
Abstract
Introduction
A potentially elevated risk for pulmonary thrombosis with Janus kinase inhibitors (JAKinibs) was identified, as well as an increased risk for portal vein thrombosis, in ruxolitinib patients. Consequently, the objective of this investigation was to repeat a comprehensive analysis of the US FDA’s Adverse Event Reporting System (FAERS) database to assess postmarketing reporting rates of thromboembolic events (TEs) in patients treated with JAKinibs.
Methods
FAERS data (1 January 2010 to 30 September 2019) were searched for reports of all FDA-approved JAKinibs across all indications. For each drug–adverse drug reaction (ADR) pair, the reporting odds ratio (ROR) [two-sided 95% confidence interval (CI)] and empirical Bayesian geometric mean (EBGM) [one-sided 95% lower bound] were calculated to detect drug–ADR pairs with higher-than-expected reporting rates within the FAERS. Significance was declared when both lower 95% CI bounds were > 1.
Results
Significantly elevated reporting rates of pulmonary thrombosis were evident with tofacitinib (ROR 2.36 [1.69–3.31]; EBGM 2.01 [1.53]), as was pulmonary embolism with baricitinib (ROR 12.23 [8.35–17.89]; EBGM 7.72 [3.82]) and portal vein thrombosis with ruxolitinib (ROR 4.16 [2.70–6.40]; EBGM 4.52 [3.11]). Deep vein thrombosis reports were increased with baricitinib (ROR 14.84 [9.64–22.84]; EBGM 9.49 [5.91]), as was thrombosis with ruxolitinib (ROR 1.40 [1.20–1.63]; EBGM 1.72 [1.52]). The relationship between the time of treatment initiation and event occurrence indicated that time to events occurred randomly.
Conclusions
This study found significant reporting rates for TEs in patients treated with JAKinibs across brands and indications, providing additional evidence that JAKinibs may be contraindicated in patients at risk of TEs.
Similar content being viewed by others


An increased reporting rate of thromboembolic events for many Janus kinase inhibitors was observed. |
In clinical practice, additional consideration should be given when prescribing Janus kinase inhibitors in patients with existing risk factors for thromboembolic events. |
1 Introduction
Thromboembolic events (TEs), including venous thromboembolism (VTE), are an important cause of preventable morbidity and mortality. In the US, VTEs alone affect over 900,000 people per year, with 10–30% dying within 1 month of diagnosis [1]. VTEs are associated with a significant economic burden, with incremental total direct medical costs of between US$12,000 and US$15,000 among first-year survivors and between US$18,000 and U$23,000 per incident case, factoring in subsequent complications [2]. In total, VTEs are estimated to cost the US healthcare system US$7–12 billion dollars each year [2].
Considerable research has identified individuals at higher risk for future TEs. Patient-related risk factors include older age, obesity, and current smoking status [3]. Diseases linked to an increased risk for TEs include certain malignancies, congestive heart failure, recent myocardial infarction (MI), immobility, and immune-mediated diseases (IMDs), including inflammatory arthritis and ulcerative colitis (UC) [4,5,6]. More recently, the use of specific immunomodulatory medications, including Janus kinase inhibitors (JAKinibs), has been identified as a potential additional risk factor for TE events [7].
JAKinibs are small molecules that block the activity of one or more of the intracellular tyrosine kinases (JAK1, JAK2, JAK3, and TYK2). In blocking these cytokines, JAKinibs interfere with the JAK-STAT signaling pathway and thereby induce immunosuppression [8, 9]. Several JAKinibs have been approved by the US FDA since 2011 for a variety of indications, including rheumatoid arthritis [RA], UC, psoriasis, psoriatic arthritis, myelofibrosis, and acute graft-versus-host disease.
Although demonstrated to be effective [10], JAKinibs have been associated with a higher risk of TE events, especially at higher doses, resulting in the FDA assigning a ‘boxed warning’ for increased risk for TE events associated with JAKinibs [11,12,13,14], and, in some cases, restricting use with regard to higher doses. This warning label applies to most of the commercially available JAKinibs, except ruxolitinib. Using real-world data from the FDA’s Adverse Event Reporting System (FAERS), Verden and colleagues found a potentially elevated risk of pulmonary thrombosis with JAKinibs, as well as a potentially increased risk of portal vein thrombosis for ruxolitinib based on available data for tofacitinib, tofacitinib extended release (XR), and ruxolitinib, from their approval dates to 31 March 2017 [7]. It should be noted that the literature remains mixed regarding the relationship between JAKinibs and increased incidence of cardiovascular (CV) events or TEs [15,16,17]. However, a recent meta-analysis found that across seven postmarketing surveillance studies of JAKinibs, pulmonary thromboembolism was the most commonly reported respiratory complication [17]. As the burden of TE events remains a substantial public health concern, it is important to continue to examine the strength of this potential safety signal in real-world data. This is particularly true for JAKinibs since they are indicated for, and commonly prescribed to, patients experiencing a broad range of IMDs, such as rheumatoid and psoriatic arthritis as well as UC, all of which already have a high background risk for TEs [18,19,20,21].
The primary objective of the current analysis was to conduct an updated and comprehensive analysis of FAERS to assess postmarketing reporting rates for TE events using the same methodology as Verden and colleagues [7]. A secondary objective was to descriptively examine the time elapsed between treatment initiation and reported TEs for each drug, as well as examining the frequency and percentage of reports, by age and sex.
2 Methods
The FAERS database, which contains information on adverse event and medication error reports submitted to the FDA, was searched (1 January 2010–30 September 2019) for reports on all FDA-approved JAKinibs (ruxolitinib, tofacitinib, tofacitinib XR, upadacitinib, fedratinib, and baricitinib) across all indications. It should be noted that baricitinib was approved 15 months prior to the end date for the current data window. As pointed out by the FDA, there are a number of limitations in FAERS data, including duplicate and incomplete reports, existence of a report does not establish causation, information in the reports has not been source-verified, and rates of occurrence cannot be established. A report may also have multiple drugs and multiple adverse drug reactions (ADRs), which are coded using Medical Dictionary for Regulatory Activities (MedDRA®) Preferred Terms (PT) [22]. Drugs are assigned a role (primary suspect, secondary suspect, concomitant, interacting) by the person reporting the ADR. Cases were only included if a JAKinib was listed as the ‘primary suspect’ of the reported ADR. ADRs of interest in this study included pulmonary thrombosis, pulmonary embolism, portal vein thrombosis, deep vein thrombosis (DVT), thrombosis, and venous thrombosis.
2.1 Data Cleaning Procedures
FAERS reports were downloaded directly from the FDA for each quarters in the study period. Quarterly files for each type of data (demographics, drugs, reactions, indications, therapies, outcomes) were inputted to SAS and combined over time. Note that certain file types had changes in format over the study period, and were therefore considered. Only the last (most current) case ID records were retained to remove duplicate records for the same report. In addition, certain specific reports indicated as erroneous in the FDA website were completely removed as recommended. Units were standardized for certain variables that could be entered with different units (e.g. age, weight). Event and reporting dates given as year or year and month were imputed to complete dates, inserting ‘01’ as the missing month or day of the month, as recommended by the FDA. A single drug record was kept for a small number of duplicate drug records that remained in the data with identical ID and drug information, prior to merging the different types of data (demographics, therapies, reactions) according to the primary ID.
To identify reports for each of the drugs of interest in this study, brand name and generic name variations were considered in combination with the New Drug Application (NDA) number. Text for drug names was read ignoring capitalization, spaces, and other extraneous characters. The distribution of string distance scores between the correct drug brand and generic names and the drug strings in the database were examined for each of the study drugs to look for the variations in spelling occurring in the data. This process facilitated the identification of drug reports with misspelled drug names. In most misspelled records, the misspelling of the drug name was obvious (omission of a letter, switch of two letters, etc.) To confirm the record pertaining to a drug of interest, other information (when reported), such as NDA number, route, and dose, were corroborated. In addition, original reporting date and event date (if available) were checked to ensure they came after the drug FDA approval date. Records with drug names that remained in doubt were not assigned to the drugs of interest.
2.2 Statistical Analysis
For each drug–event pair, the reporting odds ratio (ROR) and empirical Bayesian geometric mean (EBGM) were calculated to detect drug–ADR pairs with higher-than-expected reporting rates versus all other drugs in the FDA registry. The ROR is a transparent familiar measure that is easily interpretable/programmed (such as odds ratios used in categorical data analysis). Another advantage is that being an odds ratio, non-selective underreporting of a drug or ADR has no influence on the value of the ROR; however, the ROR is very sensitive to small samples, therefore drugs with lower overall reporting have a higher probability of disproportionate ADR detection with the ROR. To overcome the small-sample drawback, the EBGM is typically used in conjunction with the ROR. The EBGM pulls disproportionality scores toward the null, especially when there are limited data and a small number of drug–ADR reports. The EBGM uses a Poisson distribution to model the observed counts for each drug–ADR combination with a prior (Gamma mixture) distribution and to derive expected counts; however, the EBGM is hard to calculate and involves distributional assumptions and optimization of the likelihood function. Use of both statistics diminishes the effect of spuriously high ROR values, resulting in more reliable detection of safety signals.
ROR estimates with a two-sided 95% confidence interval (CI) lower bound > 1.0 were considered significant. One-sided 95% lower confidence bound of the EBGM was generated, with values > 1 considered to be significant. A strong signal of a disproportionately high event rate for a drug–event pair was declared when both the ROR and EBGM were significant. When both RORs and EBGM values were above 1, but were not significant, this was considered to indicate a non-significant trend toward higher-than-expected reporting rates.
A composite TE (CTE) was derived from the six ADRs of interest in the study (e.g. pulmonary thrombosis, pulmonary embolism, portal vein thrombosis, DVT, thrombosis, and venous thrombosis). The CTE represents any reported TE of interest and addresses the somewhat arbitrary PT used in the report, which may depend on the timing of the diagnosis. In addition, MI and cerebrovascular accident (CVA) composites were created and were derived from Standardized MedDRA Queries (SMQs). In consultation with clinical experts, PTs in the SMQs most likely to be associated with medication administration were identified and selected to create these composite events for the study. Specifically, the MI composite was derived from the MI broad SMQ, which includes 69 unique PTs; of these, 24 PTs were selected. The CVA definition was derived from the CVA broad SMQ, which includes 147 unique PTs; of these, 79 PTs were selected. Similar to the six ADRs based on the presence of the single PT in the adverse event reports, these three composite events were binary events based on the presence (= 1) of any of the selected PTs in the ADR (zero otherwise).
To assess for possible bias resulting from differences in length of time on the market for each JAKinib, we conducted a sensitivity analysis to calculate the RORs for the first year after each JAKinib FDA approval compared with the ROR in the remaining study period. We used only single PT events to simplify examination of these results. One of the JAKinib drugs involved (upadacitinib) had only been in the market for < 3 months and was excluded from this sensitivity analysis.
To assess a potential pattern in the timing of ADRs, the distribution of days from the start of therapy to the ADR was examined separately for each of the drug–ADR pairs. Mean, standard deviation (SD), and seven representative percentiles for ADR timing and age at the time of the report were included. The frequency and percentage of reports by sex were also analyzed. Values for start of therapy, age, and sex were commonly not reported, but the frequency of missing values was reported. All analysis was conducted using SAS 9.4 (SAS Institute, Inc., Cary, NC, USA).
3 Results
3.1 Pulmonary Thrombosis
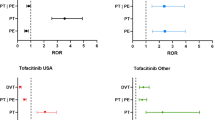
A total of 61 cases of pulmonary thrombosis were identified in the FAERS, with 13 cases reported for ruxolitinib, 48 for tofacitinib (34 immediate release [IR], 14 XR), and zero for both upadacitinib and baricitinib (Table 1). A statistically significant reporting rate for pulmonary thrombosis was observed for tofacitinib (ROR 2.36 [two-sided 95% CI 1.69–3.31]; EBGM 2.01 [one-sided 95% CI 1.53]). Furthermore, a trend toward higher-than-expected reporting rates was also observed with tofacitinib XR (ROR 1.59 [0.94–2.70]; EBGM 1.51 [0.95]).
3.2 Pulmonary Embolism
A total of 316 cases of pulmonary embolism were identified in the FAERS, with 107 cases reported for ruxolitinib, 181 for tofacitinib (154 IR, 27 XR), one for upadacitinib, and 27 for baricitinib (Table 1). A significantly increased reporting rate for pulmonary embolism was also observed for baricitinib (ROR 12.23 [8.35–17.89]; EBGM 7.72 [3.82]), while trends were observed for tofacitinib (ROR 1.08 [0.93–1.27]; EBGM 1.11 [0.97]) and upadacitinib (ROR 18.79 [2.59–136.59]; EBGM 0.71 [0.31]).
3.3 Portal Vein Thrombosis
A total of 25 cases of pulmonary embolism were identified in the FAERS, with 21 cases reported for ruxolitinib, 2 for tofacitinib (2 IR, 0 XR), zero for upadacitinib, and 2 for baricitinib (Table 1). A significant increased reporting rate for portal vein thrombosis was observed with ruxolitinib (ROR 4.16 [2.70–6.40]; EBGM 4.52 [3.11]).
3.4 Deep Vein Thrombosis
A total of 172 cases of DVT were identified in the FAERS, with 53 cases reported for ruxolitinib, 98 for tofacitinib (78 IR, 20 XR), zero for upadacitinib, and 21 for baricitinib (Table 1). A significant increased reporting rate for DVT was observed with baricitinib (ROR 14.84 [9.64–22.84]; EBGM 9.49 [5.91]).
3.5 Thrombosis
A total of 345 cases of thrombosis were identified in the FAERS, with 165 cases reported for ruxolitinib, 179 for tofacitinib (126 IR, 53 XR), zero for upadacitinib, and one for baricitinib (Table 1). A significant reporting rate for thrombosis was observed for ruxolitinib (ROR 1.40 [1.20–1.63]; EBGM 1.72 [1.52]) and tofacitinib (ROR 1.13 [0.95–1.35]; EBGM 1.15 [0.99]).
3.6 Venous Thrombosis
A total of 10 cases of venous thrombosis were identified in the FAERS, with 5 cases reported for ruxolitinib, 5 for tofacitinib (4 IR, 1 XR), and zero for upadacitinib and baricitinib (Table 1). There was no evidence of a significant increased reporting rate or trend for venous thrombosis.
3.7 Sensitivity Analysis
To evaluate the potential bias in results related to the possibly increased ADR reporting during the first year of JAKinib use after FDA approval compared with the subsequent study period, we calculated the RORs for these two periods separately. We found no tendency for increased reporting during the first year of marketing the drugs and no noticeable deviations from the overall results reported.
3.8 Composite Scores
For the CTE composite score (Table 2), a significantly increased reporting rate was observed with baricitinib (ROR 7.43 [5.45–10.13]; EBGM 4.38 [3.70]). Trends were observed with ruxolitinib (ROR 0.90 [0.81–1.00]; EBGM 1.11 [1.01]), tofacitinib (ROR 1.04 [0.94–1.16]; EBGM 1.51 [0.95]), and upadacitinib (ROR 6.83 [0.94–49.56]; EBGM 1.42 [0.23]). A trend was also observed with baricitinib for both MI (ROR 2.22 [1.19–4.13]; EBGM 1.42 [0.82]) and CVA (ROR 2.00 [1.24–3.22]; EBGM 1.15 [0.77]) composite scores, as well as upadacitinib for the CVA composite score (ROR 4.69 [0.65–34.05]; EBGM 1.16 [0.20]).
3.9 Time to Adverse Drug Reaction
The distribution of days from the start of therapy to ADR was examined for each of the drug–ADR pairs. As can be seen in Fig. 1, timing from the start of therapy to occurrence of the ADR extends from the first days or weeks to 1 year or more after the start of therapy, with mostly widespread right-skewed distributions.

Time from therapy start to ADR (in days). ADR adverse drug reaction, XR extended release
3.9.1 Distribution of Events by Age and Sex
Figure 2 contains summary statistics of the age distribution of the drug–ADR pairs. The median age of patients for drug–ADR events with sufficient sample sizes (n > 5) was > 65 years, with the exception of tofacitinib and tofacitinib XR pulmonary embolism drug–ADR events, which had median ages of 62 and 61 years, respectively. Generally, 67% of reports occurred in women.

Age distribution of adverse drug reactions, by drug. XR extended release
4 Discussion
There is a growing concern that patients treated with JAKinibs may experience an increased risk for TE, which has led the FDA to require a ‘boxed warning’ for most JAKinibs currently on the market [14]. Although data from clinical trials suggest the risk is low, they are limited by the relatively low number of patients in the pivotal studies and the exclusion of participants with a higher burden of comorbidities; however, findings from postmarketing studies suggest the risk may be significant [7, 14]. The findings from the current analysis, which includes a large population of patients treated with JAKinibs in a ‘real world’ setting, further support this observation. Specifically, an increased reporting rate for pulmonary thrombosis was found with tofacitinib, as well as a trend with tofacitinib XR. Our results are in agreement with Mease and colleagues [23] regarding the disproportionately high reporting found for pulmonary thrombosis. Specifically, in supplementary Table S12 of their study, they reported the following: pulmonary thrombosis EBGM (90% CI) of 1.76 (1.40–2.19) and ROR (90% CI) of 1.83 (1.45–2.30). Our results found significant disproportionate reporting for tofacitinib based on our rule (both ROR and EB05 lower-bound CI values were > 1.0) for pulmonary thrombosis only, as follows: EBGM (EB05) of 2.01 (1.53), and ROR (95% CI) of 2.36 (1.69–3.31). The results from Mease et al. [23] and our results are in close agreement on the estimates, but the Mease et al. signal detection rule is more stringent. In addition, there was an increased reporting rate of pulmonary embolism with baricitinib and a trend with tofacitinib. Increased reporting rates for thrombosis were found with ruxolitinib and also a trend with tofacitinib. The reporting rate of portal vein thrombosis was disproportionately high with ruxolitinib, as was the risk of DVT with baricitinib. Overall, the results of the current study appear to converge with the TE safety evidence for JAKinibs that have been recently published.
This study not only confirms but expands upon the findings previously reported by Verden and colleagues [7] in five important ways. First, like the study by Verden et al. [7], the current study found an increased risk of pulmonary thrombosis with tofacitinib and tofacitinib XR. Second, also like the study by Verden et al., the current study did not find an increased risk for pulmonary embolism with ruxolitinib, but found an increased risk for portal vein thrombosis as well as thrombosis, whereas Verden et al. only reported on trends. Third, this current study found significantly increased reporting for various TEs with baricitinib (pulmonary embolism and deep vein thrombosis) that were not included in the prior study [7]. Fourth, we expanded on the findings of Verden et al. by conducting analyses on upadacitinib, which was launched in August 2019. Although, in this current analysis, upadacinitib seems to show an increased reporting rate for pulmonary embolism, the observed results for upadacitinib must be interpreted with caution, given that it was only approved in August 2019 and the FAERS database was searched through September 2019. Fifth, this is the first analysis that examined the risk for three different composite scores and found an increased risk for total CTEs with baricitinib and a trend for MI and CVA. It is important to note that ruxolitinib does not currently have a ‘boxed warning’ for an increased risk of TE. Given the observed increased reporting risk for portal vein thrombosis as well as thrombosis with ruxolitinib found in the current study, additional analyses are warranted.
It is also important to note that the results of the analysis of ADRs based on the individual PTs do not converge with the results of the CTE analysis. With the exception of baricitinib, CTEs did not show a strong signal of disproportionality, despite the strong significant disproportionality findings for select individual PTs with ruxolitinib and tofacitinib. This might be due to the mixing of disproportionately high (thrombosis and portal vein thrombosis with ruxolitinib; thrombosis, pulmonary thrombosis and pulmonary embolism with tofacitinib) and low (venous thrombosis and deep vein thrombosis) ADRs included in the CTE resulting in an average insignificant CTE rate. The observed non-convergence in the significance of individual PE conditions and the CTE results may potentially be clinically explained by the fact that a proportion of patients who experienced DVT may be minimally symptomatic and thus do not seek care. Therefore, DVT events may be underreported. However, DVTs that initially go undetected may eventually manifest later as pulmonary events such as PE or pulmonary thrombosis, where patients will most likely be symptomatic and seek emergency care. This highlights the need for increased clinical vigilance and potentially proactive screening for DVT in patients being treated with JAKinibs.
In addition to the link between JAKinibs and an increased reporting risk for various TEs, the current study was the first study to examine time to ADR. The results of this analysis suggested no clearly discernable pattern between the occurrence of TEs and the start of therapy. The timing of ADRs appears to be widespread, suggesting that patients could potentially develop the event at any time in the course of treatment. However, for many of the drug–ADR pairs, the majority of the start of therapy dates were missing, therefore there is only limited information on the timing of the ADRs, which can be potentially biased and requires further validation. If validated, this is valuable information for physicians as it highlights the importance of continuously monitoring patients for potential TEs throughout their treatment duration. These findings may have implications for IMD patients whose underlying disease could be considered complex (i.e. patients with multiple comorbidities that collectively increase their overall risk for developing future TE), even before the addition of JAKinibs into their treatment regimen.
The descriptive analysis of the age at which patients experienced a TE suggested that a large proportion (> 50%) of JAKinib-treated patients who experience a TE in the FAERS dataset were over the age of 65 years. Confirmation of these preliminary observation analyses in other databases (e.g. commercial claims dataset or Medicare) is warranted. Furthermore, physicians may need to exercise additional caution when using JAKinibs in the elderly population, given the complex/various comorbidities that these patients typically experience.
Although the current study showed a potentially insightful relationship between the use of JAKinibs and the odds of reporting TE events in the FAERS registry, it is not without limitations. Due to the nature of the FAERS registry, there is uncertainty whether the reported events were due to the suspected drug, the underlying disease of the patient, or some other cause. Many cases are not reported to the FAERS registry, and reporting rates may not be similar across included drugs or within a given drug class. Furthermore, reporting rates may vary over time (higher post-approval and diminishing over time or peaking after the addition of labeling warnings by the FDA). However, our sensitivity analysis did not find evidence of substantial differences in the RORs for the drug–ADR pairs when comparing the first-year post-approval period with the remainder of the study period. The ‘primary suspect’ designation in the FAERS registry is subjective, and the influence of other drugs or factors cannot be ruled out from a given case report. Time to ADR, age, and sex data are limited due to missing values and should be interpreted with caution. Finally, the rates are not exposure-adjusted as these data are not publicly available. Additional real-world data, including analyses of administrative claims databases, are warranted.
5 Conclusion
The current analysis supports the growing concern regarding the increased risk of TEs in patients treated with JAKinibs across indications. Of particular concern may be patients with IMDs who are already at elevated risk for TEs [6] and who are commonly treated with JAKinibs [24]. Clinicians should carefully consider whether IMD patients have existing TE risk factors (e.g. age, obesity, and current smoking status) before adding JAKinibs, and closely monitor these patients, given that the timing of a TE from the start of therapy appears to be random. Overall, this finding converges and strengthens the results published by Verden and colleagues in 2018. Given the complex relationship between TE and JAKinibs, the results from this and the prior analysis should be validated in other real-world datasets.
References
Venous thromboembolism (blood clots): data and statistics. Centers for Disease Control and Prevention. Updated 7 February 2020. http://cdc.gov/ncbddd/dvt/data.html. Accessed 11 July 2020.
Grosse SD, Nelson RE, Nyarko KA, Richardson LC, Raskob GE. The economic burden of incident venous thromboembolism in the United States: a review of estimated attributable healthcare costs. Thromb Res. 2016;137:3–10. https://doi.org/10.1016/j.thromres.2015.11.033.
Gregson J, Kaptoge S, Bolton T, Pennells L, Willeit P, Burgess S, et al. Cardiovascular risk factors associated with venous thromboembolism. JAMA Cardiol. 2019;4(2):163–73. https://doi.org/10.1001/jamacardio.2018.4537.
Dean SM, Abraham W. Venous thromboembolic disease in congestive heart failure. Congest Heart Fail. 2010;16(4):164–9. https://doi.org/10.1111/j.1751-7133.2010.00148.x.
Sejrup JK, Børvik T, Grimnes G, Isaksen T, Hindberg K, Hansen JB, et al. Myocardial infarction as a transient risk factor for incident venous thromboembolism: results from a population-based case-crossover study. Thromb Haemost. 2019;119(8):1358–64. https://doi.org/10.1055/s-0039-1692176.
Borjas-Howard JF, Leeuw K, Rutgers A, Meijer K, Tichelaar V. Risk of recurrent venous thromboembolism in autoimmune diseases: a systematic review of the literature. Semin Thromb Hemost. 2019;45(2):141–9. https://doi.org/10.1055/s-0038-1661387.
Verden A, Dimbil M, Kyle R, Overstreet B, Hoffman KB. Analysis of spontaneous postmarket case reports submitted to the FDA regarding thromboembolic adverse events and JAK inhibitors. Drug Saf. 2018;41(4):357–61. https://doi.org/10.1007/s40264-017-0622-2.
O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med. 2013;368(2):161–70. https://doi.org/10.1056/NEJMra1202117.
Danese S, Grisham M, Hodge J, Telliez JB. JAK inhibition using tofacitinib for inflammatory bowel disease treatment: a hub for multiple inflammatory cytokines. Am J Physiol Gastrointest Liver Physiol. 2016;310(3):G155–62. https://doi.org/10.1152/ajpgi.00311.2015.
Benamu E. Infectious risks associated with biologics targeting janus kinase-signal transducer and activator of transcription signaling and complement pathway for inflammatory diseases. Infect Dis Clin North Am. 2020;34(2):271–310. https://doi.org/10.1016/j.idc.2020.02.014.
Smolen JS, Genovese MC, Takeuchi T, Hyslop DL, Macias WL, Rooney T, et al. Safety profile of baricitinib in patients with active rheumatoid arthritis with over 2 years median time in treatment. J Rheumatol. 2019;46(1):7–18. https://doi.org/10.3899/jrheum.171361.
Kremer JM, Genovese MC, Keystone E, Taylor PC, Zuckerman SH, Ruotolo G, et al. Effects of baricitinib on lipid, apolipoprotein, and lipoprotein particle profiles in a phase IIb study of patients with active rheumatoid arthritis. Arthritis Rheumatol. 2017;69(5):943–52. https://doi.org/10.1002/art.40036.
Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol. 2017;13(4):234–43. https://doi.org/10.1038/nrrheum.2017.23.
Rajasimhan S, Pamuk O, Katz JD. Safety of janus kinase inhibitors in older patients: a focus on the thromboembolic risk. Drugs Aging. 2020;37(8):551–8. https://doi.org/10.1007/s40266-020-00775-w.
Olivera PA, Lasa JS, Bonovas S, Danese S, Peyrin-Biroulet L. Safety of janus kinase inhibitors in patients with inflammatory bowel diseases or other immune-mediated diseases: a systematic review and meta-analysis. Gastroenterology. 2020;158(6):1554-73.e12. https://doi.org/10.1053/j.gastro.2020.01.001.
Xie W, Huang Y, Xiao S, Sun X, Fan Y, Zhang Z. Impact of Janus kinase inhibitors on risk of cardiovascular events in patients with rheumatoid arthritis: systematic review and meta-analysis of randomised controlled trials. Ann Rheum Dis. 2019;78(8):1048–54. https://doi.org/10.1136/annrheumdis-2018-214846.
Khoo JK, Barnes H, Key S, Glaspole IN, Östör AJ. Pulmonary adverse events of small molecule JAK inhibitors in autoimmune disease: systematic review and meta-analysis. Rheumatology (Oxford). 2020;59(9):2217–25. https://doi.org/10.1093/rheumatology/keaa117.
McCurdy JD, Kuenzig ME, Smith G, Spruin S, Murthy SK, Carrier M, et al. Risk of venous thromboembolism after hospital discharge in patients with inflammatory bowel disease: a population-based study. Inflamm Bowel Dis. 2020;26(11):1761–8. https://doi.org/10.1093/ibd/izaa002.
Cheng K, Faye AS. Venous thromboembolism in inflammatory bowel disease. World J Gastroenterol. 2020;26(12):1231–41. https://doi.org/10.3748/wjg.v26.i12.1231.
Chung WS, Peng CL, Lin CL, Chang YJ, Chen YF, Chiang JY, et al. Rheumatoid arthritis increases the risk of deep vein thrombosis and pulmonary thromboembolism: a nationwide cohort study. Ann Rheum Dis. 2014;73(10):1774–80. https://doi.org/10.1136/annrheumdis-2013-203380.
Ungprasert P, Srivali N, Spanuchart I, Thongprayoon C, Knight EL. Risk of venous thromboembolism in patients with rheumatoid arthritis: a systematic review and meta-analysis. Clin Rheumatol. 2014;33(3):297–304. https://doi.org/10.1007/s10067-014-2492-7.
International Council for Harmonisation (ICH). 2019. https://www.ich.org/page/meddra.
Mease P, Charles-Schoeman C, Cohen S, Fallon L, Woolcott J, Yun H, et al. Incidence of venous and arterial thromboembolic events reported in the tofacitinib rheumatoid arthritis, psoriasis and psoriatic arthritis development programmes and from real-world data. Ann Rheum Dis. 2020;79(11):1400–13. https://doi.org/10.1136/annrheumdis-2019-216761.
Jamilloux Y, El Jammal T, Vuitton L, Gerfaud-Valentin M, Kerever S, Sève P. JAK inhibitors for the treatment of autoimmune and inflammatory diseases. Autoimmun Rev. 2019;18(11):102390. https://doi.org/10.1016/j.autrev.2019.102390.
Acknowledgements
The authors wish to thank Jason Allaire, PhD, of Generativity Health Economics and Outcomes Research, for editorial assistance, which was funded by Arena Pharmaceuticals.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported by Arena Pharmaceuticals.
Conflict of interest
Juliana Setyawan is an employee of and owns stock in Arena Pharmaceuticals. Nassir Azimi, Vibeke Strand, Andres Yarur, and Moshe Fridman have received consulting fees from Arena Pharmaceuticals.
Ethics approval
Not applicable
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Data are available on the FAERS database.
Code availability
Not applicable.
Author contributions
All authors contributed to the writing and editing of this paper. JS and MF designed the study, and MF performed all analyses. All authors contributed to the interpretation of the results, and read and approved the final version submitted for publication.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Setyawan, J., Azimi, N., Strand, V. et al. Reporting of Thromboembolic Events with JAK Inhibitors: Analysis of the FAERS Database 2010–2019. Drug Saf 44, 889–897 (2021). https://doi.org/10.1007/s40264-021-01082-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-021-01082-y