2010;103:234–41.
Zhao X, Sun P, Zhou Y, Liu Y, Zhang H, Mueck W, Kubitza D, Bauer RJ, Zhang H, Cui Y. Safety, pharmacokinetics and pharmacodynamics of single/multiple doses of the oral, direct Factor Xa inhibitor rivaroxaban in healthy Chinese subjects. Br J Clin Pharmacol. 2009;68:77–88.
Graff J, von Hentig N, Misselwitz F, Kubitza D, Becka M, Breddin HK, Harder S. Effects of the oral, direct Factor Xa inhibitor rivaroxaban on platelet-induced thrombin generation and prothrombinase activity. J Clin Pharmacol. 2007;47:1398–407.
Mueck W, Stampfus J, Kubitza D, Becka M. Clinical pharmacokinetic and pharmacodynamic profile of rivaroxaban. Clin Pharmacokinet. 2014;53:1–16.
Kubitza D, Becka M, Mueck W, Halabi A, Maatouk H, Klause N, Lufft V, Wand DD, Philipp T, Bruck H. Effects of renal impairment on the pharmacokinetics, pharmacodynamics and safety of rivaroxaban, an oral, direct Factor Xa inhibitor. Br J Clin Pharmacol. 2010;70:703–12.
Kubitza D, Roth A, Becka M, Alatrach A, Halabi A, Hinrichsen H, Mueck W. Effect of hepatic impairment on the pharmacokinetics and pharmacodynamics of a single dose of rivaroxaban—an oral, direct Factor Xa inhibitor. Br J Clin Pharmacol. 2013;76:89–98.
Kubitza D, Becka M, Zuehlsdorf M, Mueck W. Effect of food, an antacid, and the H2 antagonist ranitidine on the absorption of BAY 59-7939 (rivaroxaban), an oral, direct Factor Xa inhibitor, in healthy subjects. J Clin Pharmacol. 2006;46:549–58.
Stampfuss J, Kubitza D, Becka M, Mueck W. The effect of food on the absorption and pharmacokinetics of rivaroxaban. Int J Clin Pharmacol Ther. 2013;51:549–61.
Kubitza D, Becka M, Roth A, Mueck W. Absence of clinically relevant interactions between rivaroxaban—an oral, direct Factor Xa inhibitor—and digoxin or atorvastatin in healthy subjects. J Int Med Res. 2012;40:1688–707.
Kubitza D, Becka M, Schwers S, Voith B. Investigation of pharmacodynamic and pharmacokinetic interactions between rivaroxaban and enoxaparin in healthy male subjects. Clin Pharmacol Drug Dev. 2013;2:270–7.
Kubitza D, Becka M, Mueck W, Zuehlsdorf M. Rivaroxaban (BAY 59-7939)—an oral, direct Factor Xa inhibitor—has no clinically relevant interaction with naproxen. Br J Clin Pharmacol. 2007;63:469–76.
Kubitza D, Becka M, Mueck W, Schwers S. Effect of co-administration of rivaroxaban and clopidogrel on bleeding time, pharmacodynamics and pharmacokinetics: a phase I study. Pharmaceuticals (Basel). 2012;5:279–96.
Kubitza D, Becka M, Mueck W, Zuehlsdorf M. Safety, tolerability, pharmacodynamics, and pharmacokinetics of rivaroxaban—an oral, direct Factor Xa inhibitor—are not affected by aspirin. J Clin Pharmacol. 2006;46:981–90.
Lang D, Freudenberger C, Weinz C. In vitro metabolism of rivaroxaban—an oral, direct Factor Xa inhibitor—in liver microsomes and hepatocytes of rat, dog and man. Drug Metab Dispos. 2009;37:1046–55.
Geerts WH, Bergqvist D, Pineo GF, Heit JA, Samama CM, Lassen MR, Colwell CW. Prevention of venous thromboembolism: American College of Chest Physicians evidence-based clinical practice guidelines (8th Edition). Chest. 2008;133:381S–453S.
Falck-Ytter Y, Francis CW, Johanson NA, Curley C, Dahl OE, Schulman S, Ortel TL, Pauker SG, Colwell CW Jr. Prevention of VTE in orthopedic surgery patients: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141:e278S–325S.
Wilke T, Muller S. Non-adherence in outpatient thromboprophylaxis after major orthopedic surgery: a systematic review. Expert Rev Pharmacoecon Outcomes Res. 2010;10:691–700.
Turpie AGG, Fisher WD, Bauer KA, Kwong LM, Irwin MW, Kälebo P, Misselwitz F, Gent M, ODXIa-Knee Study Group. BAY 59-7939: an oral, direct Factor Xa inhibitor for the prevention of venous thromboembolism in patients after total knee replacement. A phase II dose-ranging study. J Thromb Haemost. 2005;3:2479–86.
Eriksson BI, Borris LC, Dahl OE, Haas S, Huisman MV, Kakkar AK, Muehlhofer E, Dierig C, Misselwitz F, Kälebo P, ODIXa-HIP Study Investigators. A once-daily, oral, direct Factor Xa inhibitor, rivaroxaban (BAY 59-7939), for thromboprophylaxis after total hip replacement. Circulation. 2006;114:2374–81.
Eriksson BI, Borris L, Dahl OE, Haas S, Huisman MV, Kakkar AK, Misselwitz F, Kälebo P, ODIXa-HIP Study Investigators. Oral, direct Factor Xa inhibition with BAY 59-7939 for the prevention of venous thromboembolism after total hip replacement. J Thromb Haemost. 2006;4:121–8.
Eriksson BI, Borris LC, Dahl OE, Haas S, Huisman MV, Kakkar AK, Misselwitz F, Muehlhofer E, Kälebo P. Dose-escalation study of rivaroxaban (BAY 59-7939)—an oral, direct Factor Xa inhibitor—for the prevention of venous thromboembolism in patients undergoing total hip replacement. Thromb Res. 2007;120:685–93.
Mueck W, Borris LC, Dahl OE, Haas S, Huisman MV, Kakkar AK, Kälebo P, Muelhofer E, Misselwitz F, Eriksson BI. Population pharmacokinetics and pharmacodynamics of once- and twice-daily rivaroxaban for the prevention of venous thromboembolism in patients undergoing total hip replacement. Thromb Haemost. 2008;100:453–61.
Mueck W, Eriksson BI, Bauer KA, Borris L, Dahl OE, Fisher WD, Gent M, Haas S, Huisman MV, Kakkar AK, Kälebo P, Kwong LM, Misselwitz F, Turpie AGG. Population pharmacokinetics and pharmacodynamics of rivaroxaban—an oral, direct Factor Xa inhibitor—in patients undergoing major orthopaedic surgery. Clin Pharmacokinet. 2008;47:203–16.
Turpie AGG, Lassen MR, Eriksson BI, Gent M, Berkowitz SD, Misselwitz F, Bandel TJ, Homering M, Westermeier T, Kakkar AK. Rivaroxaban for the prevention of venous thromboembolism after hip or knee arthroplasty. Pooled analysis of four studies. Thromb Haemost. 2011;105:444–53.
Eriksson BI, Borris LC, Friedman RJ, Haas S, Huisman MV, Kakkar AK, Bandel TJ, Beckmann H, Muehlhofer E, Misselwitz F, Geerts W, RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med. 2008;358:2765–75.
Kakkar AK, Brenner B, Dahl OE, Eriksson BI, Mouret P, Muntz J, Soglian AG, Pap AF, Misselwitz F, Haas S, RECORD2 Investigators. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet. 2008;372:31–9.
Lassen MR, Ageno W, Borris LC, Lieberman JR, Rosencher N, Bandel TJ, Misselwitz F, Turpie AGG, RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med. 2008;358:2776–86.
Turpie AGG, Lassen MR, Davidson BL, Bauer KA, Gent M, Kwong LM, Cushner FD, Lotke PA, Berkowitz SD, Bandel TJ, Benson A, Misselwitz F, Fisher WD, RECORD4 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty (RECORD4): a randomised trial. Lancet. 2009;373:1673–80.
Eriksson BI, Kakkar AK, Turpie AGG, Gent M, Bandel TJ, Homering M, Misselwitz F, Lassen MR. Oral rivaroxaban for the prevention of symptomatic venous thromboembolism after elective hip and knee replacement. J Bone Jt Surg Br. 2009;91:636–44.
Cohen AT, Agnelli G, Anderson FA, Arcelus JI, Bergqvist D, Brecht JG, Greer IA, Heit JA, Hutchinson JL, Kakkar AK, Mottier D, Oger E, Samama MM, Spannagl M, VTE Impact Assessment Group in Europe (VITAE). Venous thromboembolism (VTE) in Europe. The number of VTE events and associated morbidity and mortality. Thromb Haemost. 2007;98:756–64.
Heit JA, Cohen AT, Anderson FA, on behalf of the VTE Impact Assessment Group. Estimated annual number of incident and recurrent, non-fatal and fatal venous thromboembolism (VTE) events in the US. Blood (ASH Annual Meeting Abstracts). 2005;106. (Abstract 910).
Torbicki A, Perrier A, Konstantinides S, Agnelli G, Galiè N, Pruszczyk P, Bengel F, Brady AJ, Ferreira D, Janssens U, Klepetko W, Mayer E, Remy-Jardin M, Bassand JP, Vahanian A, Camm J, De Caterina R, Dean V, Dickstein K, Filippatos G, Funck-Brentano C, Hellemans I, Kristensen SD, McGregor K, Sechtem U, Silber S, Tendera M, Widimsky P, Zamorano JL, Zamorano JL, Andreotti F, Ascherman M, Athanassopoulos G, De Sutter J, Fitzmaurice D, Forster T, Heras M, Jondeau G, Kjeldsen K, Knuuti J, Lang I, Lenzen M, Lopez-Sendon J, Nihoyannopoulos P, Perez Isla L, Schwehr U, Torraca L, Vachiery JL. Guidelines on the diagnosis and management of acute pulmonary embolism: the Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). Eur Heart J. 2008;29:2276–315.
Kearon C, Akl EA, Comerota AJ, Prandoni P, Bounameaux H, Goldhaber SZ, Nelson ME, Wells PS, Gould MK, Dentali F, Crowther M, Kahn SR. Antithrombotic therapy for VTE disease: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141:e419S–94S.
Buller HR, Lensing AW, Prins MH, Agnelli G, Cohen A, Gallus AS, Misselwitz F, Raskob G, Schellong S, Segers A. A dose-ranging study evaluating once-daily oral administration of the Factor Xa inhibitor rivaroxaban in the treatment of patients with acute symptomatic deep vein thrombosis: the Einstein-DVT Dose-Ranging Study. Blood. 2008;112:2242–7.
Agnelli G, Gallus A, Goldhaber SZ, Haas S, Huisman MV, Hull RD, Kakkar AK, Misselwitz F, Schellong S, ODIXa-DVT Study Investigators. Treatment of proximal deep-vein thrombosis with the oral direct Factor Xa inhibitor rivaroxaban (BAY 59-7939): the ODIXa-DVT (oral direct factor Xa Inhibitor BAY 59-7939 in patients with acute symptomatic deep-vein thrombosis) study. Circulation. 2007;116:180–7.
Fiessinger JN, Huisman MV, Davidson BL, Bounameaux H, Francis CW, Eriksson H, Lundström T, Berkowitz SD, Nyström P, Thorsén M, Ginsberg JS, THRIVE Treatment Study Investigators. Ximelagatran vs low-molecular-weight heparin and warfarin for the treatment of deep vein thrombosis: a randomized trial. JAMA. 2005;293:681–9.
van Gogh Investigators, Büller HR, Cohen AT, Davidson B, Decousus H, Gallus AS, Gent M, Pillion G, Piovella F, Prins MH, Raskob GE. Idraparinux versus standard therapy for venous thromboembolic disease. N Engl J Med. 2007;357:1094–104.
Mueck W, Lensing AW, Agnelli G, Decousus H, Prandoni P, Misselwitz F. Rivaroxaban: population pharmacokinetic analyses in patients treated for acute deep-vein thrombosis and exposure simulations in patients with atrial fibrillation treated for stroke prevention. Clin Pharmacokinet. 2011;50:675–86.
The EINSTEIN Investigators. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010;363:2499–510.
The EINSTEIN Investigators. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010;363: Supplementary Material. http://www.nejm.org/doi/suppl/10.1056/NEJMoa1007903/suppl_file/nejmoa1007903_protocol.pdf.
The EINSTEIN–PE Investigators. Oral rivaroxaban for the treatment of symptomatic pulmonary embolism. N Engl J Med. 2012;366:1287–97.
Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–245.
Camm AJ, Kirchhof P, Lip GYH, Schotten U, Savelieva I, Ernst S, Van Gelder IC, Al Attar N, Hindricks G, Prendergast B, Heidbuchel H, Alfieri O, Angelini A, Atar D, Colonna P, De Caterina R, De Sutter J, Goette A, Gorenek B, Heldal M, Hohloser SH, Kolh P, Le Heuzey JY, Ponikowski P, Rutten FH, Vahanian A, Auricchio A, Bax J, Ceconi C, Dean V, Filippatos G, Funck-Brentano C, Hobbs R, Kearney P, McDonagh T, Popescu BA, Reiner Z, Sechtem U, Sirnes PA, Tendera M, Vardas PE, Widimsky P, Vardas PE, Agladze V, Aliot E, Balabanski T, Blomstrom-Lundqvist C, Capucci A, Crijns H, Dahlof B, Folliguet T, Glikson M, Goethals M, Gulba DC, Ho SY, Klautz RJ, Kose S, McMurray J, Perrone Filardi P, Raatikainen P, Salvador MJ, Schalij MJ, Shpektor A, Sousa J, Stepinska J, Uuetoa H, Zamorano JL, Zupan I. Guidelines for the management of atrial fibrillation: The Task Force for the Management of Atrial Fibrillation of the European Society of Cardiology (ESC). Eur Heart J. 2010;31:2369–429.
Miyasaka Y, Barnes ME, Gersh BJ, Cha SS, Bailey KR, Abhayaratna WP, Seward JB, Tsang TS. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation. 2006;114:119–25.
Wang TJ, Massaro JM, Levy D, Vasan RS, Wolf PA, D’Agostino RB, Larson MG, Kannel WB, Benjamin EJ. A risk score for predicting stroke or death in individuals with new-onset atrial fibrillation in the community: the Framingham Heart Study. JAMA. 2003;290:1049–56.
Bae JP, Dobesh PP, Klepser DG, Anderson JD, Zagar AJ, McCollam PL, Tomlin ME. Adherence and dosing frequency of common medications for cardiovascular patients. Am J Manag Care. 2012;18:139–46.
Halperin JL, Executive Steering Committee SPORTIF III and V Study Investigators. Ximelagatran compared with warfarin for prevention of thromboembolism in patients with non-valvular atrial fibrillation: Rationale, objectives, and design of a pair of clinical studies and baseline patient characteristics (SPORTIF III and V). Am Heart J. 2003;146:431–8.
Executive Steering Committee of behalf of the SPORTIF III Investigators. Stroke prevention with the oral direct thrombin inhibitor ximelagatran compared with warfarin in patients with non-valvular atrial fibrillation (SPORTIF III): randomised controlled trial. Lancet. 2003;362:1691–8.
Patel MR, Mahaffey KW, Garg J, Pan G, Singer DE, Hacke W, Breithardt G, Halperin JL, Hankey GJ, Piccini JP, Becker RC, Nessel CC, Paolini JF, Berkowitz SD, Fox KAA, Califf RM, ROCKET AF Investigators. Rivaroxaban versus warfarin in non-valvular atrial fibrillation. N Engl J Med. 2011;365:883–91.
Mueck W, Schwers S, Stampfuss J. Rivaroxaban and other novel oral anticoagulants: pharmacokinetics in healthy subjects, specific patient populations and relevance of coagulation monitoring. Thromb J. 2013;11:10.
Grech ED, Ramsdale DR. Acute coronary syndrome: unstable angina and non-ST segment elevation myocardial infarction. Br Med J. 2003;326:1259–61.
Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, Neumann FJ, Ardissino D, De Servi S, Murphy SA, Riesmeyer J, Weerakkody G, Gibson CM, Antman EM. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001–15.
Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, Horrow J, Husted S, James S, Katus H, Mahaffey KW, Scirica BM, Skene A, Steg PG, Storey RF, Harrington RA, PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045–57.
Théroux P, Waters D, Lam J, Juneau M, McCans J. Reactivation of unstable angina after the discontinuation of heparin. N Engl J Med. 1992;327:141–5.
Merlini PA, Bauer KA, Oltrona L, Ardissino D, Cattaneo M, Belli C, Mannucci PM, Rosenberg RD. Persistent activation of coagulation mechanism in unstable angina and myocardial infarction. Circulation. 1994;90:61–8.
Rothberg MB, Celestin C, Fiore LD, Lawler E, Cook JR. Warfarin plus aspirin after myocardial infarction or the acute coronary syndrome: meta-analysis with estimates of risk and benefit. Ann Intern Med. 2005;143:241–50.
Andreotti F, Testa L, Biondi-Zoccai GG, Crea F. Aspirin plus warfarin compared to aspirin alone after acute coronary syndromes: an updated and comprehensive meta-analysis of 25,307 patients. Eur Heart J. 2006;27:519–26.
Mega JL, Braunwald E, Mohanavelu S, Burton P, Poulter R, Misselwitz F, Hricak V, Barnathan ES, Bordes P, Witkowski A, Markov V, Oppenheimer L, Gibson CM. ATLAS ACS-TIMI 46 study group. Rivaroxaban versus placebo in patients with acute coronary syndromes (ATLAS ACS-TIMI 46): a randomised, double-blind, phase II trial. Lancet. 2009;374:29–38.
Xu XS, Moore K, Burton P, Stuyckens K, Mueck W, Rossenu S, Plotnikov A, Gibson M, Vermeulen A. Population pharmacokinetics and pharmacodynamics of rivaroxaban in patients with acute coronary syndromes. Br J Clin Pharmacol. 2012;74:86–97.
Mega JL, Braunwald E, Wiviott SD, Bassand JP, Bhatt DL, Bode C, Burton P, Cohen M, Cook-Bruns N, Fox KA, Goto S, Murphy SA, Plotnikov AN, Schneider D, Sun X, Verheugt FW, Gibson CM, ATLAS ACS 2-TIMI 51 Investigators. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med. 2012;366:9–19.
Prins MH, Lensing AW, Bauersachs R, van Bellen B, Bounameaux H, Brighton TA, Cohen A, Davidson B, Decousus H, Raskob GE, Berkowitz SD, Wells PS, On behalf of the EINSTEIN Investigators. Oral rivaroxaban versus standard therapy for the treatment of symptomatic venous thromboembolism: a pooled analysis of EINSTEIN-DVT and PE randomized studies. Thromb J. 2013;11:21.
Camm AJ, Lip GYH, De Caterina R, Savelieva I, Atar D, Hohnloser SH, Hindricks G, Kirchhof P, Bax JJ, Baumgartner H, Ceconi C, Dean V, Deaton C, Fagard R, Funck-Brentano C, Hasdai D, Hoes A, Kirchhof P, Knuuti J, Kolh P, McDonagh T, Moulin C, Popescu BA, Reiner Z, Sechtem U, Sirnes PA, Tendera M, Torbicki A, Vahanian A, Windecker S, Vardas P, Al Attar N, Alfieri O, Angelini A, Blomstrom-Lundqvist C, Colonna P, De Sutter J, Ernst S, Goette A, Gorenek B, Hatala R, Heidbuchel H, Heldal M, Kristensen SD, Kolh P, Le Heuzey JY, Mavrakis H, Mont L, Filardi PP, Ponikowski P, Prendergast B, Rutten FH, Schotten U, Van Gelder IC, Verheugt FW. 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J. 2012;33:2719–47.
Acknowledgments and disclosures
The authors would like to acknowledge Stephen Purver, who provided editorial support, with funding from Bayer HealthCare Pharmaceuticals and Janssen Scientific Affairs, LLC.
Conflicts of interest
TT serves as a consultant for Boehringer-Ingelheim, Janssen, and BMS/Pfizer. PPD serves as a consultant for Janssen, AstraZeneca, and BMS/Pfizer.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Trujillo, T., Dobesh, P.P. Clinical Use of Rivaroxaban: Pharmacokinetic and Pharmacodynamic Rationale for Dosing Regimens in Different Indications. Drugs 74, 1587–1603 (2014). https://doi.org/10.1007/s40265-014-0278-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-014-0278-5