
Abstract
Study Design and Objective
Randomised, double-blind, crossover trial to confirm bioequivalence of somapacitan, a long-acting growth hormone (GH), in 5 mg/1.5 mL and 10 mg/1.5 mL strengths in equimolar doses.
Methods
Healthy participants were randomised (1:1:1) to subcutaneous somapacitan treatment in one dosing period with 5 mg/1.5 mL and two periods with 10 mg/1.5 mL. Eligibility criteria included age 18–45 years and body mass index 18.5–24.9 kg/m2. Exclusion criteria included history of GH deficiency, previous GH treatment, weight > 100.0 kg and participation in any clinical trial of an investigational medicinal product within 45 days or five times the half-life of the previous investigational product before screening. Area under the curve from time 0 until last quantifiable observation (AUC0–t), maximum serum concentration (Cmax), time to Cmax and terminal half-life of somapacitan and safety were assessed.
Results
In total, 33 participants were randomised. For AUC0–t, estimated treatment ratio (ETR) (5 mg/1.5 mL versus 10 mg/1.5 mL) was 0.95 (90% confidence interval [CI] 0.89–1.01). Point estimate and 90% CIs were within the acceptance range (0.80–1.25). For Cmax, ETR was 0.77 (90% CI 0.68–0.89). Point estimate and 90% CIs were outside the acceptance range (0.80–1.25). Mean insulin-like growth factor-I (IGF-I) and IGF-I standard deviation score concentration–time curves for each strength were almost identical. No new safety issues were identified.
Conclusions
Bioequivalence criterion for somapacitan 5 mg/1.5 mL and 10 mg/1.5 mL was met for AUC0–t but not for Cmax. The two strengths had equivalent IGF-I responses.
Trial Registration
ClinicalTrials.gov, NCT03905850 (3 April 2019).
Plain Language Summary
Somapacitan is a long-acting growth hormone used to treat people with growth hormone deficiency. Somapacitan is injected under the skin with an injection pen. The dose is based on a person’s body weight and how they respond to treatment. We compared two strengths of injection pen, containing either 5 or 10 mg of somapacitan per 1.5 mL. For both strengths, participants were given the same dose. We wanted to understand whether the body absorbs these different strengths into the bloodstream in the same way. We also measured levels of insulin-like growth factor-I (IGF-I), a hormone formed when growth hormone is present in the blood, to see the effect of different strengths of somapacitan on the body. In our study, 33 healthy adults received one round of injection using the somapacitan 5 mg pen and two rounds using the somapacitan 10 mg pen, all at least 3 weeks apart. We found no differences in the amount of somapacitan being absorbed into the bloodstream, nor how fast it was absorbed. The peak amount of somapacitan in the bloodstream was higher in people using the 10 mg pen. There were no differences in IGF-I levels following use of either injection pen. Overall, our results show both strengths of somapacitan lead to similar responses in the body. Having different strength options could allow doctors to adjust the dose of somapacitan more easily, depending on a patient’s response to treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The authors examine how the body is exposed to two different strengths of somapacitan (a growth hormone therapy that is administered once per week), in terms of the total amount of the drug absorbed by the body throughout treatment and the maximum amount of the drug absorbed by the body at any individual time within the treatment period. |
The results indicate that there is no difference in total exposure of the two strengths. However, there was a difference in the peak amount of drug measured in the blood. No differences in insulin-like growth factor-I level, which is an indicator of response to growth hormone therapy, were observed between the two strengths. |
The equivalent somapacitan AUC and IGF-I responses between the two strengths of somapacitan indicate they can be considered clinically equivalent, and both strengths have been approved for use by medical regulatory authorities. |
1 Introduction
Growth hormone (GH) therapy is approved for the treatment of adult and paediatric growth hormone deficiency (GHD) and is currently being investigated as a treatment for various growth-related disorders. GH therapy may benefit patients with GHD, idiopathic short stature, Turner syndrome, Noonan syndrome and those born small for gestational age and enable them to reach a final height that is within the range of their target (based on parental stature) [1, 2]. Until recently, daily subcutaneous (sc) GH replacement injections were the only treatment option for GH disorders, which can be burdensome for affected patients in terms of inconvenience, pain, and cost and may lead to non-adherence [3,4,5]. Studies in both children and adult patients with GHD receiving daily GH treatment have shown treatment adherence to be poor, leading to suboptimal therapy outcomes [3, 6,7,8,9] and increased economic costs [10]. Therefore, a long-acting GH (LAGH) preparation that requires less frequent administration may help to improve treatment adherence in children and adults with GHD.
Somapacitan (Sogroya®; Novo Nordisk, Denmark) was the first approved LAGH preparation for the treatment of AGHD in the USA, Europe, Japan, Australia and Saudi Arabia [11,12,13,14,15]. It is currently approved for GH deficiency in children in the USA, Europe, Japan and Saudi Arabia [11, 12, 14, 15]. Somapacitan is a human GH analogue with one amino acid substitution, Leu101Cys (not involved in binding to the GH receptor), and an albumin-binding moiety which prolongs half-life [11, 12]. The prolonged action of somapacitan is achieved with a similar protraction technology that has previously been used successfully to extend the half-life of insulin [16] and glucagon-like peptide-1 therapies [17, 18]. In a similar manner to daily GH, somapacitan binds to GH receptors and induces intracellular signalling to stimulate the secretion of insulin-like growth factor-I (IGF-I) [11, 12]. The level of IGF-I is an indicator for bioavailable GH and can therefore be considered as the biomarker for the efficacy of somatropins [19].
In phase 1 trials, IGF-I levels in patients with AGHD treated with somapacitan were shown to be elevated for at least 7 days after every dosing visit and were similar to levels observed for daily GH, for doses between 0.02 and 0.04 mg/kg [20]. Furthermore, the safety and efficacy of somapacitan for the treatment of AGHD have been studied in three phase 3 clinical trials: REAL 1 (NCT02229851) [21], REAL 2 (NCT02382939) [22] and REAL Japan (NCT03075644) [23]. Efficacy of somapacitan compared with placebo was demonstrated with reductions in truncal fat and visceral fat and increases in lean body mass and appendicular skeletal muscle mass [21]. Improvements indicating desired changes in body composition were seen with both somapacitan and daily GH in previously treatment-naive patients with AGHD [21], and similar efficacy of somapacitan was observed for improvements in adipose tissue as for daily GH in previously GH-treated patients [23]. A similar IGF-I standard deviation score (SDS) was observed for somapacitan as for daily GH in all three trials [21,22,23]. The safety profile of somapacitan was shown to be comparable with that of daily GH, with no new safety issues identified [21, 23], and somapacitan, delivered as a once-weekly dose, was reported to be more convenient to use than once-daily GH in these patients [22].
The recent 2-year results from the phase 3 REAL 4 trial (NCT03811535) demonstrated sustained efficacy and tolerability of somapacitan in children with GHD after switching from daily GH. Height velocity was sustained in both groups of patients (somapacitan 0.16 mg/kg/week or daily GH 0.034 mg/kg/day) between weeks 52 and 104, with comparable mean IGF-I SDS within normal range and no new safety or tolerability issues identified [24].
Different formulations of daily GH have been shown to be bioequivalent to each other [25, 26], with pharmacokinetic (PK) similarities demonstrated between doses of 5 mg/1.5 mL, 10 mg/1.5 mL and 15 mg/1.5 mL [25]. Thus, it was hypothesised that different strengths of somapacitan would also be bioequivalent. Demonstration of bioequivalence could reduce excess usage, thus the introduction of the 5 mg/1.5 mL pen was relevant, as the 10 mg/1.5 mL somapacitan injection pen holds a greater drug quantity than needed by patients on low doses of somapacitan, within the in-use expiry of the drug.
In the present trial, we assessed the bioequivalence of somapacitan 5 mg/1.5 mL sc and 10 mg/1.5 mL sc in equimolar doses of 0.04 mg/kg body weight and investigated their PK and pharmacodynamic (PD) properties.
2 Materials and Methods
2.1 Trial Design
This was an interventional, randomised, double-blind, single-centre, three-period, complete crossover trial in healthy participants (NCT03905850) conducted in Germany. The trial was designed according to European Medicines Agency (EMA) [27] and US Food and Drug Administration (FDA) [28] guidelines on bioequivalence, which require area under the curve from time 0 until the time of the last quantifiable concentration after dosing (AUC0–t) and maximum serum concentration (Cmax) to be contained within the same predefined limits for the test and reference products. The trial used a three-period complete crossover design to explore differences in PK profiles between somapacitan (Sogroya®; Novo Nordisk A/S, Denmark) 5 mg/1.5 mL and 10 mg/1.5 mL, as somapacitan has high intra-participant variability of Cmax. EMA and FDA guidelines [27, 28] allow for a widened acceptance interval for Cmax, provided that the higher within-participant variability is judged to be clinically irrelevant. Expected variability in Cmax was > 30% and judged to be not clinically relevant; hence, the acceptance interval was widened accordingly for this parameter.
As somapacitan is intended for use in both sexes, guidelines recommend that both male and female participants be involved in bioequivalence trials. Participants were healthy, with concomitant disease and medications excluded. The number of participants was chosen based on a sample size calculation derived from previous trial designs [29].
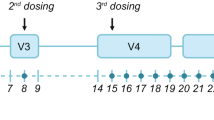
Participants were randomly allocated in a 1:1:1 ratio (by computer) to one of three treatment sequences consisting of two periods with single doses of somapacitan 10 mg/1.5 mL sc (reference product) and one period with a single dose of somapacitan 5 mg/1.5 mL sc (test product) (Fig. 1). Both strengths were administered at equimolar doses of 0.04 mg/kg, based on body weight measured at visit 2, day 1. A dose of 0.04 mg/kg was selected, as this was the mean dose administered to adults in the somapacitan phase 3 AGHD clinical development programme and has been established as within the therapeutic AGHD dose range [20, 21, 30]. Both strengths of somapacitan were subject to the same storage conditions in a refrigerator (from 2 to 8 °C) and protected from light. No co-administration with other drugs occurred and participants were fasting prior to dosing.

Trial design. All dosing visits were in-house and conducted over a 5-day period. Dosing visits took place on days 1, 22 and 43, with a + 7-day dosing visit window. Each dosing visit included PK/PD sampling and was followed by PK/PD sampling over a 3-day period and again 1 week later. Participants checked into each in-house visit 1 day before dosing. PD pharmacodynamic, PK pharmacokinetic
Each participant attended three in-house dosing and PK/PD sampling visits, each with a duration of 5 days. PK and PD samples were stored at the study site until shipment on dry ice to the central lab at − 30 °C (± 10 °C) and − 80 °C (± 10 °C), respectively. PK assessments were based on serum concentrations of somapacitan and PD analysis was based on serum IGF-I concentrations. Participants received doses on days 1, 22 and 43 (a window of + 7 days was allowed for each dosing visit), and PK/PD samples were taken during the full duration of each in-house visit. After each 5-day in-house visit, PK/PD samples were taken for an additional period of 3 days (i.e. if a dose was given on day 1, then PK/PD sampling would take place additionally on days 6, 7 and 8), with another PK/PD sampling visit occurring 1 week later (day 15 according to the example of dosing on day 1). Each dose was administered with the participant in a fasting state and was followed by a 3-week observation/washout period. The three dosing periods were followed by a final end-of-trial visit on days 64–71 (Fig. 1).
PK/PD sampling was performed 10 min prior to each dose, and again 15 min, 1, 2, 4, 6, 8, 12, 16, 20, 24, 26, 28, 30, 36, 42, 48, 50, 56, 64, 72, 74, 80, 86, 96, 120, 144, 168 and 336 h (i.e. 15 min to 14 days) after the latest dose given. Follow-up samples were then taken 504–672 h (on days 64–71) (Fig. 1) relative to dosing on day 43 (last dose). The maximum amount of blood collected for sampling from each subject over the duration of the trial did not exceed 400 mL.
The full analysis set (FAS) was defined as all randomised participants who received both products (two injections of the reference product [somapacitan 10 mg/1.5 mL] and one injection of the test product [somapacitan 5 mg/1.5 mL]). If an endpoint could not be calculated for a participant, they were excluded from the FAS for analyses of that endpoint. The safety analysis set (SAS) was defined as all randomised participants who received at least one of the two products.
2.2 Participants
2.2.1 Inclusion and Exclusion Criteria
Eligible participants were aged between 18 and 45 years at the time informed consent was sought and signed. Participants had a body mass index (BMI) between 18.5 and 24.9 kg/m2 and were considered to be generally healthy based on their medical history, physical examination and clinical parameters such as vital signs, electrocardiogram results and laboratory tests performed during the screening visit, as judged by the investigators. Key exclusion criteria included participation in any clinical trial of an approved or non-approved investigational medicinal product within 45 days or five times the half-life of the previous investigational medicinal product, whichever was longer, before screening. In addition, participants with a known history of GH deficiency or those who were non-naive to GH treatment (both as declared by the participant), or participants who were > 100.0 kg in body weight, were excluded.
2.3 Endpoints
Co-primary endpoints were area under the somapacitan serum concentration–time curve from time 0 until the time of the last quantifiable concentration after dosing (AUC0–t) and the maximum serum concentration of somapacitan (Cmax), derived from the somapacitan serum concentration–time curves from time 0 to 504 h (21 days) after dosing.
Supportive secondary PK endpoints derived from the somapacitan serum concentration–time curve included AUC0–168 h (from 0–168 h [7 days] after dosing), AUC0–inf (from time 0 to infinity after dosing), time to maximum serum concentration of somapacitan (tmax) and the terminal half-life of somapacitan (t½).
Further supportive secondary PD endpoints were derived from the serum IGF-I concentration–time curve: AUCIGF-I 0–168 h, area under the IGF-I serum concentration–time curve from time 0 to 168 h after dosing; Cmax IGF-I, maximum serum concentration of IGF-I after dosing; tmax IGF-I, time to maximum serum concentration of IGF-I after dosing.
2.4 Laboratory Analyses and Safety Evaluations
A validated somapacitan specific luminescent oxygen channelling immunoassay (AlphaLISA®, PerkinElmer, Waltham, MA) was used to analyse somapacitan serum concentrations. Analysis of somatropin in human serum was performed via enzyme-linked immunosorbent assay and chemiluminescent immunoassay [30].
Analysis of IGF-I was performed by the special laboratory using a commercially available assay (Immuno Diagnostic Systems immunoassay [ISYS assay]), which was validated and showed acceptable accuracy, precision, dilution linearity and parallelism.
An adverse event (AE) was defined as any untoward medical occurrence in a clinical trial participant that was temporally associated with the administration of either of the investigational products. The event may or may not have been considered related to the treatment. Injection-site reactions were evaluated by the investigator after dosing.
2.5 Pharmacokinetic and Statistical Analyses
AUC0–t and Cmax were derived from the serum somapacitan concentration–time curve. AUC0–t, AUC0–168 h and AUCIGF-I 0–168 h of somapacitan were approximated by a linear trapezoidal technique. AUC0–t, AUC0–inf, AUC0–168 h, AUCIGF-I 0–168 h and Cmax of somapacitan were log-transformed prior to analysis using an analysis of variance (ANOVA) model. The model included product, period, sequence and participant within sequence as fixed effects. From this model, the product difference was estimated, and the estimate was back-transformed to original scale and presented as ratios with corresponding 90% confidence intervals (CIs). Analysis of Cmax, using an ANOVA model, and assessment of bioequivalence were performed, following the EMA [27] and FDA [28] approaches. AUC0–inf was calculated as AUC0–t + AUCextrapolated, where AUCextrapolated = \(\hat{C}_{t} /\lambda z\), t was the time of the last quantifiable concentration and \(\hat{C}_{t}\) was the estimated concentration at the time of the last quantifiable concentration. T½ was calculated as t½ = log (2)/λz, where λz was estimated as the slope from a linear regression on the terminal phase data with the logarithm to the concentration as the response variable and time as the explanatory variable. Valid observations from the final part of the curve, which were approximately linear, were used for the analysis.
Pharmacokinetic and statistical analyses were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC, USA).
2.6 Ethics
The trial was conducted in accordance with the Declaration of Helsinki [31] and ICH Good Clinical Practice guidelines [32]. Prior to the start of the trial, the protocol, the consent form and the participant information sheet were reviewed and approved according to local regulations by appropriate health authorities and by the Ethics Committee of the North Rhine Medical Association. The participants were informed of the risks and benefits of the trial, and that they could withdraw from the trial at any time for any reason. Consent was obtained in writing prior to any trial-related activities.
3 Results
3.1 Baseline Characteristics
A total of 33 healthy participants were randomised in the trial, with 11 participants in each of the three treatment sequences. One participant (in sequence 10 mg/1.5 mL, 5 mg/1.5 mL and 10 mg/1.5 mL) withdrew from the trial before receiving the third treatment, resulting in a total of 32 completers in the trial. The final FAS included 32 participants, while the SAS included 33 participants. Baseline demographic characteristics are summarised in Supplementary Table 1. The majority of the participants were white (96.8%) and male (84.3%). The mean (standard deviation [SD]) age and BMI in study participants were 35.1 (6.6) years and 22.8 (1.8) kg/m2, respectively.
3.2 Co-Primary Endpoints
The geometric mean concentration–time curves for somapacitan 5 mg/1.5 mL and 10 mg/1.5 mL showed similar profiles, with some variability around Cmax, and no large differences between the strengths were observed during the elimination phase. Values fell below the lower limit of quantification after 336 h (Fig. 2).

Geometric mean curves of full PK profile of somapacitan by treatment (FAS). End of period is the sample taken just before the next dosing or at the follow-up visit if the treatment is the last in the participant’s treatment sequence. Dashed reference line represents the lower limit of quantification; values below the lower limit of quantification are imputed. FAS full analysis set, PK pharmacokinetic
The geometric mean of AUC0–t for somapacitan 5 mg/1.5 mL (820 ng × h/mL) was comparable to those for the first and second somapacitan 10 mg/1.5 mL treatments (839 and 894 ng × h/mL, respectively). The geometric mean of Cmax for somapacitan 5 mg/1.5 mL (10.61 ng/mL) was lower than those for the first and second somapacitan 10 mg/1.5 mL treatments (14.09 and 13.18 ng/mL, respectively). The coefficient of variation (CV) of Cmax was above 50% and higher than for AUC0–t for all three treatments (Supplementary Table 2).
The estimated treatment ratio (ETR) of AUC0–t (90% CI) for somapacitan 5 mg/1.5 mL versus somapacitan 10 mg/1.5 mL of 0.95 (0.89–1.01) was within the bioequivalence acceptance range (0.80–1.25); hence, the bioequivalence criterion for AUC0–t was met. For Cmax, the within-participant coefficient of variation and the within-participant SD for somapacitan 10 mg/1.5 mL was estimated as 54.3% and 0.52, respectively. A widened acceptance range was defined for Cmax in line with EMA and FDA guidelines on bioequivalence due to high within-participant variability of Cmax [27, 28]. The Cmax treatment ratio was 0.77 (0.68–0.89). This point estimate was outside the EMA and FDA acceptance range of 0.80‒1.25 and the lower limit of the 90% CI was outside the EMA widened acceptance range (0.70–1.43); hence, the EMA and FDA bioequivalence criteria for Cmax were not met.
3.3 Secondary PK Endpoints
ETRs (90% CI) of the two strengths for AUC0–168 h and AUC0–inf were 0.93 (0.84–1.02) and 0.95 (0.89–1.01), respectively. The geometric mean of t½ was comparable for somapacitan 5 mg/1.5 mL (52.7 h) and somapacitan 10 mg/1.5 mL delivered as a first and second treatment (53.9 and 50.5 h, respectively). The median of tmax was comparable for somapacitan 5 mg/1.5 mL (10 h) and somapacitan 10 mg/1.5 mL delivered as a first and second treatment (8 h, respectively) (Supplementary Table 2).
3.4 Secondary PD Endpoints: IGF-I
Mean IGF-I and IGF-I SDS concentration–time curves for the two strengths were almost identical (Fig. 3), with the AUCIGF-I 0–168 h ETR between the doses being 1.00 (90% CI 0.98–1.02). Median tmax IGF-I values for the two strengths were identical (96 h). The geometric mean of Cmax IGF-I was similar for somapacitan 5 mg/1.5 mL (322.1 ng/mL) and somapacitan 10 mg/1.5 mL delivered as a first and second treatment (316.4 and 323.8 ng/mL, respectively) (Supplementary Table 2).

Geometric mean curves of a the full profile of IGF-I (ng/mL) and b the IGF-I SDS full profile. End of period is the sample taken just before the next dosing or at the follow-up visit if the treatment is the last in the participant’s treatment sequence. IGF-1 insulin-like growth factor-I, SDS standard deviation score
3.5 Safety
Seventeen (51.5%) participants experienced 34 AEs, most of which were mild (30 events); no serious AEs were experienced, and all participants recovered (Supplementary Table 3). The most common AEs (≥ 5% of the participants) were headache (eight AEs in five participants [15.2%]), back pain (three AEs in three participants [9.1%]) and vomiting (two AEs in two participants [6.1%]). There was no apparent pattern of difference in the distribution of AEs between the two strengths of somapacitan.
4 Discussion
This trial investigated the bioequivalence of somapacitan 5 mg/1.5 mL and 10 mg/1.5 mL in healthy participants, according to EMA [27] and FDA [28] guidelines. The primary objective was not met; although the AUC0–t ETR fulfilled the EMA and FDA bioequivalence criteria, the Cmax ETR did not. However, difference in Cmax for different strengths is also seen in daily GH preparations [26]. In the present study, Cmax was estimated to be approximately 20% lower for somapacitan 5 mg/1.5 mL compared with somapacitan 10 mg/1.5 mL.
However, when evaluating bioequivalence criteria of AUC and Cmax, it should be noted that somapacitan has a known high intra-participant variability in Cmax (> 30%) [30]. EMA and FDA guidelines [27, 28] allow for a widened acceptance interval when there is wide variability in Cmax, as is the case for somapacitan. A possible explanation for the ~ 20% difference in Cmax is that the concentration gradient between somapacitan at the injection site and the surrounding tissue, lymph and capillaries draining the site was hypothetically lower for the 5 mg/1.5 mL strength, where the dose of 0.04 mg/kg was administered with a higher injection volume compared with the 10 mg/1.5 mL strength, leading to a slightly lower rate of absorption.
The mean concentration-time curves of IGF-I and IGF-I SDS were almost identical. As IGF-I is considered a biomarker for the efficacy of GH [19], this suggests equivalent efficacy for the two strengths of somapacitan.
The overall clinical response can be expected to be the same between the two strengths, despite the observed difference in Cmax, as Cmax is less likely to be indicative of clinical responses to growth hormone products compared with overall exposure (AUC) and IGF-I. IGF-I has been shown to be the primary mediator of the clinical response to somapacitan [33]. The data in this study indicates equivalent IGF-I response and, therefore, supports equivalent IGF-I mediated clinical responses between the two strengths.
It should be noted that, in this trial, a dose of 0.04 mg/kg was selected, as this dose (equivalent to 2.9 mg at the mean weight in the trial) has been established as being within the therapeutic AGHD dose range (0.1–8.0 mg) [20, 21, 30]. The PK and PD of 0.04 mg/kg somapacitan have been characterised in healthy adult participants, patients with AGHD and children with GHD [20, 34,35,36]. Non-linear and body-weight dependent PK was observed, and similar PK of somapacitan was found in healthy participants and patients with AGHD and GHD when accounting for body weight [34].
In patients with AGHD, 0.04 mg/kg somapacitan was associated with an increase from baseline in IGF-I levels over a week with mean IGF-I levels reaching a peak within normal range from − 2 to 2 SDS [20]. In healthy participants, 0.04 mg/kg somapacitan led to a similar increase in IGF-I from baseline, as in patients with AGHD [34], but with higher baseline IGF-I levels and peaks IGF-I reaching close to 3 SDS [36], matching what has been found in this trial.
No new safety issues were observed, and we consider the two different strengths of somapacitan to have similar safety profiles. This is in keeping with previous studies that demonstrated a similar safety profile across different doses and formulations of somapacitan [20, 35, 36].
A limitation of this study is that somatostatin was not used to suppress endogenous GH production prior to GH administration. Previous studies have described the use of somatostatin when testing bioequivalence of recombinant GH formulations as being an efficient and effective way to test recombinant GH in relative isolation [25, 37]. However, in this study, somatostatin was not used as participants were in a fasting state prior to their in-house visit; thus, endogenous GH would be negligible due to suppression by negative feedback. Furthermore, endogenous GH is not measured by the somapacitan PK assay.
5 Conclusions
From a clinical point of view, we consider the two strengths of somapacitan, 5 mg/1.5 mL and 10 mg/1.5 mL, to have equivalent IGF-I-mediated responses, as well as somapacitan (AUC) profiles. Even though the bioequivalence criterion was only met for AUC0–t and not for Cmax, we do not consider the observed difference in Cmax to have any clinical significance due to the similar AUC0–t profiles and almost identical IGF-I responses between the two strengths. Availability of both strength formulations of somapacitan will be beneficial for the individual dose titration and to tailor treatment in patients with GHD.
References
Miller BS, Blair JC, Rasmussen MH, Maniatis A, Kildemoes RJ, Mori J, et al. Weekly somapacitan is effective and well tolerated in children with GH deficiency: the randomized phase 3 REAL4 trial. J Clin Endocrinol Metab. 2022;107:3378–88.
Juul A, Backeljauw P, Højby M, Kawai M, Kildemoes RJ, Linglart A, et al. Somapacitan in children born small for gestational age: a multi-centre, open-label, controlled phase 2 study. Eur J Endocrinol. 2023;188:lvac008.
Christiansen JS, Backeljauw PF, Bidlingmaier M, Biller BM, Boguszewski MC, Casanueva FF, et al. Growth Hormone Research Society perspective on the development of long-acting growth hormone preparations. Eur J Endocrinol. 2016;174:C1-8.
Yuen KCJ, Miller BS, Biller BMK. The current state of long-acting growth hormone preparations for growth hormone therapy. Curr Opin Endocrinol Diabetes Obes. 2018;25:267–73.
Haverkamp F, Johansson L, Dumas H, Langham S, Tauber M, Veimo D, et al. Observations of nonadherence to recombinant human growth hormone therapy in clinical practice. Clin Ther. 2008;30:307–16.
Johannsson G, Ragnarsson O. Growth hormone deficiency in adults with hypopituitarism—what are the risks and can they be eliminated by therapy? J Intern Med. 2021;290:1180–93.
Johansson JO, Wirén L, Oscarsson J, Bengtsson BA, Johannsson G. Growth hormone (GH) replacement in GH-deficient adults: a crossover trial comparing the effect on metabolic control, well-being and compliance of three injections per week versus daily injections. Growth Horm IGF Res. 2003;13:306–15.
Rosenfeld RG, Bakker B. Compliance and persistence in pediatric and adult patients receiving growth hormone therapy. Endocr Pract. 2008;14:143–54.
Yuen KCJ, Llahana S, Miller BS. Adult growth hormone deficiency: clinical advances and approaches to improve adherence. Expert Rev Endocrinol Metab. 2019;14:419–36.
Cutfield WS, Derraik JG, Gunn AJ, Reid K, Delany T, Robinson E, et al. Non-compliance with growth hormone treatment in children is common and impairs linear growth. PLoS One. 2011;6: e16223.
Novo Nordisk. Sogroya PI. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761156s000lbl.pdf. Accessed Jan 23, 2023.
Novo Nordisk. Sogroya summary of product characteristics. https://www.ema.europa.eu/en/documents/product-information/sogroya-epar-product-information_en.pdf/. Accessed Jan 23, 2023.
Therapeutic Goods Administration. Australian Public Assessment Report for Sogroya; 2022. https://www.tga.gov.au/sites/default/files/2022-10/auspar-sogroya-20221007.pdf. Accessed Aug 17, 2023.
Saudi Drugs Information System. Sogroya Patient Information Leaflet; 2022. https://sdi.sfda.gov.sa/Home/Result?drugId=11495. Accessed Aug 17, 2023.
Pharmaceuticals and Medical Devices Agency. Report on Investigation Results; 2022. https://www.pmda.go.jp/files/000246397.pdf. Accessed Aug 17, 2023.
Kurtzhals P, Havelund S, Jonassen I, Kiehr B, Ribel U, Markussen J. Albumin binding and time action of acylated insulins in various species. J Pharm Sci. 1996;85:304–8.
Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem. 2000;43:1664–9.
Lau J, Bloch P, Schäffer L, Pettersson I, Spetzler J, Kofoed J, et al. Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide. J Med Chem. 2015;58:7370–80.
Johannsson G, Bidlingmaier M, Biller BMK, Boguszewski M, Casanueva FF, Chanson P, et al. Growth Hormone Research Society perspective on biomarkers of GH action in children and adults. Endocr Connect. 2018;7:R126–34.
Rasmussen MH, Janukonyté J, Klose M, Marina D, Tanvig M, Nielsen LF, et al. Reversible albumin-binding GH possesses a potential once-weekly treatment profile in adult growth hormone deficiency. J Clin Endocrinol Metab. 2016;101:988–98.
Johannsson G, Gordon MB, Højby Rasmussen M, Håkonsson IH, Karges W, Sværke C, et al. Once-weekly somapacitan is effective and well tolerated in adults with GH deficiency: a randomized phase 3 trial. J Clin Endocrinol Metab. 2020;105:e1358–76.
Johannsson G, Feldt-Rasmussen U, Håkonsson IH, Biering H, Rodien P, Tahara S, et al. Safety and convenience of once-weekly somapacitan in adult GH deficiency: a 26-week randomized, controlled trial. Eur J Endocrinol. 2018;178:491–9.
Otsuka F, Takahashi Y, Tahara S, Ogawa Y, Højby Rasmussen M, Takano K. Similar safety and efficacy in previously treated adults with growth hormone deficiency randomized to once-weekly somapacitan or daily growth hormone. Clin Endocrinol. 2020;93:620–8.
Miller BS, Blair JC, Rasmussen MH, Maniatis A, Mori J, Böttcher V, et al. Effective GH replacement with somapacitan in children with GHD: REAL4 2-year results and after switch from daily GH. J Clin Endocrinol Metab. 2023. https://doi.org/10.1210/clinem/dgad394.
Jacobsen LV, Rolan P, Christensen MS, Knudsen KM, Rasmussen MH. Bioequivalence between ready-to-use recombinant human growth hormone (rhGH) in liquid formulation and rhGH for reconstitution. Growth Horm IGF Res. 2000;10:93–8.
Vahl N, Jensen SB, Rasmussen MH, Susgaard S, Jørgensen JO, Christiansen JS, et al. Bioavailability of recombinant human growth hormone in different concentrations and formulations. Pharmacol Toxicol. 1996;79:144–9.
European Medicines Agency. Guideline on the investigation of bioequivalence (CPMP/QWP/EWP/1401/98 Rev. 1); 2010. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf. Accessed Jan 23, 2023.
U.S. Department of Health and Human Services FaDA, Center for Drug Evaluation and Research (CDER). Guidance for industry. Bioavailability and bioequivalence studies submitted in NDAs or INDs—general considerations. Draft Guidance, Biopharmaceutics; 2014. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioavailability-and-bioequivalence-studies-submitted-ndas-or-inds-general-considerations. Accessed Jan 23, 2023.
Tothfalusi L, Endrenyi L. Sample sizes for designing bioequivalence studies for highly variable drugs. J Pharm Pharm Sci. 2012;15:73–84.
European Medicines Agency. Sogroya Assessment Report; 2021. https://www.ema.europa.eu/en/documents/assessment-report/sogroya-epar-public-assessment-report_en.pdf. Accessed Jan 23, 2023.
World Medical Association. Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310:2191–4.
International Conference on Harmonisation. ICH Harmonised Tripartite Guideline for Good Clinical Practice E6 (R2); 2016. https://www.ema.europa.eu/en/ich-e6-r2-good-clinical-practice-scientific-guideline. Accessed Jan 23, 2023.
Kildemoes RJ, Backeljauw PF, Højby M, Blair JC, Miller BS, Mori J, et al. Model-based analysis of IGF-I response, dosing, and monitoring for once-weekly somapacitan in children with GH deficiency. J Endocr Soc. 2023;7:bvad115.
Juul RV, Rasmussen MH, Agersø H, Overgaard RV. Pharmacokinetics and pharmacodynamics of once-weekly somapacitan in children and adults: supporting dosing rationales with a model-based analysis of three phase I trials. Clin Pharmacokinet. 2019;58:63–75.
Battelino T, Rasmussen MH, De Schepper J, Zuckerman-Levin N, Gucev Z, Sävendahl L. Somapacitan, a once-weekly reversible albumin-binding GH derivative, in children with GH deficiency: a randomized dose-escalation trial. Clin Endocrinol. 2017;87:350–8.
Rasmussen MH, Olsen MW, Alifrangis L, Klim S, Suntum M. A reversible albumin-binding growth hormone derivative is well tolerated and possesses a potential once-weekly treatment profile. J Clin Endocrinol Metab. 2014;99:E1819–29.
Rasmussen MH, Jøns K, Christiansen T, Madsen JL. Growth hormone bioavailability, insulin-like growth factor-I and IGF binding-protein-3 release in Japanese and Caucasian subjects. J Bioequivalence Bioavailability. 2015;7:1–5.
Acknowledgements
The authors are grateful to the people who participated in this study, and to Navid Nedjatian, Novo Nordisk, for his review of and input to the manuscript. Medical writing and editing support for the development of this manuscript, under the direction of the authors, was provided by Amy Hepple, Malgorzata Urbacz, Beverly La Ferla and Helen Marshall, of Ashfield MedComms, and Katy Adams, formerly of Ashfield MedComms, and funded by Novo Nordisk Health Care AG.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The trial was sponsored by Novo Nordisk A/S.
Conflict of Interest
S.L.D., B.B.D., M.H.R., C.S. and R.J.K. are employees of and hold stocks in Novo Nordisk.
Data Availability
The data that support the findings of this study are available on request from the corresponding author.
Ethics Approval
The trial was conducted in accordance with the Declaration of Helsinki and ICH Good Clinical Practice guidelines. Prior to the start of the trial, the protocol, the consent form and the participant information sheet were reviewed and approved according to local regulations by appropriate health authorities and by the Ethics Committee of the North Rhine Medical Association.
Consent to Participate
The participants were informed of the risks and benefits of the trial, and that they could withdraw from the trial at any time for any reason. Consent was obtained in writing prior to any trial-related activities.
Consent for Publication
Not applicable.
Code Availability
Not applicable.
Author Contributions
All authors were involved in the study conceptualisation and methodology, data review, and analysis of results and clinical trial report. S.L.D. and B.B.D. were responsible for the study conduct. All authors agreed to be accountable to all aspects of the work and to ensure any questions related to the accuracy and integrity of the work are fully investigated and resolved. All authors read and approved the final version for submission to the journal.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Dombernowsky, S.L., Damholt, B.B., Højby Rasmussen, M. et al. Investigating the Bioavailability and Insulin-like Growth Factor-I Release of Two Different Strengths of Somapacitan: A Randomised, Double-Blind Crossover Trial. Clin Pharmacokinet 63, 1015–1024 (2024). https://doi.org/10.1007/s40262-024-01395-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-024-01395-y