
Abstract
Rett syndrome (RTT) has enjoyed remarkable progress in achieving specific therapies. RTT, a unique neurodevelopmental disorder first described in 1966, progressed slowly until the landmark paper of Hagberg and colleagues in 1983. Thereafter, rapid advances were achieved including the development of specific diagnostic criteria and the active search for a genetic etiology, resulting 16 years later in identification of variants in the methyl-CpG-binding protein (MECP2) gene located at Xq28. Shortly thereafter, the NIH Office of Rare Diseases funded the RTT Natural History Study (NHS) in 2003, initiating the acquisition of natural history data on clinical features from a large population of individuals with RTT. This information was essential for advancement of clinical trials to provide specific therapies for this disorder. In the process, the International Rett Syndrome Association (IRSA) was formed (now the International Rett Syndrome Foundation—IRSF), which participated directly in encouraging and expanding enrollment in the NHS and, subsequently, in developing the SCOUT program to facilitate testing of potential therapeutic agents in a mouse model of RTT. The overall objective was to review clinical characteristics developed from the NHS and to discuss the status of specific therapies for this progressive neurodevelopmental disorder. The NHS study provided critical information on RTT: growth, anthropometrics, longevity, key comorbidities including epilepsy, breath abnormalities, gastroesophageal dysfunction, scoliosis and other orthopedic issues, puberty, behavior and anxiety, and progressive motor deterioration including the appearance of parkinsonian features. Phenotype–genotype correlations were noted including the role of X chromosome inactivation. Development of clinical severity and quality of life measures also proved critical for subsequent clinical trials. Further, development of biochemical and neurophysiologic biomarkers offered further endpoints for clinical trials. Initial clinical trials prior to the NHS were ineffective, but advances resulting from the NHS and other studies worldwide promoted significant interest from pharmaceutical firms resulting in several clinical trials. While some of these have been unrewarding such as sarizotan, others have been quite promising including the approval of trofinetide by the FDA in 2023 as the first agent available for specific treatment of RTT. Blarcamesine has been trialed in phase 3 trials, 14 agents have been studied in phase 2 trials, and 7 agents are being evaluated in preclinical/translational studies. A landmark study in 2007 by Guy et al. demonstrated that activation of a normal MECP2 gene in a null mouse model resulted in significant improvement. Gene replacement therapy has advanced through translational studies to two current phase 1/2 clinical trials (Taysha102 and Neurogene-401). Additional genetic therapies are also under study including gene editing, RNA editing, and X-chromosome reactivation. Taken together, progress in understanding and treating RTT over the past 40 years has been remarkable. This suggests that further advances can be expected.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Rett syndrome (RTT) is a rare, progressive neurodevelopmental disorder occurring mainly in females but also noted in males. |
Methyl-CpG-binding protein 2 (MECP2) variants underly > 95% of individuals with RTT including classic and atypical RTT as well as males with a progressive neonatal encephalopathy and classic RTT due to somatic mosaicism or Klinefelter syndrome (47XXY). |
Clinical trials have been conducted since the 1990s but emerged dramatically in the last 10–15 years including numerous pharmaceuticals, two gene-based replacement therapies, and several other gene therapies including DNA and RNA editing and X chromosome reactivation. |
Trofinetide is the first US Food and Drug Administration approved product for the specific treatment of RTT. |
1 Introduction
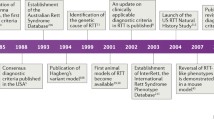
Specific treatment for rare neurodevelopmental disorders has been slow to develop. Rett syndrome (RTT) may represent an exception. First recognized in the late 1950’s by Andreas Rett, a developmental pediatrician in Vienna, Austria and by Bengt Hagberg, a child neurologist in Uppsala, Sweden, RTT languished in medical literature after the original description by Rett in 1966 [1]. Sufficient differences existed between the individuals described in this report and those seen by Hagberg that further information did not appear until the latter was encouraged by other child neurologists in Europe to describe his experience and that of fellow child neurologists. Following a chance meeting of Hagberg and Rett around 1980, Hagberg decided that this unique neurodevelopmental disorder should be called Rett syndrome. The landmark publication of Hagberg et al. in the Annals of Neurology in 1983 [2] resulted in the worldwide recognition of RTT, and with it, an intensive search for a definitive etiology was launched. As RTT was seen almost exclusively in females, these efforts pointed to a genetic causation and focusing on the X chromosome. In stepwise fashion over the next sixteen years, the search was increasingly restricted to the distal portion of the long arm of the X chromosome at Xq28 [3, 4]. In 1999, Ruthie Amir et al. [5] presented their findings which linked RTT to variations in the gene, methyl-CpG-binding protein 2 (MECP2) at Xq28.
In the intervening years between 1983 and 1999, much emphasis was placed on identifying individuals with RTT, aided by the first set of clinical criteria generated at a conference organized by Rett in 1984 [6]. Further, recognition of variant presentations became evident differentiating classical (typical) RTT and variant (atypical) RTT. In addition, other presentations emerged including an early-onset seizure variant [7] and a variant with exceptionally early neurodevelopmental delay [8]. These latter two were subsequently associated with their own individual genetic signatures: CDKL5 variants were identified in the early-onset seizure variant [9], now called CDKL5 deficiency disorder and FOXG1 variants [10], were identified in those with early developmental delay, now called FOXG1 disorder. A much smaller number of males was also recognized, expressing a wide variation in features ranging from early onset neonatal encephalopathy [11] to significant developmental delay. Some of these males appeared to have features typical of RTT. This is now known to result from somatic mosaicism or Klinefelter syndrome (47XXY), each expressing two X chromosomes with one having an MECP2 variant [12].
Even before identification of MECP2 variants, treatment trials were developed for RTT syndrome, but with the expansion of clinical diagnoses throughout the world and the identification of a genetic marker by Amir et al. [5], a search for effective therapies rapidly intensified including both pharmaceutical products and specific gene strategies. Increased diagnosis of RTT was aided by progressive revisions of the diagnostic criteria in 2002 [13] and 2010 [14], the first responding to the recent identification of the specific gene, and the second representing a simplification and stratification of the prior criteria based on the rapid expansion in clinical diagnoses over the intervening eight years and advocated by the need to be certain that the terminology used was clearly understood across the world.
Simultaneously with the Hagberg report in 1983, diagnoses were made for the first time in the US: in Washington DC, Seattle, and Houston. The diagnosis of RTT in three girls in Washington led their mothers to form the International Rett Syndrome Association [IRSA, now the International Rett Syndrome Foundation (IRSF)]. IRSA rapidly mobilized parent engagement and patient recruitment, which created a national presence in the US and greatly facilitated, through the support of travel clinics across the US, the subsequent US Natural History Study (USNHS), which was founded in 2003 and began enrollment in 2006. IRSF also developed a drug-testing feasibility program called the Steve Kaminsky Scout program, which facilitated the testing of potential therapeutic agents in an animal (mouse) model of RTT (Rettsyndrome.org foundation Scout Program and performed by PsychoGenics, Inc.). This program, which was begun in 2013, has been utilized as an essential forerunner in moving pharmaceutical agents to human trials and has tested more than 33 compounds (see below).
The objective of this review was to understand the clinical features of RTT derived from the USNHS, the largest study to date, and to provide an overview on the status of specific pharmacologic and genetic therapies for RTT.
2 Literature Search
This review covers the history of Rett syndrome from its first recognition by Andreas Rett and Bengt Hagberg more than 60 years ago through the initial studies assessing clinical criteria and genetic etiology to the USNHS of RTT. The NIH-sponsored RTT USNHS provided the largest clinical repository of individuals with RTT and RTT-related disorders gathered over 16 years. Individuals were seen semiannually, annually, or biannually depending on their age. More than 1600 individuals with RTT were evaluated, with a mean of 5.4 visits and a range of 1 to 18 visits. Literature derived from the USNHS was surveyed to address the relevant issues related to RTT including clinical trial readiness. With these advances in understanding RTT, specific therapeutic initiatives were also reviewed. These included a search of literature relative to translational studies and clinical trials for specific agents, assessment of ClinicalTrials.gov, the International Rett Syndrome Foundations report on “Companies Investing in Clinical Trials for Rett” (rettsyndrome.org), and the 2023 report from Medgadget, “Rett Syndrome Pipeline Insights” (https://www.medgadget.com/2023/rett-syndrome-pipeline-insights-20-key-companies-investigating-their-lead-candidates-to-improve-the-treatment-space-by-DelveInsight).
3 Rett Syndrome Natural History Study
The RTT USNHS was funded by the Office of Rare Diseases at the NIH in 2003, with data first being acquired in 2006. The resulting data were utilized to underscore the accuracy and suitability of the 2010 diagnostic criteria revision and created a rich reservoir of clinical information essential for developing subsequent clinical trials. The USNHS ended in 2021 with more than 1600 individuals with RTT having been enrolled.
The information derived from this study together with a broad increase in publications worldwide greatly expanded the depth and breadth of understanding of RTT and encouraged advances in drug discovery and clinical trials, as described below.
Growth failure was recognized as an early feature of RTT. Abnormal deceleration in the rate of head growth was noted as early as the second month of life and overall reduction in height and weight growth rates was remarkable in comparison with those in typically developing females. Data from the USNHS underscored these features and led to the development of specific growth charts (height, weight, body mass index, and head circumference) for RTT [15]. The initial clinical criteria for RTT included microcephaly, which was subsequently removed as a criterion, as up to 20% may have head circumferences in the normal range despite undergoing an abnormal deceleration in head growth after birth. Anthropometric measurements were provided on 1154 females with RTT demonstrating significant reductions compared to typically developing individuals [16]. Of interest, muscle area measurements were significantly greater in those individuals with independent arm and hand use and gait.
Longevity was assessed using Kaplan–Meier methodology in the North American database [17] and in the USNHS [18] indicating that 50% survival exceeded 50 years of age. When the original group described by Rett was reassessed in 2010 [19], the median age of survival was less than 14 years and the likelihood of reaching 25 years of age was 21%. These dramatic differences in survival over the last 30 years were related both to earlier diagnosis and to improved healthcare including better dietary management, increasing recognition of the need for improved overall health maintenance, the expansion of the clinical care teams, and the increased emphasis on physical and occupational therapies.
4 MECP2 Variants
The discovery of MECP2 variants as the principal etiology for RTT by Amir et al. [5] was fundamental in the expansion of clinical investigations in RTT. This gene product predominantly activates or inhibits methyl-binding domains in the nuclei of neurons and glia in the CNS [20, 21]. The complete roster of genes regulated by MECP2 is not completely evident, but this ability to regulate other genes is crucial in producing the neurodevelopmental abnormalities in RTT.
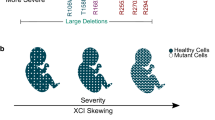
The identification of MECP2 variants in > 96% of those meeting the clinical criteria for RTT led to the identification of clear genotype–phenotype correlations with three specific recurrent point variations (R133C, R294X, and R306C) and distal carboxy-terminal truncations having overall milder features than four recurrent point mutations (R106W, R168X, R255X, and R270X), deletions or insertions, or the so-called large deletions involving one or more exons [22,23,24].
In most individuals with RTT, the MECP2 variant is de novo, arising as a new mutation in the rapidly dividing germinal cells of the sperm. However, the identification of mothers who shared the same variant with their daughters but were phenotypically normal or had mild-to-moderate developmental delays suggested that variations in X chromosome inactivation (XCI) could be responsible [3, 25, 26]. In addition, the occurrence of identical twins with the same variant and differing clinical features could be explained by similar variations. Fang et al. [27] noted significant skewing of XCI in blood in up to 36% of individuals in the NHS. Clearly, these analyses do not necessarily reflect the XCI pattern in the CNS but do suggest a possible explanation for the observed clinical variability.
The distribution of the MECP2 variant in the CNS is a random process and could vary from individual to individual, also serving as a basis for clinical variation.
Another issue is the potential association between parental age and the increased incidence of RTT. However, detailed analysis of the NHS data failed to identify a relationship between parental age and the diagnosis of RTT [28]. Interestingly, other X-linked disorders (Duchenne muscular dystrophy and hemophilia A and B) also do not show a relationship with parental age [29,30,31].
Males with MECP2 variants were also enrolled in the NHS. Thirty males were reported with the broad array of clinical features noted above including the early neonatal encephalopathy. Two males had somatic mosaicism and met clinical criteria for RTT, whereas more than a dozen males had the male RTT encephalopathy [12].
4.1 Clinical Features
Seizures and breathing issues have also received increased diagnostic and management attention over the past 30 years. Seizures in RTT are relatively infrequent at least until 30 months of age but do increase in frequency over the next 8–10 years. The presence of seizures not only was assessed by EEG reports in most cases but also were accepted by parent report of physician’s assessment. It is recognized in RTT that breath holding events can be interpreted as seizures. Where a question arises, the recommendation is for a video-EEG assessment. When viewed across time, seizures were noted in the NHS in about 90% of individuals but did occur in varying patterns [32, 33]. Some individuals had seizures that were relatively short term and did not recur, other individuals had seizures that waxed and waned, and still others had seizures that remained lifelong. Overall, management of seizures with pharmaceutical agents has been effective. In a small percentage (~5%), antiseizure medications have been insufficiently effective, that is, the individuals were refractory to medical management. Their treatment was supplemented by the ketogenic diet or by installation of a vagal nerve stimulator.
Awake breathing issues, that is breath holding or hyperventilation or both, are also extremely prominent and may be quite disruptive, perhaps more so in specific settings as they can interfere with daily living such as feeding, therapy sessions, and general interaction with parents or caregivers [34]. These breathing issues were typically not prominent before age 5 years; although, some individuals demonstrated these before this. When assessed longitudinally, these breathing issues may occur in 95% of individuals with RTT. Remissions were common, often lasting up to 1 year, and terminal remission was seen in 15%. The greatest difficulty was between age 5–15 years, often interfering with eating, therapy sessions, and general communication with family and therapists. After age 15 years, the breathing issues tended to be less prominent. The lack of effective treatment is overwhelming in many, indicating the need for effective therapies.
The assessment of purposeful hand function underscores the slowly progressive nature of RTT as these abilities decrease steadily during childhood and adolescence [35]. Specific MECP2 variants may result in significantly different skill levels, but over time, a steady decline in functional skills is noted with the steepest decline being seen in the variant group with the highest initial skill level.
Hand skills are significantly affected by the presence of the characteristic stereotypies. These occur in 100% of those with classic RTT but are variable from individual to individual and may change in frequency in each individual due to many factors including mood and environment [36]. Specific stereotypies may include hand wringing, hand tapping or clapping, finger rubbing, or hand mouthing. Hand mouthing was reported in nearly 50% at their baseline assessment. These stereotypies interfere with activities of daily living that include hand use specifically. Overall, increased hand stereotypies were associated with worse voluntary hand function. When viewed across time, these stereotypic activities remained relatively unchanged even as hand function, as noted above, declined.
4.2 Quality of Life Assessments
Individuals with RTT and their parents or caregiver have specific alterations in the quality of life (QOL). In the NHS, 97% of individuals with RTT lived at home although for a small number their care was provided by other siblings or relatives. QOL assessments of individuals with RTT revealed that those with worse motor skill had lower behavioral issues whereas those with better motor skills were able to cause their parents more difficulties due to worse behavioral issues. Behavioral challenges include being physically aggressive or the ability to create significant safety issues such as turning on the stove or water, climbing on furniture or even exiting the house [37]. This led to the concern that any therapy that improved motor performance could potentially create other issues for the parents.
QOL of the parents or caregivers also revealed specific issues. For most, the mental or psychological aspects of dealing with their child with RTT greatly exceeded the physical issues, at least in the early years after diagnosis [38]. Factors such as dealing with the diagnosis itself plus the worry and anxiety about what was in store for them were almost overwhelming. However, as time passed, the parents/caregivers gradually adapted or accommodated to these issues, but their own physical health became increasingly problematic, particularly as they had to lift or move their daughter multiple times from place to place throughout the day. Thus, the burden of illness had a definite negative impact on their overall QOL [39].
4.3 Multiple Comorbidities
The many medical comorbidities of RTT raised significant issues. Among these gastrointestinal (GI) problems, disrupted sleep, progressive scoliosis, and occasional cardiologic problems stand out.
GI issues can span the length of the digestive tract [40]. Chewing and swallowing are often quite difficult, gastroesophageal (GE) reflux is very common, and constipation is present in most. In addition, gastroparesis and gall bladder dysfunction [40] may occur. These require constant vigilance.
Sleep concerns include both difficulty going to sleep and maintaining sleep [41]. Often, the sleep difficulties reflect the many GI issues in that GE reflux, constipation, or even hunger may be involved. Before utilizing sleep aids, these specific issues should be addressed.
Scoliosis is present in 85% or more of individuals with RTT and is generally more severe in those with lower muscle tone or inability to ambulate [42, 43]. Scoliosis may begin in the preschool period and may progress steadily through adolescence. Surgery may be required in nearly 20% due to progression of the curvature greater than 40°. Scoliosis is often accompanied by truncal rotation, which also requires vigilance. Bracing and better positioning may be helpful in reducing the rate of progression. When surgical correction is required, the outcome is strongly endorsed by the parents or caregivers in improving the outlook of their daughters.
In addition, joint contractures, dystonia, and hip deformities also require attention. Currently, the degree of skeletal deformities is markedly less than seen 30 years ago or more, due to an increase in vigilance and earlier intervention by orthopedists and physiatrists.
Cardiologic issues are less common but could represent a significant problem if not assessed at least annually [44,45,46]. Prolongation of the corrected QT (QTc) interval beyond 450 ms is seen in about 20% of individuals with RTT [47]. If this is noted, a cardiologist should be consulted. Medications utilized include beta blockers and mexiletine. An animal study in a RTT mouse model indicated that the sodium channel blocker, phenytoin, was more effective than beta-blockers, but this has not been trialed in humans [44]. Extreme prolongation beyond 500 ms is unusual but in some has led to installation of a cardiac pacemaker. Multiple medications are associated with the potential for extending the QTc interval. However, this should not be regarded as an absolute prohibition and the use of these medications should be monitored by ECG assessments.
Anxiety and other behavioral issues have received increasing attention over the past twenty years [48, 49]. Behavioral issues include both internalizing (anxiety and social withdrawal) and externalizing (self-abuse and physical aggression) components. The former are very common, seen in virtually all with RTT, whereas the latter are generally less common and milder but still deserve attention. Anxiety is particularly problematic when the individual is out in crowds such as at a mall or restaurant or simply riding in a car. Selective serotonin reuptake inhibitors such as escitalopram or sertraline have been effective in improving such problems [49].
4.4 Caregiver Concerns and Clinical Trial Metrics
An important aspect of care for individuals with RTT is based on the top concerns as perceived by their parents or caregivers. In the last segment of the NHS, the top five concerns of those with RTT were assessed at each visit [50]. These assessments revealed the top concern to be the inability to communicate with their daughters in terms of what they would like or what they were requesting. The next four were seizures, inability or impairment in walking, poor hand use, and constipation. Two of the top concerns, seizures and constipation, are the most concerning comorbidities. Overall, these five factors serve as important outcome markers in judging the efficacy of specific treatments for these individuals.
In this regard, the development of clinical trial metrics is crucial. The Clinical Global Inventory of Severity (CGI-S) and of Improvement (CGI-I) were developed for assessment of clinical trial outcomes [51]. Specific RTT benchmarks were utilized for the CGI-S and both measures utilized a seven-point Likert scale. The CGI-S scales rated levels of severity from normal or 1 to most significantly affected or 7. The CGI-I was based on improvement or decline, with 4 being no change; 3, 2, and 1 being progressively better; and 5, 6, and 7 being progressively worse. The CGI scales were initially utilized by clinician raters following careful training and assessment of ability to judge the potential changes. More recently, the CGI-I has been utilized by parents/caregivers.
For the caregivers, the Rett Syndrome Behavioral Questionnaire (RSBQ) has been utilized widely. The RSBQ was not developed as an outcome measure and offers some challenges that require specific education of the caregivers who will be using this questionnaire. The RSBQ is not without its concerns, as reported recently, with nearly half the questions revealing floor or ceiling effects [52]. Importantly, however, both the CGI scales and the RSBQ are accepted by the FDA as suitable outcome measures.
In addition, other outcome measures have been developed and could be employed in future following suitable assessment. In the USNHS, the Motor Behavioral Assessment (MBA), originally developed by Fitzgerald et al. [53, 54] in the 1990s, was assessed at each visit. The outcomes were assessed for core validity via factor analysis [55]. The result was a five-factor grouping along with three specific RTT behaviors that constituted the revised MBA (R-MBA) [55]. The R-MBA still needs to be reassessed in a separate group of individuals with RTT to establish validity and reliability.
More recently a separate caregiver reported assessment has been reported [56]. This measure provides parent or caregiver assessment of problem severity in RTT, referred to as the Rett Syndrome Caregiver Assessment of Symptom Severity (RCASS). The RCASS will also require additional study to establish validity and reliability.
In addition to the development of outcome measures, the NHS also engaged in work to identify neurophysiological [57, 58] and molecular biomarkers [59]. Although additional validation work is needed to determine if they are suitable biomarkers, eventually such biomarkers could be incorporated into clinical trials in RTT.
5 Assessment of Specific Pharmacologic Agents
RTT is a complex disorder with multiple touchpoints for intervention. It is likely that a combination of therapies will be necessary to address these issues. The progress noted below underscores this perception in view of the incremental improvements noted with trofinetide.
The current treatment options for the common clinical issues related to RTT are summarized in Table 1. These include specific pharmaceutical agents for epilepsy, gastroesophageal reflux, delayed stomach emptying, constipation, sleep, anxiety, self-abuse, muscle rigidity/hypertonia, drooling, pain, prolonged QTc, bone health, menstrual management, and health maintenance. These products have proved effective overall but may require continued oversight to be certain that they remain effective over the lifetime.
In terms of specific agents directed at improving overall outcomes in RTT, an increasing number of pharmacologic agents have entered testing at the translational or clinical level over the past twenty years. To some extent, this was aided by the Scout program developed by the IRSF in 2013, initially under the initiative of Dr Steve Kaminsky, IRSF’s chief scientific officer at that time and named for him.
The agents are described in three sections. The first deals with early clinical trials with two conducted before the identification of MECP2 variants as the principal etiologic cause of RTT. The second deals with more recent clinical trials and is separated into three sections. The first covers completed trials, the second deals with a number of agents being studied at the preclinical level, and the third involves the current gene therapy trials as well as the preclinical studies of gene editing, X-chromosome reactivation, and RNA editing.
5.1 Clinical Trials
5.1.1 Early Trials
Three investigator-initiated early trials were conducted, one involving the opiate antagonist (naltrexone), one involving ʟ-carnitine prior to the identification of MECP2 variants, and the third utilizing folate-betaine shortly after the identification of MECP2 variants.
5.1.2 Naltrexone
Naltrexone, an opiate antagonist, was utilized in a double-blind, crossover phase 2 trial in RTT prior to the identification of MECP2 variants [60]. Twenty-five individuals meeting the established criteria for RTT were entered into this trial, which involved a 4-month trial of naltrexone or placebo, a 1-month washout period, and a second 4-month period featuring a crossover to the alternative of naltrexone or placebo. The hypothesis being tested was that naltrexone would improve breathing characteristics, one of the principal comorbidities in RTT, based on the finding of elevated ß-endorphins in cerebrospinal fluid (CSF) of individuals with RTT [61]. Data from the first period not only suggested improvements in disorganized breathing during wakefulness but also indicated that four individuals receiving naltrexone progressed one or more clinical stages versus none receiving placebo. The more rapid progression including a decline in motor function suggested a deleterious effect. However, this decline could have been due to failure to control for clinical progression in the study design. Another potential issue was that the FDA Investigational New Drug application (IND) limited the amount of naltrexone allowed to 1 mg/kg/day. Nevertheless, naltrexone has not proved to be consistently beneficial subsequently.
5.1.3 ʟ-Carnitine
A trial of ʟ-carnitine was conducted in 35 individuals with RTT due to the recognition of plasma carnitine deficiency in some individuals with RTT [62]. This double-blind, placebo-controlled study involved eight weeks each for placebo and ʟ-carnitine. Improvements were noted but was limited by the study power and the specific benefits were limited to a subset of individuals. The study failed to identify significant improvement in functional ability but raised the question whether a specific subset of individuals could benefit from ʟ-carnitine.
5.1.4 Folate-Betaine
This trial was based on the hypothesis that features of RTT could be improved by increasing methyl group availability in affected individuals through the administration of the folate and betaine combination [63]. A total of 68 participants who were randomized in this placebo-controlled, double-blind, phase 2 study completed the 12-month protocol with assessments at baseline, 3, 6, and 12 months. No objective improvement was noted in any of the outcome measures: breathing, hand movements, growth, anthropometry, the Rett syndrome Motor Behavioral Assessment, parent questionnaires, or EEGs. One potential adverse effect in study design was the failure to balance individuals in the placebo and folate-betaine study groups based on their specific MECP2 variant or maintain similar levels of clinical severity between the two groups.
5.2 Completed Trials (Table 2)
5.2.1 Trofinetide
Trofinetide, the first drug approved by the FDA (March 2023) for treatment of individuals with RTT, was the result of a series of studies first initiated more than fifteen years ago with insulin-like growth factor (IGF-1).
The initial translational (preclinical) study utilized the terminal tripeptide of IGF-1, the active component of IGF-1 known to be important in developmental biology in the brain [64]. In a mouse model of RTT, this tripeptide was shown to increase survival, reduce breathing and cardiac issues, and improve motor performance. At the biologic level, the tripeptide yielded partial increases in spine density, synaptic amplitude, and cortical plasticity. Based on the hypothesis that synaptic development and maturation is deficient in RTT, the results of this translational research provided the initiative to test both IGF-1 as well as the terminal tripeptide in humans with RTT.
Based on the ground-breaking results of Tropea et al. [64], recombinant IGF-1 (mecasermin) [65, 66] was evaluated in a phase 1 trial in 12 individuals with MECP2 variants, 9 of whom met diagnostic criteria for RTT (NCT012253317). The trial, which consisted of 4-week ascending dose and 20-week open label components revealed improvements in apnea and mood and revealed no adverse effects. This led to a double-blind, placebo-controlled crossover study in 30 girls with RTT, which revealed no differences between placebo and control groups and, thus, was deemed to indicate no overall improvement despite demonstrating safety (NCT01777542) [66].
The pharmaceutical company, Neuren, then developed a synthetic terminal tripeptide of IGF-1, trofinetide, which increased the half-life compared with the unmodified IGF-1 tripeptide and could be given by the oral or gastrostomy routes [67]. Informed by the findings of Tropea et al., phase 2 trials of trofinetide were initiated in adolescents and adults (NCT01703533) and children (NCT02715115). The first trial involved 56 adolescent and adult females with RTT syndrome and variants in MECP2 in a double-blind, placebo-controlled trial involving a 2:1 ratio of individuals receiving 35 mg/kg of trofinetide for 14 or 28 days or 70 mg/kg twice daily for 28 days [68]. Those receiving trofinetide had no safety or tolerability issues and those at the higher dose demonstrated efficacy. The second phase 2 trial was conducted in girls or adolescents from 5 to 15 years of age and involved a double-blind, placebo-controlled study of safety, tolerability, and efficacy [69]. A total of 62 females were randomized equally to placebo for 14 days and then placebo, 50, 100, or 200 mg/kg twice daily for 42 days. A further 20 individuals, 10 each receiving placebo or 200 mg/kg twice daily, were studied for 42 days after review of safety data in the original groups. Outcome measures included the CGI-I completed by the clinicians and the RSBQ completed by the parents of caregivers. Again, safety and tolerability were acceptable. Trofinetide at the 200 mg/kg twice daily dose showed statistically significant improvement in both the CGI-I and the RSBQ.
The absence of safety and tolerability issues in the two phase 2 trials is remarkable considering the issues of diarrhea, vomiting, and occasional weight loss noted in the phase 3 trial. Possible reasons could be the much shorter treatment period or the lower doses utilized in the phase 2 trials or possibly a change in the medication vehicle in the phase 3 trials as some increases in diarrhea and vomiting were also noted in the placebo group.
To mount a phase 3 study involving sufficient subjects, Neuren Pharmaceuticals entered into an agreement with Acadia Pharmaceuticals to allow the latter to conduct this phase 3 trial in the USA. The initial trial called Lavender (NCT04181723) involved 187 females, aged 5–20 years, in a randomized double-blind, placebo-controlled study with the CGI-I and RSBQ as coprimary outcome measures, along with secondary outcomes measures of hand function, ambulation, expressive and receptive communication, quality of life and caregiver burden [70, 71]. The dose was weight-based and involved BID dosing. The double-blind portion of the study was 12 weeks followed by a 40 open label follow-on study [70]. After 12 weeks, both the CGI-I (p = 0.003; effect size 0.47) and RSBQ (p = 0.0175; effect size 0.37) revealed statistically significant differences for trofinetide versus placebo. Further, the secondary outcome measure of communication and symbolic behaviors was significantly improved (p = 0.0064; effect size 0.43) [72]. While trofinetide demonstrated acceptable safety, the incidence of diarrhea (80.6% for trofinetide and 19.1% for placebo) provided some concerns and led to withdrawal of 14 participants. Increased vomiting was also noted in 26.0%. Lilac open label trials (NCT04279314 and NCT04776746) were also conducted until trofinetide received FDA approval (Cell Press in press, 2024). In addition, an open label Daffodil trial was initiated in 15 females aged 2–5 years (NCT04988867) using the same weight-based, twice daily dosing (manuscript in preparation). These latter studies continued to demonstrate persistent, even increased efficacy, until termination and transition to the FDA approved product.
Subsequently, Health Canada has recently agreed to accept a New Drug Submission for trofinetide, granting it priority review for the treatment of RTT. In Europe, Neuren retains the rights to trofinetide and the path forward for RTT there is currently unclear.
5.2.2 Blarcamesine
Blarcamesine is a sigma 1 receptor agonist that was shown to have positive effects in the mouse model of RTT tested through the IRSF Scout program (Rettsyndrome.org foundation Scout Program and performed by PsychoGenics, Inc.). In animal models, the sigma 1 receptor agonist improved homeostasis in the CNS with increased synaptic development, increased GABA (gamma aminobutyric acid) and BDNF (brain-derived neurotrophic factor), and led to a reduction in seizures and markers of neurodegeneration [73].
This led to three human trials: a phase 2 trial in adults in the US (NCT03758924), a phase 3 trial named the AVATAR trial in adults in Australia (NCT03941444) and a phase 2/3 trial in children, the EXCELLENCE trial, in Australia, UK, and Canada (NCT04304482). The AVATAR trial involved 20 adults receiving Anavex 2-73 and 13 receiving the placebo. The primary outcome measure was the RSBQ scale and secondary measures included the CGI-I and the ADAMS scale of emotional behavior. The RSBQ demonstrated clinically meaningful improvement [(p = 0.037; Cohen’s d effect size of 1.91 (very large)] in 72.2% on Anavex 2-73 versus 38.5% on placebo. In terms of the secondary endpoints, the ADAMS scales showed meaningful improvement in 52.9% on treatment versus 8.3% on placebo (p = 0.010; Cohen’s d effect size of 0.61). The CGI-I showed similar meaningful improvement to the RSBQ (p = 0.037; Cohen’s d effect size of 1.91). Safety data revealed no significant adverse effects versus placebo other than lethargy or sedation.
The third trial with this product, the EXCELLENCE trial in children with RTT, was a phase 2/3 study conducted in Australia, UK, and Canada. The primary outcome measure was the RSBQ completed by the caregivers whereas the secondary outcome measure was the clinician-completed CGI-I. This trial ended in June 2023 and according to a media release on 2 January 2024, the RSBQ failed to achieve significant differences between placebo and study groups, perhaps related to a higher response in the placebo group (https://www.anavex.com/post/Anavex-life-sciences).
5.2.3 Sarizotan
Sarizotan binds to serotonin and dopamine receptors and in mouse models of RTT was shown to increase serotonin levels and significantly improve breathing patterns known to be common in RTT [74]. That is, sarizotan showed a 75% reduction in apnea and a substantial correction of periodic breathing patterns in an animal model. Produced by Newron Pharmaceuticals, sarizotan was utilized in a phase 2 trial (NCT02790034) termed the STARS study in adolescents and adults with RTT in the US, Italy, India, UK, and Australia. As a randomized, double-blind study, the goal was to examine sarizotan’s effect on respiratory issues including its efficacy, safety, and tolerability. Subjects with RTT were required to demonstrate at least 10 episodes per hour of breathing abnormalities in the form of episodes of breath holding (apnea) lasting ≥ 10 s. Unfortunately, the final results revealed no reduction in apnea with sarizotan versus placebo. In addition, secondary outcomes related to caregiver response of effectiveness and motor performance in those on sarizotan were also not different from those on placebo. Thus, this study was deemed a failure.
5.2.4 Ketamine
Ketamine is a nonselective N-methyl-d-aspartate receptor (NMDAR) antagonist shown to improve features of RTT in female mouse models of RTT and extend their lifespan [75]. These improvements were related to inhibition of GABAergic interneurons and reduction of adverse synaptic activity in brainstem centers affecting respirations and autonomic control [76]. An initial exploratory trial (NCT02562820) was not initiated, but a subsequent investigator-initiated phase 2 trial (NCT03633058) was begun in children aged 6–12 years. This trial involved a 4-week placebo-controlled, crossover design with 12 unique individuals with RTT in each cohort and four ascending dose levels (0.75, 1.5, 3.0, and 4.5 mg/kg BID) at four sites in the USA. Outcome measures included the CGI-I, the Motor Behavior Assessment scale, a separate clinician and caregiver seven-point Likert scale clinical measure assessing domains such as hand function, walking, behavior, and communication skills, the caregiver-completed RSBQ, sleep habits, Caregiver Burden of Illness scale, and biosensor and EEG assessments at selected sites. The trial enrollment was limited by the coronavirus disease 2019 (COVID-19) pandemic and enrollment was stopped after the first two dose cohorts were completed. No significant adverse effects were noted at either dose. The outcome analysis of these results is currently pending.
A new study is planned in combination with donepezil and vorinostat supported by Department of Defense funding, but FDA approval is needed.
5.2.5 Triheptanoin
Triheptanoin (Dojolvi) has been utilized in two clinical trials in RTT. Triheptanoin is a medium chain triglyceride approved for use in individuals with long chain fatty acid oxidation defects. It was utilized in individuals with RTT based on information that mitochondrial function is diminished in RTT. The current status of the two trials listed below is described as unknown in ClinicalTrials.gov.
The first trial (NCT02696044) was an open label study in females with RTT aged 3–21 years in a single site in the USA [77]. Nine were enrolled based on the criteria of four seizures/month and/or four episodes of dystonia in the month prior to enrollment. The dose was 1–4 mg/kg/day based on age of the participant. One participant stopped due to vomiting and diarrhea, and one stopped due to diarrhea. Of the four individuals with intractable seizures, two had greater than 50% reduction in seizures, one was unchanged, and one was worse. Two participants had dystonia; one improved and one was unchanged. No signs of adverse effects were noted. This trial was supported by Ultragenyx Pharmaceuticals. Further study is unknown.
The second trial (UX007) was investigator initiated in Israel and was also supported by Ultragenyx Pharmaceuticals. This was also a single site study involving ten individuals with RTT, aged 5–18 years (NCT03059160). Criteria included either two seizures per month or breathing abnormalities and ability to ambulate with or without support. Individuals were scheduled for a 4-week baseline assessment, a 20-week drug exposure, and a 4-week washout. Triheptanoin was given three times per day. No results are available.
5.2.6 Fingolimod
Fingolimod (Gilenya) was employed in a phase 1 trial (NCT02061137) in Basel, Switzerland. Based on evidence that fingolimod increased BDNF levels and volume of deep gray and cortical gray matter in a mouse model of RTT [78], this trial involved six girls with mean age of 11.3 years in a 1-year study supported by Novartis. Using a dose of 0.25–0.5 mg/day based on weight, BDNF levels in blood and CSF and gray matter volume by MRI were assessed [79]. Despite excellent safety and tolerability responses, this trial failed to demonstrate an increase either in BDNF levels (blood and CSF) or increases in gray matter volume. Interestingly, BDNF levels were correlated inversely with the Rett Syndrome Severity Scale (RSSS) and the Hand Apraxia Scale with worse (higher) scores associated with lower BDNF levels. The Vineland Adaptive Behavior Scale was directly related to the BDNF levels, that is, higher BDNF levels were associated with higher scores on the Vineland.
5.2.7 Cannabidiol
Cannabidiol (Epidiolex) was utilized in two phase 3 trials (NCT03844832 and NCT4252586), the first being a placebo-controlled, double-blind study with three study groups (5 mg/kg/day, 15 mg/kg/day, and placebo) and the second being an open label follow-on study. Both studies were supported by Jazz Pharmaceuticals (initially GW pharmaceuticals prior to being acquired by Jazz). While the study aim was to address the overall clinical issues in RTT, the principal impact of this agent had been on seizure control. The outcome measures included the CGI-I, the CGI-S, the RSBQ, the Children Sleep Habit Questionnaire, and the Motor Behavioral Assessment-9 (MBA-9). The first study succeeded in enrolling 11 participants in the 5 mg/kg/day group, 9 participants in the 15 mg/kg/day group, and 10 participants in the placebo group; the second study enrolled 21 individuals. Although some results were achieved, both studies were terminated due to diminished enrollment amidst the COVID-19 pandemic. Further, diminished enrollment could also have been due to competition with the on-going trofinetide trial. No publication emerged from this study.
In a separate study [80] from France, ten participants were enrolled in a study of cannabidiol with a median duration of 13 months and a median dose of 15 mg/kg/day at the last visit. Five of these participants were treated simultaneously with clobazam. Seizure frequency was reduced in seven subjects, one being seizure-free and six having seizure reduction of > 75% (two subjects) or > 50% (four subjects). No adverse effects were noted. Additional improvements were noted in reduced agitation or anxiety (five subjects) and reduction in spasticity (four subjects). Additionally, it was suggested that cannabidiol improved the efficacy of clobazam.
5.2.8 Cannabidivarin
Cannabidivarin is a phytocannabionoid and analogue of cannabidiol. This compound, which does not have psychoactive properties seen with tetrahydrocannabinol, was shown to be efficacious in animal models of RTT. In a study with male Mecp2-null mice, cannabidivarin improved memory deficits and delayed neurological features in association with normalizing BDNF and IGF-1 levels along with the PI3K/AKT/mTOR pathway [81]. A prior study showed similar results in male Mecp2-deficient mice [82].
Based on these animal findings, a small phase 1 trial was initiated in five individuals, aged 6 years and older with RTT in Australia [83]. Each subject was treated with a maximum dose of 10 mg/kg/day resulting in a 79% reduction in monthly seizure frequency. Outcome measures included seizure type and frequency, EEG, the RSBQ, the RSSS, and adverse events. Side effects were mild or moderate, none associated with cessation of the medication, and included increased sleepiness and drooling. Neither EEG recordings nor clinical features of RTT were altered. Further study seems warranted, but nothing suggests this.
5.2.9 Glatiramer acetate
Glatiramer acetate (Copaxone), a peptide copolymer involving a random sequence of four amino acids, was studied in two clinical trials, one in Israel (NCT02023424) and one in New York at the Montefiore Medical Center (NCT02153723). These studies were based on the findings that glatiramer acetate is known to increase BDNF levels in individuals with multiple sclerosis and was shown to do the same in a null-male mouse model of RTT [84]. The two trials had dramatically different results.
The first (NCT02023424) involved a phase 1 open label trial of this agent in 14 girls of 5–15 years of age with RTT. The primary outcome measure was improvement in the 24-h EEG and the secondary outcome measures were breath-holding and hyperventilation, behavior, communication, motor skills, and feeding; decreased seizure frequency, improved sleep; and changes in height and weight. The trial was halted after four individuals developed significant postinjection reactions. One individual had a few minutes of difficulty breathing and mild edema whereas the three other subjects had more significant reactions ultimately leading to the decision to halt the trial [85].
The second (NCT02153723) was a phase 2 open label trial involving ten individuals aged 10 years of age or older. Nine were < 18 years and one was > 18 years. The clinical endpoints were gait velocity, breath-holding, and visual memory by eye tracking. All ten subjects had improved gait velocity ranging from 13% to 95% (p = 0.03). Breath-holding and visual memory also improved (p = 0.03). While these results suggested the need for a larger trial, no formal report was provided, and nothing more has developed over the past 8 years, perhaps related to the conflicting outcomes of the two trials.
5.2.10 Dextromethorphan
Dextromethorphan is an approved pharmaceutical used in cough suppressant medications. It is known to be an NMDA receptor antagonist. As NMDA receptor density is known to be increased in the prefrontal cortex in girls aged 2–8 years with RTT, while being reduced in those older than 10 years, the subsequent finding of similar features in female mice heterozygous for MeCP2 confirmed the regional and age-dependent findings. As such, dextromethorphan was trialed in females with RTT. Three trials were conducted at the Kennedy Krieger Institute in Baltimore.
The initial trial (NCT0069550) involving children and adolescents aged 1–15 years also included the medication, donepezil, approved for use in individuals with dementia. Donepezil was included as a cholinesterase inhibitor to counter the known abnormality of reduced choline acetyltransferase activity in individuals with RTT. Unfortunately, no information is available on this open label trial.
A second trial (NCT00593957) in children and adolescents aged from 2 to less than 15 years examined the effects of escalating does of dextromethorphan in three groups: 0.25 mg/kg/day, 2.5 mg/kg/day, and 5.0 mg/kg/day. The trial covered 6 months with 13 completing the lowest dose, 12 completing the intermediate dose, and 10 completing the highest dose. The primary outcome measure was EEG spike frequency. Individuals in each of the groups failed to show improvement. In addition, receptive language assessment failed to demonstrate any improvement.
The third trial (NCT01520363) involving dextromethorphan was also limited to testing in girls from 1 to less than 10 years with RTT. This trial was a placebo-controlled, double-blind study lasting 3 months with dextromethorphan provided at 5 mg/kg/day in divided doses. A total of 26 individuals began the trial; of whom, 24 completed it. The primary outcome measure was the Mullen scale of visual reception, fine motor, receptive language, and expressive language. Secondary outcome measures were the RSBQ and the Vineland. In the outcome analysis, those completing the study failed to demonstrate improvement in any of the four Mullen elements. No publications resulted from any of the three trials.
5.2.11 Desipramine
Desipramine is a tricyclic antidepressant approved for treatment of depression. In a male mouse model of RTT, abnormal breathing patterns were seen by 1 month of age and reduced tyrosine hydroxylase (TH) neurons as well as lower norepinephrine (NE) levels [86]. In this model, desipramine improved both to wild-type levels [87]. These results led to a 6-month phase 2 clinical trial (NCT00990691) in 36 girls and adolescents with RTT, ages 4–18 years, conducted in France. The study examined changes in respiratory alterations in a randomized, placebo-controlled double-blind study [88]. Desipramine was dosed on a weight-based strategy using 2–3 mg/kg/day, 1–2 mg/kg/day, or placebo. The primary outcome measure was the Apnea hypopnea index (AHI). The high dose group had an AHI of −31, the low dose AHI was −17.5, and the placebo group was −13. The changes were not significant (p = 0.78); although, the plasma concentration of desipramine and the AHI registered a significant inverse correlation (p = 0.0002). The investigators suggest that the significant relationship between desipramine concentration and improved AHI provided a rationale for further studies of the NE pathway in RTT.
5.2.12 Statins
Buchovecky et al. [89], using a suppressor screen, identified elevated cholesterol as a potential issue in a mouse model of RTT. Based on this information that excess neuronal cholesterol is involved in the pathophysiology of RTT, it was postulated that inhibiting cholesterol synthesis would benefit individuals with RTT. This led to a single site (Montefiore Medical Center) phase 2 open label of lovastatin in girls with RTT (NCT02563860). This study involved girls aged 3 years and older who were ambulatory, in a dose escalating trial of 10 mg for eight weeks, 20 mg for eight weeks, and 40 mg for 16 weeks. A total of 20 girls began, and 19 completed the study. The primary outcome measure was gait velocity with the secondary measure being visual memory via eye tracking. Unfortunately, no results have been provided. Further, Villani et al. [90] reported subsequently that lovastatin failed to affect motor skills and survival in a mouse model of RTT.
5.2.13 Antioxidant cocktail
De Felice et al. [91] reported significant evidence of oxidative stress in RTT. Considering mitochondrial dysfunction to have an important role in the pathogenesis of RTT, a randomized double-blind crossover trial was initiated in Toronto (NCT04041713). This trial involved girls and women with RTT, aged 2–21 years, who were at least partially ambulatory. The subjects were treated with an antioxidant cocktail, RETT-T, at 4 g for those < 30 kg and 8 g for those > 30 kg. This was a double-blind crossover design of 8 weeks on RETT-T or placebo, a 2-week washout period, and then a further 8 weeks on the alternate arm (RETT-T or placebo). The primary outcome measure was the RTT Motor Behavioral Assessment, the secondary measures being the Rett Syndrome Gross Motor Scale, the CGI-I, the Top 3 concerns, the RSBQ, the Children’s Sleep Habits Questionnaire, and the Anxiety, Depression, and Mood Scale (ADAMS). No results are available from the trial. However, Baroncelli et al. [92] recently reported that a cocktail of vitamin E, N-acetylcysteine, and α-lipoic acid in male and female mice did improve head growth and hippocampal synaptic plasticity but failed to improve behavior, breathing issues, or overall survival.
5.2.14 Vatiquinone
Edison Pharmaceuticals announced a phase 2a randomized, placebo-controlled trial of vatiquinone conducted in Siena, IT (NCT01822249). This was a 6-month trial of individuals with RTT in stage 1 or 2 and abnormalities of at least two disease biomarkers. The trial was 6 months in duration. The treatment group received 15 mg/kg three times per day. The primary outcome measure was the Rett Syndrome Clinical Severity Score (CSS). The secondary measures were the oxidative stress biomarkers (although not specified in ClinicalTrials.gov), head circumference, RSBQ, PedsQL, adverse events, and respiratory disturbance index (RDI) determined at polysomnography. No results are reported in clinical trials.gov, but the company noted that the product did not meet the efficacy outcome measures. Further, Hayek presented an abstract at the 4th European Congress on Rett Syndrome (1 November 2015; Rome, Italy) noting that the primary outcome measure was not met but did note better head growth in this trial [93].
5.2.15 Donepezil
Donepezil was proposed in NCT05625568, but no sites are listed and no results reported. This was to be a phase 2, multicenter double-blind, placebo-controlled trial involving adults, aged 18–45 years, with CSS between 10 and 35. A total of 48 subjects were anticipated, 16 each in placebo, 5 mg/day or 10 mg/day for 14 weeks. The primary outcome measure was safety and tolerability, and the secondary measures included the RSBQ, MBA, CGI-S, CGI-I, quantitative EEGs, and evoked responses. This product was discontinued as the parent company, Vyant Bio, was dissolved in December 2023.
A new study is planned in combination with ketamine and vorinostat supported by Department of Defense funding, but FDA approval is needed.
5.2.16 Creatine Supplementation
Based on the importance of DNA methylation on the role of MECP2 in RTT, a trial of creatine supplementation was initiated in Vienna (NCT01147575). The study was a double-blind, crossover design of creatine monophosphate (200 mg/kg/day) in three doses compared with placebo. The trial involved 6 months on drug or placebo, a 4-week washout, and a second 6 months on the alternative preparation. Individuals receiving folic acid, B12, or vitamin fortified food were excluded. The primary outcome measure was DNA methylation; the secondary measures were methionine, homocysteine, S-adenosyl methionine, and S-adenosyl homocysteine, as well as the MBA. No results were reported in clinicaltrials.gov. However, Freilinger et al. [94] reported results of the trial in 18 individuals with RTT, stages 3 and 4 and aged 3–25 years. Creatine supplementation did provide a statistically significant increase in DNA methylation, but the total and subscores of the MBA did not improve significantly. The authors did propose longer term use of creatine in a multicenter effort.
5.3 Preclinical Studies
The following agents, AMO-04, BHV 5000, GXV 001, NLX 101, RVL001, REL 1017, and pridopidine, have been mentioned in relation to RTT treatment strategies, all in the preclinical space for RTT. AMO-04, a product of AMO Pharma, is a glutamate modulator that showed promise in the IRSF-sponsored Scout program. BHV 5000, a product of Biohaven, is a glutamate receptor antagonist. GXV 001, a product of GEXVal, modulates the G-protein coupled receptor (GPCR). NLX101, a product of Neurolixis, interacts with the 5-hydroxytryptamine 1A receptor and improves breathing in a RTT mouse model [93]. RVL001, a product of Unravel Therapeutics, is a small molecule with an unknown target at present. REL 1017, a product of Relmada Therapeutics, is a d-methadone, a nonopioid analgesic, and an NMDA receptor antagonist. Pridopidine, a product of Prilenia, a dopamine modulator that selectively inhibits D2 dopamine receptors, is currently being trialed with amyotrophic lateral sclerosis and has been mentioned in relation to potential therapy for RTT.
5.3.1 Genetic Intervention Therapy
The goal of genetic intervention therapy has been under consideration for more than 15 years since the landmark paper of Guy et al. [96]. Using a mouse model of RTT, reactivation of an MECP2 gene under the control of a tamoxifen promoter reversed many of the features of RTT. Interestingly, it appeared that the reversal was independent of age in the animal studies, that is, whether the animals were presymptomatic or were older animals with significantly abnormal features. This result led to the crafting of a replacement MECP2 gene in the AAV9 vector to correct the abnormal gene in individuals with RTT. However, MECP2 is a tightly modulated gene in that over-expression can lead to the well-known MECP2 duplication disorder. This is an equally difficult disorder. To prevent such over-expression, methodologies were developed to limit the gene expression within a safety margin that would prevent such unwanted consequences.
Initially developed by AveXis (subsequently acquired by Novartis), this platform demonstrated good expression of the AAV-vector gene transmission in animals, including nonhuman primates, and led to the initial gene therapy clinical trial (NCT03633058) in RTT. However, for independent reasons, Novartis elected to discontinue this program. Subsequently, the gene replacement model was elaborated separately by Taysha Gene Therapies in Dallas and Neurogene in New York. Steven Gray (Taysha Gene Therapies) and Stuart Cobb (Neurogene) were strongly involved in developing the initial methodologies under AveXis and are currently leading these two clinical trials.
5.3.2 Taysha 102
In late 2022, Taysha Gene Therapies activated their gene replacement clinical trial (NCT05606614) in adults in Canada. More recently, a separate clinical trial (NCT061522237) has been activated in children in the USA.
The first trial, NCT05606614, was initiated in Canada as a phase 1/2, open-label, dose escalation study of TSHA-102. This REVEAL study was approved for adults as a safety and tolerability study examining the efficacy of TSHA-102 at two sequential dose levels. This study, with an estimated enrollment of 18 participants, is limited to adults who have classic RTT and whose parents agreed for them to receive blood or blood products, if necessary. The agent is a recombinant, nonreplicating, self-complementary AAV9 (scAAV9) vector carrying a miniMECP2 gene [95] and 3’ UTR features to limit overexpression of the virally delivered miniMECP2. This is a single, one-time administration of the minigene product. The mode of entry is via intrathecal delivery. The primary outcome measures are adverse events/serious adverse events, the CGI-I, the revised Motor Behavioral Assessment (R-MBA), the Rett Syndrome Hand Function Scale (RSHFS), the CGI-S, seizure frequency, adaptive behavior (Vineland-3), quantitative EEG from visual evoked responses, and auditory evoked responses. Initial treatment was begun in 2023.
Subsequently, Taysha gained approval for NCT061522237, the REVEAL Pediatric Study, to study children aged 5–8 years in the USA (the pediatric trial may expand to children aged 3–8 years). Up-to-date immunizations are required at least 42 days in advance of entry into the trial. The same requirement regarding parents’ approval for subjects to receive blood or blood products is stated. As with the adult trial, this is a phase 1/2 open-label trial involving a single, one-time administration of the minigene. Two dose levels will be trialed sequentially in separate subjects depending on the outcome of the first dose level. The primary outcome measures are nearly the same as in the adult trial except for exclusion of the Hand Function Scale.
According to a recent press release, the first subject began treatment at the end of 2023 and three subjects have now been enrolled, three adults and one child.[97]
5.3.3 Neurogene
In 2023, Neurogene activated a clinical trial (NCT05898620). This trial, NGN-401, was approved for children aged 4–10 years. It is a phase 1/2 safety, tolerability, and efficacy open-label trial using the AAV9 vector with a proprietary transgene regulation technology. In this trial, the MECP2 gene is full length, designed to provide therapeutic levels without over-expression provided in a single, one-time administration. In this trial, the mode of treatment is via the intracerebroventricular (ICV) delivery route. Two dose levels are anticipated with approval to the higher dose depending on the outcome in the lower dose group. The primary outcome measures are treatment-emergent adverse events, serious adverse events, adverse events, clinical laboratory abnormalities, and any change in the physical and neurological examination.
According to a press release, the first treatment began in 2023 [98]. In addition, Neurogene was selected by the FDA for the Start program to provide enhanced communication with the regulatory agency [99].
5.3.4 Additional Genetic Interventions in Preclinical Studies
Additional genetic interventions are under study, but all remain in the preclinical arena. These include gene editing, X chromosome reactivation, and RNA editing [100,101,102,103]. Additionally, read-through therapy to address nonsense mutations by correcting missing amino acid has been studied, but, to date, no positive outcomes have been reported.
One gene editing study is listed under Clinicaltrials.gov (NCT05740761). This is being conducted in Siena, Italy, utilizing the CRISPR/Cas9 methodology combined with the AAV9 vector mediated delivery aimed at correcting the four most common MECP2 point variants: c.473C>T, p.T158M; c.503C>T, p.R168X; c.763, p.R255X; and c.916C>T, p.R306C. The methodology is being testing in vitro in human fibroblast models from individuals with these MECP2 variants. The primary aim is to determine the percentage of gene editing obtained in cells representing the above variants. The secondary aim is to determine what, if any, off-target editing is noted. Namely, this seeks to determine the specificity of the methodology. The protocol was initiated in 2023. No results have been presented.
Gene editing is also being explored by BEAM Therapeutics. X chromosome reactivation is being examined by Alcyone Therapeutics (ACTX-101) and Herophilus (HRP-12975). RNA editing is being studied by Shape Therapeutics in collaboration with Roche and by VICO Therapeutics.
Although these efforts are in the preclinical arena, we do anticipate continued progress in bringing these potential therapies to human trial.
6 Conclusions
The last quarter century has brought a virtual explosion of new therapies being examined in clinical trials for RTT. One product, trofinetide, was approved by the FDA in March 2023. In as much as trofinetide produces incremental improvement rather than completely reversing the adverse effects of RTT completely, it is anticipated that other agents will be necessary to lead to even better treatment outcomes.
At the same time, the long-anticipated advent of gene therapy trials has now begun with two separate treatment strategies being explored. It is recognized that these trials may require substantial patience before they can be fully deployed. Further, the study of additional gene replacement strategies is even farther in the future as they attempt to move from preclinical study to active clinical trials.
Nevertheless, the relatively rapid advance in therapeutic intervention in RTT is noteworthy. One can only anticipate further advances in the coming years to achieve the goals of parents, caregivers, scientists, and physicians.
References
Rett A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien Med Wochenschr. 1966;116(37):723–6.
Hagberg B, Aicardi J, Dias K, et al. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann Neurol. 1983;14(4):471–9.
Schanen NC, Dahle EJ, Capozzoli F, et al. A new Rett syndrome family consistent with X-linked inheritance expands the X chromosome exclusion map. Am J Hum Genet. 1997;61(3):634–41.
Schanen NC. Molecular approaches to the Rett syndrome gene. J Child Neurol. 1999;14(12):806–14.
Amir RE, Van den Veyver IB, Wan M, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–8.
Hagberg B, Goutieres F, Hanefeld F, et al. Rett syndrome: criteria for inclusion and exclusion. Brain Dev. 1985;7(3):372–3.
Hanefeld F. The clinical pattern of the Rett syndrome. Brain Dev. 1985;7(3):320–5.
Rolando S. Rett syndrome: report of eight cases. Brain Dev. 1985;7(3):290–6.
Tao J, Van Esch H, Hagedorn-Greiwe M, et al. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am J Hum Genet. 2004;75(6):1149–54.
Ariani F, Hayek G, Rondinella D, et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am J Hum Genet. 2008;83(1):89–93.
Kankirawatana P, Leonard H, Ellaway C, et al. Early progressive encephalopathy in boys and MECP2 mutations. Neurology. 2006;67(1):164–6.
Neul JL, Benke TA, Marsh ED, et al. The array of clinical phenotypes of males with mutations in Methyl-CpG binding protein 2. Am J Med Genet B Neuropsychiatr Genet. 2019;180(1):55–67.
Hagberg B, Hanefeld F, Percy A, et al. An update on clinically applicable diagnostic criteria in Rett syndrome Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur J Paediatr Neurol. 2002;6(5):293–7.
Neul JL, Kaufmann WE, Glaze DG, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68(6):944–50.
Tarquinio DC, Motil KJ, Hou W, et al. Growth failure and outcome in Rett syndrome: specific growth references. Neurology. 2012;79(16):1653–61.
Motil KJ, Geerts S, Annese F, et al. Anthropometric measures correspond with functional motor outcomes in females with Rett syndrome. J Pediatr. 2022;244:169-177.e3.
Kirby RS, Lane JB, Childers J, et al. Longevity in Rett syndrome: analysis of the North American Database. J Pediatr. 2010;156(1):135-138.e1.
Tarquinio DC, Hou W, Neul JL, et al. The changing face of survival in Rett syndrome and MECP2-related disorders. Pediatr Neurol. 2015;53(5):402–11.
Freilinger M, Bebbington A, Lanator I, et al. Survival with Rett syndrome: comparing Rett’s original sample with data from the Australian Rett Syndrome Database. Dev Med Child Neurol. 2010;52(10):962–5.
Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56(3):422–37.
Chahrour M, Jung SY, Shaw C, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320(5880):1224–9.
Neul JL, Fang P, Barrish J, et al. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology. 2008;70(16):1313–21.
Bebbington A, Anderson A, Ravine D, et al. Investigating genotype-phenotype relationships in Rett syndrome using an international data set. Neurology. 2008;70(11):868–75.
Cuddapah VA, Pillai RB, Shekar KV, et al. Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J Med Genet. 2014;51(3):152–8.
Schanen NC, Kurczynski TW, Brunelle D, et al. Neonatal encephalopathy in two boys in families with recurrent Rett syndrome. J Child Neurol. 1998;13(5):229–31.
Augenstein K, Lane JB, Horton A, et al. Variable phenotypic expression of a MECP2 mutation in a family. J Neurodev Disord. 2009;1(4):313.
Fang X, Butler KM, Abidi F, et al. Analysis of X-inactivation status in a Rett syndrome natural history study cohort. Mol Genet Genomic Med. 2022;10(5): e1917.
Fang X, Baggett LM, Caylor RC, et al. Parental age effects and Rett syndrome. Am J Med Genet A. 2024;194(2):160–73.
Barrai I, Cann HM, Cavalli-Sforza LL, et al. The effect of parental age on rates of mutation for hemophilia and evidence for differing mutation rates for hemophilia A and B. Am J Hum Genet. 1968;20(3):175–96.
Ketterling RP, Vielhaber E, Li X, et al. Germline origins in the human F9 gene: frequent G:C–>A: T mosaicism and increased mutations with advanced maternal age. Hum Genet. 1999;105(6):629–40.
Grimm T, Kress W, Meng G, et al. Risk assessment and genetic counseling in families with Duchenne muscular dystrophy. Acta Myol. 2012;31(3):179–83.
Glaze DG, Percy AK, Skinner S, et al. Epilepsy and the natural history of Rett syndrome. Neurology. 2010;74(11):909–12.
Tarquinio DC, Hou W, Berg A, et al. Longitudinal course of epilepsy in Rett syndrome and related disorders. Brain. 2017;140(2):306–18.
Tarquinio DC, Hou W, Neul JL, et al. The course of awake breathing disturbances across the lifespan in Rett syndrome. Brain Dev. 2018;40(7):515–29.
Neul J, Benke TA, Marsh ED, Lane JB, Lieberman DN, Skinner SA, Glaze DG, Suter B, Heydemann PT, Beisang AA, Standridge SM, Ryther RCC, Haas RH, Edwards LJ, Ananth A, Percy AK. Distribution of hand function by age in individuals with Rett syndrome. Ann Child Neurol Soc. 2023;1(3):228–38.
Stallworth JL, Dy ME, Buchanan CB, et al. Hand stereotypies: lessons from the Rett syndrome natural history study. Neurology. 2019;92(22):e2594–603.
Lane JB, Lee HS, Smith LW, et al. Clinical severity and quality of life in children and adolescents with Rett syndrome. Neurology. 2011;77(20):1812–8.
Killian JT Jr, Lane JB, Lee HS, et al. Caretaker quality of life in Rett syndrome: disorder features and psychological predictors. Pediatr Neurol. 2016;58:67–74.
Lane JB, Salter AR, Jones NE, et al. Assessment of caregiver inventory for Rett syndrome. J Autism Dev Disord. 2017;47(4):1102–12.
Motil KJ, Caeg E, Barrish JO, et al. Gastrointestinal and nutritional problems occur frequently throughout life in girls and women with Rett syndrome. J Pediatr Gastroenterol Nutr. 2012;55(3):292–8.
Veatch OJ, Malow BA, Lee HS, et al. Evaluating sleep disturbances in children with rare genetic neurodevelopmental syndromes. Pediatr Neurol. 2021;123:30–7.
Percy AK, Lee HS, Neul JL, et al. Profiling scoliosis in Rett syndrome. Pediatr Res. 2010;67(4):435–9.
Killian JT, Lane JB, Lee HS, et al. Scoliosis in Rett syndrome: progression, comorbidities, and predictors. Pediatr Neurol. 2017;70:20–5.
Herrera JA, Ward CS, Pitcher MR, et al. Treatment of cardiac arrhythmias in a mouse model of Rett syndrome with Na+-channel-blocking antiepileptic drugs. Dis Model Mech. 2015;8(4):363–71.
Sekul EA, Moak JP, Schultz RJ, et al. Electrocardiographic findings in Rett syndrome: an explanation for sudden death? J Pediatr. 1994;125(1):80–2.
Ellaway CJ, Sholler G, Leonard H, et al. Prolonged QT interval in Rett syndrome. Arch Dis Child. 1999;80(5):470–2.
McCauley MD, Wang T, Mike E, et al. Pathogenesis of lethal cardiac arrhythmias in Mecp2 mutant mice: implication for therapy in Rett syndrome. Sci Transl Med. 2011;3(113):113–25.
Buchanan CB, Stallworth JL, Scott AE, et al. Behavioral profiles in Rett syndrome: data from the natural history study. Brain Dev. 2019;41(2):123–34.
Buchanan CB, Stallworth JL, Joy AE, et al. Anxiety-like behavior and anxiolytic treatment in the Rett syndrome natural history study. J Neurodev Disord. 2022;14(1):31.
Neul JL, Benke TA, Marsh ED, et al. Top caregiver concerns in Rett syndrome and related disorders: data from the US natural history study. J Neurodev Disord. 2023;15(1):33.
Neul JL, Glaze DG, Percy AK, et al. Improving treatment trial outcomes for Rett syndrome: the development of Rett-specific anchors for the clinical global impression scale. J Child Neurol. 2015;30(13):1743–8.
Hou W, Bhattacharya U, Pradana WA, et al. Assessment of a clinical trial metric for Rett syndrome: critical analysis of the Rett syndrome behavioural questionnaire. Pediatr Neurol. 2020;107:48–56.
FitzGerald PM, Jankovic J, Glaze DG, et al. Extrapyramidal involvement in Rett’s syndrome. Neurology. 1990;40(2):293–5.
FitzGerald PM, Jankovic J, Percy AK. Rett syndrome and associated movement disorders. Mov Disord. 1990;5(3):195–202.
Raspa M, Bann CM, Gwaltney A, et al. A psychometric evaluation of the motor-behavioral assessment scale for use as an outcome measure in Rett syndrome clinical trials. Am J Intellect Dev Disabil. 2020;125(6):493–509.
Raspa M, Gwaltney A, Bann CM, von Hehn J, Benke TA, Marsh ED, Peters SU, Ananth A, Percy AK, Neul JL. Psychomotor assessment of the rett syndrome caregiver assessment of symptom severity (RCASS). J Autism Dev Disord. 2024. https://doi.org/10.1007/s10803-024-06238-0.
Saby JN, Peters SU, Roberts TPL, et al. Evoked potentials and EEG analysis in Rett syndrome and related developmental encephalopathies: towards a biomarker for translational research. Front Integr Neurosci. 2020;14:30.
Saby JN, Benke TA, Peters SU, et al. Multisite study of evoked potentials in Rett syndrome. Ann Neurol. 2021;89:790–802.
Neul JL, Skinner SA, Annese F, et al. Metabolic signatures differentiate Rett syndrome from unaffected siblings. Front Integr Neurosci. 2020;14:7.
Percy AK, Glaze DG, Schultz RJ, et al. Rett syndrome: controlled study of an oral opiate antagonist, naltrexone. Ann Neurol. 1994;35(4):464–70.
Brase DA, Myer EC, Dewey WL. Possible hyperendorphinergic pathophysiology of the Rett syndrome. Life Sci. 1989;45(5):359–66.
Ellaway C, Williams K, Leonard H, et al. Rett syndrome: randomized controlled trial of l-carnitine. J Child Neurol. 1999;14(3):162–7.
Glaze DG, Percy AK, Motil KJ, et al. A study of the treatment of Rett syndrome with folate and betaine. J Child Neurol. 2009;24(5):551–6.
Tropea D, Giacometti E, Wilson NR, et al. Partial reversal of Rett syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci U S A. 2009;106(6):2029–34.
Khwaja OS, Ho E, Barnes KV, et al. Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc Natl Acad Sci U S A. 2014;111(12):4596–601.
O’Leary HM, Kaufmann WE, Barnes KV, et al. Placebo-controlled crossover assessment of mecasermin for the treatment of Rett syndrome. Ann Clin Transl Neurol. 2018;5(3):323–32.
Collins BE, Neul JL. Treatment of Rett syndrome. Drugs Future. 2021;46:29.
Glaze DG, Neul JL, Percy A, et al. A double-blind, randomized, placebo-controlled clinical study of trofinetide in the treatment of Rett syndrome. Pediatr Neurol. 2017;76:37–46.
Glaze DG, Neul JL, Kaufmann WE, et al. Double-blind, randomized, placebo-controlled study of trofinetide in pediatric Rett syndrome. Neurology. 2019;92(16):e1912–25.
Neul JL, Percy AK, Benke TA, et al. Trofinetide for the treatment of Rett syndrome: a randomized phase 3 study. Nat Med. 2023;29(6):1468–75.
Kennedy M, Glass L, Glaze DG, et al. Development of trofinetide for the treatment of Rett syndrome: from bench to bedside. Front Pharmacol. 2023;14:1341746.
Neul JL, Percy AK, Benke TA, et al. Trofinetide treatment demonstrates a benefit over placebo for the ability to communicate in Rett syndrome. Pediatr Neurol. 2023;152:63–72.
Kaufmann WE, Sprouse J, Rebowe N, et al. ANAVEX(R)2–73 (blarcamesine), a Sigma-1 receptor agonist, ameliorates neurologic impairments in a mouse model of Rett syndrome. Pharmacol Biochem Behav. 2019;187: 172796.
Abdala AP, Lioy DT, Garg SK, et al. Effect of Sarizotan, a 5-HT1a and D2-like receptor agonist, on respiration in three mouse models of Rett syndrome. Am J Respir Cell Mol Biol. 2014;50(6):1031–9.
Katz DM, Dutschmann M, Ramirez JM, et al. Breathing disorders in Rett syndrome: progressive neurochemical dysfunction in the respiratory network after birth. Respir Physiol Neurobiol. 2009;168(1–2):101–8.
Katz DM, Menniti FS, Mather RJ. N-methyl-d-aspartate receptors, ketamine, and Rett syndrome: something special on the road to treatments? Biol Psychiatry. 2016;79(9):710–2.
Shakil S, O’Leary H, Tarquinio D. Treatment of mitochondrial dysfunction in Rett syndrome with triheptanoin. Amercain Epilepsy Society; 2018.
Deogracias R, Yazdani M, Dekkers MP, et al. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2012;109(35):14230–5.
Naegelin Y, Kuhle J, Schadelin S, et al. Fingolimod in children with Rett syndrome: the FINGORETT study. Orphanet J Rare Dis. 2021;16(1):19.
Desnous B, Beretti T, Muller N, et al. Efficacy and tolerance of cannabidiol in the treatment of epilepsy in patients with Rett syndrome. Epilepsia Open. 2024;9(1):397–403.
Zamberletti E, Gabaglio M, Piscitelli F, et al. Cannabidivarin completely rescues cognitive deficits and delays neurological and motor defects in male Mecp2 mutant mice. J Psychopharmacol. 2019;33(7):894–907.
Vigli D, Cosentino L, Raggi C, et al. Chronic treatment with the phytocannabinoid Cannabidivarin (CBDV) rescues behavioural alterations and brain atrophy in a mouse model of Rett syndrome. Neuropharmacology. 2018;140:121–9.
Hurley EN, Ellaway CJ, Johnson AM, et al. Efficacy and safety of cannabidivarin treatment of epilepsy in girls with Rett syndrome: a phase 1 clinical trial. Epilepsia. 2022;63(7):1736–47.
Ben-Zeev B, Aharoni R, Nissenkorn A, et al. Glatiramer acetate (GA, Copolymer-1) an hypothetical treatment option for Rett syndrome. Med Hypotheses. 2011;76(2):190–3.
Nissenkorn A, Kidon M, Ben-Zeev B. A potential life-threatening reaction to glatiramer acetate in Rett syndrome. Pediatr Neurol. 2017;68:40–3.
Viemari JC, Roux JC, Tryba AK, et al. Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J Neurosci. 2005;25(50):11521–30.
Roux JC, Dura E, Moncla A, et al. Treatment with desipramine improves breathing and survival in a mouse model for Rett syndrome. Eur J Neurosci. 2007;25(7):1915–22.
Mancini J, Dubus JC, Jouve E, et al. Effect of desipramine on patients with breathing disorders in RETT syndrome. Ann Clin Transl Neurol. 2018;5(2):118–27.
Buchovecky CM, Turley SD, Brown HM, et al. A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat Genet. 2013;45(9):1013–20.
Villani C, Sacchetti G, Bagnati R, et al. Lovastatin fails to improve motor performance and survival in methyl-CpG-binding protein2-null mice. Elife. 2016;5: 22409.
De Felice C, Signorini C, Leoncini S, et al. The role of oxidative stress in Rett syndrome: an overview. Ann N Y Acad Sci. 2012;1259:121–35.
Hayek. J. EPI-743 in Rett syndrome: improved head growth in a randomized double-blind placebo-controlled trial. Paper presented at: 4th European Congress on Rett Syndrome: November 1, 2015; Rome, Italy.
Baroncelli L, Auel S, Rinne L, et al. Oral feeding of an antioxidant cocktail as a therapeutic strategy in a mouse model of Rett syndrome: merits and limitations of long-term treatment. Antioxidants (Basel). 2022;11(7):1406.
Freilinger M, Dunkler D, Lanator I, et al. Effects of creatine supplementation in Rett syndrome: a randomized, placebo-controlled trial. J Dev Behav Pediatr. 2011;32(6):454–60.
Levitt ES, Hunnicutt BJ, Knopp SJ, et al. A selective 5-HT1a receptor agonist improves respiration in a mouse model of Rett syndrome. J Appl Physiol. 2013;115(11):1626–33.
Guy J, Gan J, Selfridge J, et al. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315(5815):1143–7.
Taysha Gene Therapies. Taysha Gene Therapies Announces Regenerative Medicine Advanced Therapy (RMAT) Designation Granted by U.S. FDA for TSHA-102 in Rett Syndrome [press release 05/02/24]. https://ir.tayshagtx.com/new-release-details/tayshagtx Accessed 6/28/2024.
Neurogene. Neurogene announces expansion and plans for more rapid patient enrollment of Rett syndrome gene therapy clinical trial [press release 03/04/24]. https://ir.neurogene.com/news-release-details/neurogene Accessed 6/28/2024.
Neurogene. Neurogene announces NGN-401 gene therapy for Rett syndrome selected by FDA for START Pilot Program [press release 06/03/24]. Accessed 6/28/2024.
Sadhu C, Lyons C, Oh J, et al. The efficacy of a human-ready miniMECP2 gene therapy in a pre-clinical model of rett syndrome. Genes (Basel). 2023;15(1):31.
Panayotis N, Ehinger Y, Felix MS, et al. State-of-the-art therapies for Rett syndrome. Dev Med Child Neurol. 2023;65(2):162–70.
Sinnamon JR, Jacobson ME, Yung JF, et al. Targeted RNA editing in brainstem alleviates respiratory dysfunction in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2022;119(33): e2206053119.
Grimm NB, Lee JT. Selective Xi reactivation and alternative methods to restore MECP2 function in Rett syndrome. Trends Genet. 2022;38(9):920–43.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding supported production of this manuscript.
Conflict of Interest
A.K.P. has a consulting relationship with Acadia Pharmaceuticals, Anavex, Ionis, Neurogene, and Taysha, has previously served on the DSMB for Taysha, has previously had a contract researcher relationship with Acadia Pharmaceuticals, and has participated in the preparation of CME activities for the France Foundation, MedEdicus, Medscape, PeerView Institute, TotalCME. A.A. has research support from Anavex, Acadia Pharmaceuticals, and UCB. J.L.N. has received research funding from the National Institutes of Health, the International Rett Syndrome Foundation, and Rett Syndrome Research Trust; served as investigator for clinical trials conducted by Acadia Pharmaceuticals, GW Pharmaceuticals, Neuren Pharmaceuticals, and Newron; received personal consultancy for Acadia Pharmaceuticals Inc, Analysis Group, Anavex, AveXis, GW Pharmaceuticals, Hoffmann-La Roche, Ionis Pharmaceuticals, Myrtelle, Neurogene, Newron Pharmaceuticals, Signant Health, and Taysha Gene Therapies, and the preparation of CME activities for the France Foundation, MedEdicus, Medscape, PeerView Institute, TotalCME; served on the scientific advisory board of Alcyone Lifesciences; was a scientific cofounder of LizarBio Therapeutics; and a member of a data safety monitoring board for clinical trials conducted by Ovid Therapeutics and Ultragenix.
Ethical Approval
This review required no ethics approval. The Natural History Study (NHS) had ethics approval from the relevant institutions during its active period from 2003–2021.
Consent to Participate
All individuals enrolled in the NHS were provided informed consent by their parent(s) or guardian and where possible gave assent to be included. Similarly, consent was provided to participate in therapeutic trials.
Consent to Publish
Not applicable.
Availability of Data
Data from the US NHS is now managed by the International Rett Syndrome Foundation (IRSF). Access to this information may be obtained by contacting IRSF at rettsyndrome.org.
Code Availability
Not applicable.
Author Contributions
Alan Percy developed the review and provided the initial draft, performed the literature or on-line searches. Amitha Ananth and Jeffrey Neul critically reviewed and commented on the draft contributed to and approved the final document. All three authors had roles in analysis of the NHS data. All three authors participated in one or more of the clinical trials.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Percy, A.K., Ananth, A. & Neul, J.L. Rett Syndrome: The Emerging Landscape of Treatment Strategies. CNS Drugs (2024). https://doi.org/10.1007/s40263-024-01106-y
Accepted:
Published:
DOI: https://doi.org/10.1007/s40263-024-01106-y