
Abstract
Background and Objective
Overactivation of the PI3K/AKT pathway can occur in many cancers. Capivasertib is a potent, selective pan-AKT inhibitor. The objectives of this analysis were to develop a population pharmacokinetic model for capivasertib and to quantitatively assess the impact of intrinsic and extrinsic factors on the pharmacokinetics of capivasertib.
Methods
Pharmacokinetic data from four phase I and II studies were combined. Capivasertib was administered orally at a dose range of 80–800 mg twice daily over 28-day and 21-day cycles as monotherapy or in combination with paclitaxel or fulvestrant, using continuous dosing or one of two intermittent dosing schedules: either 4 days on, 3 days off (4/3) or 2 days on, 5 days off (2/5). Several models and approaches were tested for their ability to describe capivasertib disposition. The covariates assessed included dose, schedule, age, body weight, race, sex, creatinine clearance, hepatic function, renal function, smoking status, food effect, formulation, and concomitant use with paclitaxel, fulvestrant, cytochrome P450, family 3, subfamily A (CYP3A) inducers, CYP3A inhibitors and acid-reducing agents.
Results
A total of 3963 capivasertib plasma concentrations from 441 patients were included. Capivasertib pharmacokinetics was adequately described by a three-compartment model where the apparent clearance (CL/F) presented a moderate time-dependent and dose-dependent clearance. Following oral administration of multiple doses of capivasertib (400 mg twice daily; [4/3]), the initial CL/F was 62.2 L/h (between-subject variability 39.3%), and after approximately 120 hours, CL/F decreased by 18%. The effective half-life was 8.34 h. Steady state was predicted to be reached on every third and fourth dosing day each week from the second week with exposure levels that produced robust inhibition of AKT but not of other related kinases. The area under the plasma concentration–time curve and maximum plasma concentration of capivasertib were proportional between the dose levels of 80–480 mg after multiple doses but more than proportional beyond 480 mg. Schedule, age, race, sex, creatinine clearance, hepatic function, renal function, smoking status and concomitant use with fulvestrant, CYP3A inducers, CYP3A inhibitors or acid-reducing agents were not significant covariates for capivasertib pharmacokinetics. Concomitant use of paclitaxel, food effect and formulation statistically significantly affected capivasertib pharmacokinetics, but the effect was low. Body weight was statistically significantly related to capivasertib CL/F, with a 12% reduction in CL/F at steady state and a 14% increase in the area under the curve for 12 hours at steady state and maximum concentration at steady state at a lower body weight (47 kg vs 67 kg reference).
Conclusions
Capivasertib pharmacokinetics showed moderate between-subject variability, and most covariates assessed had no significant impact. Body weight, dose, concomitant use of paclitaxel, food effect and formulation showed statistically significant effects. However, these were predicted to impact exposure to capivasertib by <20% and were not expected to be clinically relevant. Based on the population pharmacokinetics, no a priori dose adjustment is needed for intrinsic and extrinsic factors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The pharmacokinetics of capivasertib, including the effects of capivasertib dose and patient baseline characteristics, have been evaluated by population pharmacokinetic modelling of 3963 plasma concentrations from 441 patients across four phase I and II studies. |
After multiple doses, capivasertib had a moderate time-dependent and dose-dependent pharmacokinetic profile, and body weight was shown to have a statistically significant impact on clearance. |
Neither the extent of the time-dependent and dose-dependent nature of capivasertib pharmacokinetics nor the degree of impact of body weight on capivasertib pharmacokinetics is expected to be clinically relevant in the capivasertib-treated patient; no dose adjustment of capivasertib is needed based on the population pharmacokinetic modelling. |
1 Background
The phosphatidylinositol 3-kinase (PI3K)/protein kinase (AKT)/phosphatase and tensin homologue (PTEN) signalling has crucial roles in cell growth and survival, and this pathway is dysregulated in many cancers. AKT is a central node in the PI3K/AKT pathway, and overexpression or activation of AKT has been observed in all major cancers, making it a promising target for the development of new therapies [1]. Capivasertib is first-in-class, potent, selective inhibitor of all three AKT isoforms (AKT1/2/3) [2]. Primary analysis of the phase III CAPItello-291 study has demonstrated that the addition of capivasertib (400 mg twice daily [b.i.d.], 4 days on, 3 days off [4/3]) to fulvestrant significantly improved progression-free survival in patients with locally advanced (inoperable) or metastatic hormone receptor-positive (HR+)/human epidermal growth factor receptor-negative (HER2−) breast cancer following recurrence or progression on or after aromatase inhibitor therapy [3]. These data led to the first regulatory approval of capivasertib plus fulvestrant in patients with HR+/HER2− advanced breast cancer and one or more tumour biomarker alterations (PIK3CA, AKT1, or PTEN) [4]. Simultaneous inhibition of AKT and cyclin-dependent kinase 4/6 (CDK4/6) pathways in HR+/HER2− advanced breast cancer is being examined in the phase Ib/III CAPItello-292 study, where the tolerability of capivasertib and fulvestrant plus palbociclib (a CDK4/6 inhibitor) has been demonstrated, and preliminary response data indicate evidence of clinical activity [5]. Several other phase III trials are also ongoing, including the CAPItello-290 study investigating capivasertib in combination with paclitaxel as first-line treatment of patients with metastatic triple-negative breast cancer [6] as well as studies investigating capivasertib for advanced prostate cancer in combination with docetaxel (CAPItello-280 [7]) or abiraterone (CAPItello-281 [8]).
The pharmacokinetics (PK) of capivasertib have been examined in several phase I and II studies of patients with advanced or metastatic solid tumours [9,10,11,12]. Phase I/II studies have also established the recommended phase II/III intermittent dosing regimen of capivasertib in combination with other anti-cancer therapies [11, 13,14,15]. Capivasertib plasma exposure was approximately dose proportional in the dose range of 80–800 mg, with a terminal half-life (t½) of approximately 10 hours (range, 7–15 h) [9]. In addition, capivasertib has been evaluated in clinical pharmacology studies in healthy subjects. These studies have shown that capivasertib can be given with or without food and with acid-reducing agents [16] and that it is eliminated primarily by metabolism as renal clearance was 21% of total clearance [17]. Overall, the available in vivo [17] and in vitro (data on file) data suggest that the metabolism is primarily mediated by CYP3A4 and UGT2B7 enzymes. Following single-dose administration of capivasertib 80 mg, the strong CYP3A4 inhibitor itraconazole increased capivasertib AUC by 95% [18]. An up to 75% increase in midazolam exposure indicates capivasertib is a weak CYP3A inhibitor at 400 mg b.i.d. on an intermittent schedule [19].
The objectives of this analysis were to develop a population pharmacokinetic model for capivasertib using data from four phase I and II clinical studies in patients with solid tumours and to quantitatively assess the impact of several intrinsic and extrinsic factors on the PK of capivasertib.
2 Methods
2.1 Clinical Studies
Pharmacokinetic data from four phase I and II studies (Study 1, BEECH, Study 4 and OAK) were used for this analysis [9,10,11,12]. Table 1 provides an overview of the four studies and Table 1 of the Electronic Supplementary Material (ESM) provides detail of the capivasertib dose/schedule and plasma sampling for the four studies. Study 1 was a phase I, first-in-human, dose-escalation study to assess the safety, tolerability, PK and preliminary anti-tumour activity of capivasertib in patients with advanced solid malignancies. This study also included intermittent capivasertib dosing in combination with fulvestrant background therapy [9]. BEECH was a phase I/II study with a safety run-in of capivasertib plus paclitaxel in patients with advanced or metastatic breast cancer, followed by a randomised expansion component of multiple cycles of capivasertib plus paclitaxel or placebo plus paclitaxel, in patients with oestrogen receptor-positive advanced or metastatic breast cancer [11]. Study 4 was a phase I dose-escalation study to assess the safety, tolerability, PK and preliminary anti-tumour activity of capivasertib in Japanese patients with advanced solid malignancies [10]. OAK was a phase I study comparing two dosage formulations of capivasertib and exploring the effect of food on the pharmacokinetic exposure, safety and tolerability of capivasertib in patients with advanced solid malignancies [12]. Across the four studies, capivasertib was administered orally at a dose range of 80–800 mg b.i.d. over 21-day and 28-day cycles as monotherapy or in combination with the anti-cancer therapies paclitaxel or fulvestrant, using continuous dosing or one of two intermittent dosing schedules (either [4/3] or 2 days on, 5 days off [2/5]).
2.2 Pharmacokinetic Assessment
Serial venous blood samples were collected up to 48 hours after the first capivasertib dose and up to 4 weeks after treatment start following multiple-dose administration for pharmacokinetic assessment (Table 1 of the ESM). Plasma concentrations of capivasertib were determined using a liquid chromatography-tandem mass spectrometry method, validated with a range of 1.00–1000 ng/mL and with a lower limit of quantification of 1.00 ng/mL.
2.3 In Vitro Cellular Kinase Inhibition
In vitro experiments aimed at quantifying the cellular kinase inhibition potentially targeted by capivasertib were conducted in various cell lines (see details in the ESM and methods). The results were overlaid with the predicted clinical exposure levels of capivasertib to assess whether the levels achieved would effectively inhibit the targeted cellular kinases.
2.4 Model Development
2.4.1 Structural Model
A three-compartment model was evaluated to describe systemic distribution and elimination. Several models and approaches, including first-order, zero-order, double absorption pathways and transit compartments, were tested for their ability to describe capivasertib absorption and apparent clearance (CL/F).
2.4.2 Scope and Parametrisation of Covariates
Potential covariate relationships were explored graphically; the influence of continuous and categorical covariates on selected model parameters was tested for statistical significance and clinical relevance. Continuous covariate effects were tested in the linear model as follows:
Additionally, continuous covariate effects were tested in the power model as follows:
where θ0 denotes the population value of the parameter when x = xmedian. The parameter θ denotes the population value conditional on the value of x, which is proportional to the power θx estimated from the model.
Categorical covariate effects were tested in the linear model only, as follows:
where θ0 denotes the population value of the parameter at the most common value for that characteristic (reference). The parameter θx denotes the fractional change in θ0 for a group of the population within that category in such a manner that the sum of all the groups within a category represents the whole study population.
Based on the mechanism rationale and visual inspection on the plots of empirical Bayes estimate versus covariates, potential covariates were selected and tested for significance in a stepwise manner using stepwise covariate model building procedure from Perl-speaks-NONMEM® (PsN) [20] with statistical criteria of p = 0.005 for the forward inclusion step and p = 0.001 for the backward elimination step.
2.4.3 Covariate Building
The covariates assessed included dose, schedule, age, body weight, race, sex, creatinine clearance (CRCL), hepatic function, renal function, smoking status, food effect, formulation and concomitant use with paclitaxel, fulvestrant, CYP3A inducers, CYP3A inhibitors and acid-reducing agents.The resulting random effects accounting for between-subject variability (BSV) in pharmacokinetic parameters were examined graphically by plotting each one against all others with covariates. The covariance step was examined, and the asymptotic standard errors of fixed and random effects produced by NONMEM were used to construct the asymptotic 95% confidence intervals (CIs). In addition, correlations between population parameters, eigenvalues and the condition number were evaluated to ensure the model was not ill-conditioned.
2.4.4 Model Discriminations and Evaluation
The performance of the model was evaluated by simulating data using parameter estimates (fixed and random effects) and conducting visual predictive checks (VPCs) and prediction-corrected VPCs with 400 replicates. In both the VPC and prediction-corrected VPC, the 5th, 50th and 95th percentiles of the observed data were compared with the model-based predicted percentiles for each bin across time and replicates. Uncertainty in parameter estimates was not considered in this analysis. In addition, a non-parametric bootstrap analysis was performed using PsN as an internal model evaluation technique. The original dataset was replicated using 2000 random draws of individual data (with replacement).
2.4.5 Simulations and Covariate Assessments
Deterministic simulations of concentration–time profiles were developed at different dose levels and schedules. Additionally, the impact of selected covariates over the concentration–time profiles was evaluated. The simulations were performed by changing one covariate at a time and keeping the others constant to investigate the relative change caused by a covariate compared with the reference subject.
2.4.6 Missing Data
All patients with at least one post-dose capivasertib plasma concentration value were included in the analysis. Plasma concentration values below the limit of quantification were excluded as they comprised < 5% of the dataset. Missing plasma concentration values were treated as missing and not replaced with estimated values. Multiple methods were used to address missing covariate values. Missing values for covariates that had values from at least 90% of patients in the population were imputed with the median (for continuous covariates) or a special category (for categorical covariates) to indicate unknown covariates.
2.4.7 Software
Dataset preparation was performed using entimICE® version 4.4 (Entimo AG, Berlin, Germany) and R Project for Statistical Computing, version 4.0.2 (Comprehensive R Network, http://cran.r-project.org). Non-linear mixed-effect modelling of concentration–time data was performed using NONMEM version 7.3.0 (ICON, Ellicott City, MD, USA) [21] and supported by PsN version 4.4.8 [20]. The NONMEM analysis was performed in a validated environment, based on Good Automated Manufacturing Practice and in accordance with 21 Code of Federal Regulations Part 11 and Good Clinical Practice regulations. Modifications to the analysis dataset, exploratory analysis, diagnostic graphics, post-processing of NONMEM analysis results and statistical analysis were performed using R.
3 Results
3.1 Summary of Patient Data
Results from 441 patients (79.6% female; 74.1% white), with a median age of 56 years (range 27–87 years) and a median body weight of 67 kg (range 32–129 kg) were included in the evaluation. Median CRCL was 97 mL/min (range 35–304 mL/min); renal function was normal (CRCL ≥ 90 mL/min) in 58.0% of patients, mildly impaired (CRCL 60–89 mL/min) in 32.9% of patients, and moderately impaired (CRCL 30–59 mL/min) in 8.8% of patients (no patients had severe impairment [CRCL 15–29 mL/min]). Hepatic function was normal (bilirubin ≤ upper limit of normal [ULN] and aspartate aminotransferase [AST] ≤ ULN) in 67.1% of patients, mildly impaired (bilirubin ≤ ULN and AST > ULN or bilirubin > 1.0–1.5 × ULN and AST of any value) in 31.3% of patients and moderately impaired (bilirubin > 1.5–3.0 × ULN and AST of any value) in 1.4% of patients (no patients had severe impairment [bilirubin > 3.0 × ULN and AST of any value]). Almost one-third (31.5%) of patients had concomitant use of an acid-reducing agent. Out of the 441 patients, 4522 capivasertib pharmacokinetic samples were reported. The analysis included 3963 observations with a median of six observations (range 2–21) per patient, while 559 observations were excluded for various reasons, such as being below the limit of quantification (141), being pre-treatment observations (411), missing observations (four) or having unexpectedly high values (three). Summary statistics of patient demographics and clinical covariates at baseline are provided in Table 2 (key covariates) and Table 2 of the ESM (other covariates). Figure 1 shows concentration–time profiles and the individual predictions for each study.

Individual capivasertib plasma concentration–time profiles after administration in the final model
3.2 PK of Capivasertib
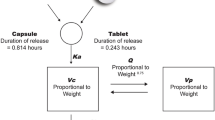
The PK of capivasertib was adequately described by a three-compartment model with two parallel first-order and zero-order absorption mechanisms with lag time and with auto-inhibition of CL/F managed by a sigmoid Emax process (Fig. 2). The model was parameterised in terms of CL/F, inter-compartmental clearances of peripheral compartments (Q3/F and Q/F), V2/F, apparent volumes of distribution of peripheral compartments (V3/F and V4/F), Ka and absorption lag time (Lag). Capivasertib linear elimination was initially implemented from the central V2/F compartment. However, it was later identified that CL/F was time- and dose-dependent. An Emax exponential function was employed, defined by maximal inhibition of CL/F (Imax), the time at which inhibition of CL0/F is reduced by half and a Hill parameter fixed at five to account for this. An exponential model was used to describe the BSV except for the fraction of dose absorbed as first order (F1), which was described using an additive model. The residual variability was modelled using log-transformed data with a combined proportional and additive residual error. The BSV and the proportional residual error were expressed as a coefficient of variation.

Capivasertib pharmacokinetic model. CL/F apparent clearance, CL0/F initial apparent clearance, D2 duration of zero-order absorption, F1 fraction absorbed as first order, Imax maximal inhibition of CL/F, Ka absorption rate constant, Lag1 lag time for absorption, Q3/F and Q4/F inter-compartmental clearances of peripheral compartments, T50 time at which inhibition of CL0/F is reduced by half, V2/F apparent volume of distribution of central compartment, V3/F and V4/F apparent volumes of distribution of peripheral compartments
As capivasertib was distributed into three compartments, three t½ were calculated corresponding to t½α, t½β and t½γ of 0.37, 4.2 and 31 h, respectively, for the first dose and of 0.42, 4.4 and 31 h, respectively, after multiple doses at 400 mg. For the administration schedule of 400 mg b.i.d. (4/3), the accumulation ratio was 1.58 and the effective t½ was 8.34 h [22]. Therefore, although it was predicted that the steady state would be reached on day 3 (after 41 h), owing to the time-dependent clearance during the first week of treatment and the intermittent (4/3) schedule, the steady state was predicted to be reached on every third and fourth dosing day each week from the second week at 400 mg b.i.d. The apparent volume of distribution at steady state was 255.6 L at 400 mg, suggesting that capivasertib distributes well in the body.
The absorption mechanisms component of the model estimated that 80% of the dose was absorbed using the first-order mechanism and the remaining 20% by a zero-order process where time to maximum concentration was reached at 1.4 h with slight variations depending on the dose level, body weight and food status. There was no Lag when capivasertib was administered orally as a tablet after an overnight fast. However, there was a short lag time when capivasertib was administered orally as a tablet or capsule under semi-fasted (no food intake from 2 h prior to 1 h after) or fed conditions, which was estimated to be 0.46 h for the capsule and 0.21 hours for the tablet. No other differences were detected between tablets and capsules. Diagnostic plots showed adequate fit to the data, with no apparent trends of residuals over time (data not shown).
3.3 Covariate Models
The model included significant covariates on the structural parameters of CL/F (body weight), absorption time lag ([ALAG], food effect and formulation), F1 (body weight) and Imax (dose and paclitaxel concomitant use), where the i suffix corresponds to the individual value for the ith individual. The equations that defined the structural parameters were as follows:
-
(a)
\(F_{1i} = \frac{{e^{{LogitF_{1}\left( {\frac{BBW}{{67}}} \right)^{F1\_BBW} }} + \eta LogitF_{1}}}{{1 + e^{{LogitF1\left( {\frac{BBW}{{67}}} \right)^{F1\_BBW} }} + \eta LogitF_{1} }},\)
-
(b)
\(F_{2i} = 1 - F_{1i} ,\)
-
(c)
\({\it{ALAG}}_{1i} = \left( {Lag_{{1\_{\it{tab}}}} \times \left( {1 - {\it{CAP}}} \right) + Lag_{{1\_{\it{cap}}}} \times {\it{CAP}}} \right)\left( {1 - {\it{FASTED)}}} \right)e^{{\eta {\it{ALAG}}1}} ,\)
-
(d)
\({\it{CL}}_{0} /{\it{F}}_{i} = {\it{CL}}_{0} /{\it{F}}\left( {1 + \left( {{\it{BBW}} - 67} \right) \times {\it{CL}}0\_{\it{BBW}}} \right)e^{{\eta {\it{CL}}_{0} /{\it{F}}}} ,\)
-
(e)
\({\it{CL}}_{} /{\it{F}}_{i} = {\it{CL}}_{0} /{\it{F}}_{i} \left( {1 - \frac{{e^{{I{\it{max}}_{i} }} \times {\it{Time}}^{5} }}{{T_{50}^{5} + {\it{Time}}^{5} }}} \right),\)
-
(f)
\(I_{\max i} = I_{\max } \left( {1 + \left( {{\it{DOSE}} - 480} \right) \times I{\it{max}}\_{\it{dose}}} \right)\left( {1 + {\it{PACL}} \times I{\it{max}}\_{\it{pacl}}} \right)e^{{\eta I_{max} }} ,\)
where CL0/F denotes the initial clearance after the first dose and CL/F refers to the time-varying CL managed by the sigmoid Emax function. Goodness-of-fit plots (Fig. 3) indicated a good fit of the model for most data where the observed capivasertib concentrations satisfactorily matched the predicted population concentrations or individual prediction concentrations. The observed-versus-predicted plots (Fig. 3a and b) showed random normal scatter around the identity line, indicating the absence of systematic bias and the adequacy of the model to describe the data. The conditional weighted residuals plots (Fig. 3c and d) showed random normal scatter around zero with no specific pattern to suggest model misspecification and a fairly constant distribution versus time with only eight observations of absolute value higher than six. The normalised prediction distribution error plots (Fig. 3e and f) showed a random normal scatter around zero. The correlation between parameter estimates in the correlation matrix lies between − 0.95 and 0.95 for all parameters. The estimated random-effect values were adequately centred around zero as the reported p values in the NONMEM output for CL/F, V2/F, F1, Imax and ALAG were larger than 0.05.

Goodness-of-fit graphs for the capivasertib population pharmacokinetic model. (a) Observed versus population predicted capivasertib concentrations; (b) observed versus individual predicted capivasertib concentrations; (c) conditional weighted residuals (CWRES) error versus population predicted capivasertib concentrations; (d) CWRES error versus time; (e) normalised prediction distribution error (NPDE) versus population-predicted concentrations; and (f) NPDE versus time. The solid black line or red dashed line is the line of identity, and the blue line is the locally estimated scatterplot smoothing line. Outliers (|CWRES| > 6) IDs are identified in red in panels c and d
In the model, the between-subject random effects, CL/F, V3/F, F1 and Imax, demonstrated a centred distribution, whereas ALAG, which depended on food status, presented a more dispersed distribution. Shrinkages were moderate, by only 15% for CL/F, which was deemed adequate to generate individual predictions. No remarkable relationships were observed among between-subject random effects and structural parameters (data not shown). All other parameters were estimated with good precision, as relative standard errors of the parameter estimates were < 25% of the estimated value. The model parameters are described in Table 3.
3.4 Model Assessment
The median values of parameters and 95% CIs obtained from the converged bootstrap runs for capivasertib are presented in Table 3. The median values of parameters were in close agreement with the population estimates in the model, suggesting that the NONMEM parameter estimates were unbiased. The model was evaluated by VPC and prediction-corrected VPC pooling all the studies, which showed adequate predictions (Figs. 1 and 2 of the ESM). Most observed concentrations were within the 95% prediction interval, indicating that the predicted variability did not exceed the observed variability.
3.5 Model Simulation
No differences in CL/F were detected among the dose levels after the first dose; however, a moderate dose-dependent and time-dependent CL/F was observed after multiple doses, as indicated by an initial CL/F of 62.2 L/h (BSV 39.3%), which decreased by 18%, 22% and 54% after approximately 120 h at 400, 480 and 800 mg (Fig. 4). The time at which inhibition of CL/F is reduced by half was 67 h. The median area under the curve for 12 h at steady state (AUC12h,ss) and the maximum concentration at steady state (Cmax,ss) for capivasertib were 7730 μg h/L and 1340 μg/L, respectively, at 400 mg b.i.d. (4/3) after multiple doses. Simulations for the three schedules explored at different dose levels show how the time-dependent inhibition and the dose levels modify the shape of the concentration–time profiles with steady state achieved after approximately 120 h (Fig. 5).

Apparent clearance (CL/F) time course after twice-daily dosing of capivasertib. h hours

Deterministic concentration–time profiles at different schedules and twice-daily dose levels (2/5), 2 days on/5 days off; (4/3) 4 days on/3 days off
Forest plots were used to assess the effect of covariates included in the model for AUC12h,ss, Cmax,ss, apparent clearance at steady state (CLss/F) and time to maximum concentration at steady state after multiple doses. The reference population had a body weight of 67 kg, with no paclitaxel usage, administered semi-fasted at 400 mg b.i.d (4/3) with tablets (Fig. 6 and Fig. 3 of the ESM). Capivasertib AUC12h,ss and Cmax,ss increased approximately proportionally between the dose levels of 80 mg and 480 mg after multiple doses but more than proportionally at doses beyond 480 mg (Fig. 6a and b). The CLss/F median ratio for a patient with a body weight of 47 kg (5th percentile) was 0.88 (95% CI 0.80–0.94), and with a body weight of 99 kg (95th percentile) was 1.20 (95% CI 1.09–1.31), against the 67-kg reference (Fig. 3a of the ESM). Patients with concomitant paclitaxel had 20% higher CLss/F (median ratio 1.20, 95% CI 1.12–1.29) [Fig. 3a of the ESM], and AUC12h,ss and Cmax,ss reduced by more than 10% (median ratio 0.84, 95% CI 0.78–0.89 and median ratio of 0.89, 95% CI 0.85–0.93, respectively; Fig. 6a and b) against the reference of no paclitaxel, after multiple doses.

Forest plot for (a) area under the plasma concentration–time curve (AUC) and (b) maximum plasma concentration. (a) Median AUC for 12 hours at steady state (AUC12h,ss) (μg.h/L) and AUC12h,ss fraction relative to the reference (body weight of 67 kg, dosed at 400 mg twice daily with tablets, semi-fasted food status and no use of paclitaxel). (b) Median maximum concentration at steady state (Cmax,ss) [μg/L] and Cmax,ss fraction relative to the reference (body weight of 67 kg, dosed at capivasertib 400 mg twice daily with tablets, semi-fasted food status and no use of paclitaxel). CI confidence interval
3.6 In Vitro Cellular Kinase Inhibition
After establishing the pharmacokinetic model for capivasertib, the relationship between exposure and kinases potentially targeted by capivasertib was analysed. At the cellular level, capivasertib has the potential to inhibit a number of kinases in the same broad AGC kinase family of which AKT (Fig. 4 of the ESM) is a member including p70S6K (Fig. 5 of the ESM), PKA (Fig. 6 of the ESM) and ROCK (Fig. 7 of the ESM) [2]. Across multiple cell lines, capivasertib has greatest activity versus AKT1, 2 and 3 reducing phosphorylation of its direct and indirect substrates PRAS40 with IC50 values of 0.31–2.33 µM, GSK3β with IC50 values of 0.08–0.76 µM and S6 with IC50 values of 0.07–1.41 µM (Figs. 8 and Fig. 9 and Table 4 of the ESM). This suggests that within the clinical exposure achieved at steady state, capivasertib (400 mg b.i.d.; [4/3]) achieves robust cover over the IC50s required to inhibit AKT1, 2 and 3 signalling during the treatment periods.
Capivasertib has some activity versus p70S6K, PKA and ROCK in kinase assays [2] but potency in cells is reduced relative to the concentration at which it inhibits AKT. In TSC null cells, in which p70S6K is constitutively active, capivasertib reduced p70S6K-mediated phosphorylation of S6 with IC50 values of 0.67–2.52 µM (Table 5 of the ESM). Capivasertib was even less effective versus PKA reducing phosphorylation of its substrate VASP with IC50 values of 1.1–10.37 µM (Table 6 of the ESM), and it was not effective at inhibiting ROCK-mediated phosphorylation of its substrate cofilin (Table 7 of the ESM). Mapping these IC50 values onto the model-predicted capivasertib plasma concentrations indicates that capivasertib at 400 mg b.i.d. (4/3) does not achieve exposures to be effective versus these additional kinases.
4 Discussion
In this analysis, the PK of capivasertib and the effect of dose and other covariates were evaluated using a population pharmacokinetic modelling approach based on data from four clinical studies. The model was found to be adequate for deriving exposure metric estimates to be used in a subsequent exposure–response analysis relating the exposure of capivasertib to clinical outcomes.
Different absorption models were tested, such as first-order, zero-order and sequential absorption models, including absorption transit compartments, among others. The absorption of capivasertib was characterised by a parallel zero-order and first-order absorption rate constant with lag time, which was slightly longer with food intake. Capivasertib clearance was modelled using a sigmoid Emax model to describe time-dependent PK. The model did not identify pharmacokinetic differences between the schedules of administration, and it was able to adequately describe the concentration–time profiles, including the pre-dose concentrations collected up to week 4 of cycle 1. Apparent clearance was constant after the first dose but time and dose dependent after multiple doses. The model predicted that CL/F decreased to a constant value after about 120 hours and that the extent of time dependency increased with increasing dose. This is consistent with in vitro and in vivo data, which showed that capivasertib is a CYP3A4 substrate and a time-dependent inhibitor of CYP3A4, suggesting that the dose-dependent and time-dependent CL/F is caused by auto-inhibition. Capivasertib AUC and maximum plasma concentration were proportional between the dose levels of 80–480 mg after multiple doses but more than proportional beyond 480 mg. The minor time-dependent PK of capivasertib at the therapeutic dose (400 mg) is not expected to be clinically relevant in the capivasertib-treated patient. However, the identification of the time-dependent and dose-dependent nature has aided in the design and interpretation of clinical pharmacology studies and is crucial for designing optimal pharmacokinetic sampling schedules in upcoming trials.
The assessment of body weight on capivasertib PK showed that patients with a lower body weight (47 kg) had a 12% reduction in CLss/F and a 14% increase in AUC12h,ss and Cmax,ss compared with patients with the reference body weight of 67 kg. The concomitant use of paclitaxel showed an increase in CLss/F (20%) and reductions in AUC12h,ss and Cmax,ss (>10%) compared with no paclitaxel. However, it is important to acknowledge that paclitaxel was exclusively used in the BEECH trial, which included a different patient population and a limited sampling schedule (only pre-dose samples were collected after 144 h [steady state expected to have been achieved after approximately 120 h]). This limitation in the pharmacokinetic schedule may restrict the ability of the model to identify the time-dependent PK. The effect of paclitaxel on capivasertib PK is being evaluated further in the ongoing CAPItello-290 phase III trial for patients with triple-negative breast cancer [6]. Assessments from patients receiving concomitant fulvestrant did not show a significant effect on capivasertib PK; the addition of capivasertib to fulvestrant has also been assessed in the CAPItello-291 phase III trial in patients with HR+/HER2− advanced breast cancer where significantly longer progression-free survival was observed over fulvestrant alone [3]. The model also showed that food versus overnight fasted and capsule formulation versus tablet slightly delayed the time to maximum concentration at steady state without any impact on AUC12h,ss or Cmax,ss. The lack of food effect is consistent with the results from a dedicated clinical pharmacology study in healthy subjects [16]. Schedule, age, race, sex, CRCL, hepatic function, renal function, smoking status and concomitant use with fulvestrant, CYP3A inducers, CYP3A inhibitors or acid-reducing agents were not significant covariates for the PK of capivasertib. Overall, none of the covariates explored in the model was predicted to have a clinically relevant impact on the PK of capivasertib.
Collectively, in vitro cellular kinase experiments indicated that the capivasertib exposure achieved at 400 mg b.i.d. (4/3) results in robust inhibition of AKT, but no other related kinases. This observation is consistent with pharmacodynamic data derived from clinical samples [9].
5 Conclusions
This data analysis from four phase I and phase II studies found that a three-compartment model adequately described capivasertib PK. The model provided individual exposure metrics to perform exposure–response analyses. Moderate BSV was observed with capivasertib. Schedule, age, race, sex, CRCL, hepatic function, renal function, smoking status and concomitant use with fulvestrant, CYP3A inducers, CYP3A inhibitors or acid-reducing agents were not significant covariates for the PK of capivasertib. Body weight and paclitaxel showed statistically significant effects on capivasertib PK; however, the effects on the exposure were predicted to be < 20% and are therefore not expected to be clinically relevant. Based on the population PK, no a priori dose adjustment is needed for intrinsic and extrinsic factors.
References
Lindsley CW. The Akt/PKB family of protein kinases: a review of small molecule inhibitors and progress towards target validation: a 2009 update. Curr Top Med Chem. 2010;10(4):458–77. https://doi.org/10.2174/156802610790980602.
Davies BR, Greenwood H, Dudley P, Crafter C, Yu DH, Zhang J, et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther. 2012;11(4):873–87. https://doi.org/10.1158/1535-7163.MCT-11-0824-T.
Turner NC, Oliveira M, Howell SJ, Dalenc F, Cortes J, Gomez Moreno HL, et al. Capivasertib in hormone receptor-positive advanced breast cancer. N Engl J Med. 2023;388(22):2058–70. https://doi.org/10.1056/NEJMoa2214131.
US Food and Drug Administration: FDA approves capivasertib with fulvestrant for breast cancer. 2023. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-capivasertib-fulvestrant-breast-cancer. Accessed 27 Feb 2024.
Neven P, Hamilton E, Pistilli B, Borges V, Campone M, Foukakis T, et al. 206P—Capivasertib (C) + palbociclib (P) and fulvestrant (F) in patients (pts) with HR+/HER2- advanced breast cancer (ABC): phase 1b data from CAPItello-292. In: Poster presented at ESMO Breast Cancer; 11–13 May 2023; Berlin.
Schmid P, Cortes J, Robson ME, Iwata H, Hegg R, Nechaeva M, et al. A phase III trial of capivasertib and paclitaxel in first-line treatment of patients with metastatic triple-negative breast cancer (CAPItello290). J Clin Oncol. 2020;38 (15 Suppl.):TPS1109-TPS. https://doi.org/10.1200/JCO.2020.38.15_suppl.TPS1109.
Crabb SJ, Ye DW, Uemura H, Morris T, Gresty C, Logan J, et al. TPS287 CAPItello-280: A phase III study of capivasertib and docetaxel versus placebo and docetaxel in metastatic castration-resistant prostate cancer. J Clin Oncol. 2023;41:TPS287-TPS. https://doi.org/10.1200/JCO.2023.41.6_suppl.TPS287.
Fizazi K, George DJ, Santis MD, Clarke N, Fay AP, Uemura H, et al. A phase III trial of capivasertib and abiraterone versus placebo and abiraterone in patients with de novo metastatic hormone-sensitive prostate cancer characterized by PTEN deficiency (CAPItello-281). J Clin Oncol. 2021;39(6 Suppl.):TPS178-TPS. https://doi.org/10.1200/JCO.2021.39.6_suppl.TPS178.
Banerji U, Dean EJ, Pérez-Fidalgo JA, Batist G, Bedard PL, You B, et al. A phase I open-label study to identify a dosing regimen of the pan-AKT inhibitor AZD5363 for evaluation in solid tumors and in PIK3CA-mutated breast and gynecologic cancers. Clin Cancer Res. 2018;24(9):2050–9. https://doi.org/10.1158/1078-0432.Ccr-17-2260.
Tamura K, Hashimoto J, Tanabe Y, Kodaira M, Yonemori K, Seto T, et al. Safety and tolerability of AZD5363 in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2016;77(4):787–95. https://doi.org/10.1007/s00280-016-2987-9.
Turner NC, Alarcon E, Armstrong AC, Philco M, Lopez Chuken YA, Sablin MP, et al. BEECH: a dose-finding run-in followed by a randomised phase II study assessing the efficacy of AKT inhibitor capivasertib (AZD5363) combined with paclitaxel in patients with estrogen receptor-positive advanced or metastatic breast cancer, and in a PIK3CA mutant sub-population. Ann Oncol. 2019;30(5):774–80. https://doi.org/10.1093/annonc/mdz086.
Dean E, Banerji U, Schellens JHM, Krebs MG, Jimenez B, van Brummelen E, et al. A phase 1, open-label, multicentre study to compare the capsule and tablet formulations of AZD5363 and explore the effect of food on the pharmacokinetic exposure, safety and tolerability of AZD5363 in patients with advanced solid malignancies: OAK. Cancer Chemother Pharmacol. 2018;81(5):873–83. https://doi.org/10.1007/s00280-018-3558-z.
Crabb SJ, Birtle AJ, Martin K, Downs N, Ratcliffe I, Maishman T, et al. ProCAID: a phase I clinical trial to combine the AKT inhibitor AZD5363 with docetaxel and prednisolone chemotherapy for metastatic castration resistant prostate cancer. Invest New Drugs. 2017;35(5):599–607. https://doi.org/10.1007/s10637-017-0433-4.
Jones RH, Casbard A, Carucci M, Cox C, Butler R, Alchami F, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2020;21(3):345–57. https://doi.org/10.1016/s1470-2045(19)30817-4.
Shore N, Mellado B, Shah S, Hauke R, Costin D, Adra N, et al. A phase I study of capivasertib in combination with abiraterone acetate in patients with metastatic castration-resistant prostate cancer. Clin Genitourin Cancer. 2023;21(2):278–85. https://doi.org/10.1016/j.clgc.2022.11.017.
Miller C, Sommavilla R, Murphy D, Morris T, Khatun M, Cullberg M. The effect of food and acid-reducing agents on the pharmacokinetic profile of capivasertib: results from a randomised, cross-over study. Br J Clin Pharmacol. 2023;89(11):3330–9. https://doi.org/10.1111/bcp.15831.
Miller C, Wild M, Zhang Z, Sommavilla R, Shanahan D, Bailey C, et al. A phase I study to determine the absolute bioavailability and absorption, distribution, metabolism, and excretion of capivasertib in healthy male participants. Drug Metab Distrib. 2024 (in press).
Miller C, Sommavilla R, Barry ST, Eberlein C, Morris T, Wadsworth I, et al. Pharmacokinetics of the Akt serine/threonine protein kinase inhibitor, capivasertib, administered to healthy volunteers in the presence and absence of the CYP3A4 inhibitor itraconazole. Clin Pharmacol Drug Dev. 2023;12(9):856–62. https://doi.org/10.1002/cpdd.1307.
Miller C, Sommavilla R, O’Bryant CL, Barve M, Dowlati A, Luke JJ, et al. Pharmacokinetic study of capivasertib and the CYP3A4 substrate midazolam in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2024. https://doi.org/10.1007/s00280-024-04667-3.
Lindbom L, Pihlgren P, Jonsson EN. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241–57. https://doi.org/10.1016/j.cmpb.2005.04.005.
Beal S, Sheiner LB, Boeckmann A, Bauer RJ (2009) NONMEM user’s guides (1989–2009). Icon Development Solutions, Ellicott City.
Gidal BE, Clark AM, Anders B, Gilliam F. The application of half-life in clinical decision making: Comparison of the pharmacokinetics of extended-release topiramate (USL255) and immediate-release topiramate. Epilepsy Res. 2017;129:26–32. https://doi.org/10.1016/j.eplepsyres.2016.10.020.
Acknowledgements
Capivasertib was discovered by AstraZeneca after a collaboration with Astex Therapeutics (and its partnership with the Institute of Cancer Research and Cancer Research Technology Limited). The authors thank all the patients and their families, and the investigators, trial-site coordinators and nurses, from the capivasertib phase I and II clinical studies. The authors also thank Amy Cheung, Philip Delff and Helen Tomkinson of AstraZeneca R&D at the time of study conduct, who participated in the clinical trials design, and Mine de Kock of AstraZeneca R&D, who provided quality-control support for the study. Medical writing support, including developing drafts under the control and direction of the authors, was provided by Elena Sugrue, PhD, of Oxford PharmaGenesis, Oxford, UK, and Suzanne Patel, PhD, from BOLDSCIENCE Inc., funded by AstraZeneca.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by AstraZeneca, who also sponsored the open-access publication of the manuscript.
Conflict of interest
Carlos Fernandez-Teruel, Marie Cullberg, Cath Eberlein, Simon T. Barry and Diansong Zhou were employees of AstraZeneca and may hold stocks and/or shares.
Ethics approval
All studies included in this analysis were approved by appropriate ethics committees and were conducted in accordance with the principles of the Declaration of Helsinki and the International Council on Harmonisation Good Clinical Practice guidelines.
Consent to participate
Written informed consent was obtained from all participants of the studies included in this analysis.
Consent for publication
Not applicable.
Availability of data and material
Data underlying the findings described in this article may be obtained in accordance with AstraZeneca’s data-sharing policy, described at: https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
Code availability
Not applicable.
Author contributions
CF, MC, CE, SB and DZ designed the research, performed the research, analysed the data and wrote the manuscript. All authors read and approved the final version of the manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Fernandez-Teruel, C., Cullberg, M., Eberlein, C. et al. Population Pharmacokinetics of Capivasertib in Patients with Advanced or Metastatic Solid Tumours. Clin Pharmacokinet (2024). https://doi.org/10.1007/s40262-024-01407-x
Accepted:
Published:
DOI: https://doi.org/10.1007/s40262-024-01407-x