
Abstract
Naloxone is a World Health Organization (WHO)-listed essential medicine and is the first choice for treating the respiratory depression of opioids, also by lay-people witnessing an opioid overdose. Naloxone acts by competitive displacement of opioid agonists at the μ-opioid receptor (MOR). Its effect depends on pharmacological characteristics of the opioid agonist, such as dissociation rate from the MOR receptor and constitution of the victim. Aim of treatment is a balancing act between restoration of respiration (not consciousness) and avoidance of withdrawal, achieved by titration to response after initial doses of 0.4–2 mg. Naloxone is rapidly eliminated [half-life (t1/2) 60–120 min] due to high clearance. Metabolites are inactive. Major routes for administration are intravenous, intramuscular, and intranasal, the latter primarily for take-home naloxone. Nasal bioavailability is about 50%. Nasal uptake [mean time to maximum concentration (Tmax) 15–30 min] is likely slower than intramuscular, as reversal of respiration lag behind intramuscular naloxone in overdose victims. The intraindividual, interindividual and between-study variability in pharmacokinetics in volunteers are large. Variability in the target population is unknown. The duration of action of 1 mg intravenous (IV) is 2 h, possibly longer by intramuscular and intranasal administration. Initial parenteral doses of 0.4–0.8 mg are usually sufficient to restore breathing after heroin overdose. Fentanyl overdoses likely require higher doses of naloxone. Controlled clinical trials are feasible in opioid overdose but are absent in cohorts with synthetic opioids. Modeling studies provide valuable insight in pharmacotherapy but cannot replace clinical trials. Laypeople should always have access to at least two dose kits for their interim intervention.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Naloxone is a safe and effective drug in opioid overdose. Its efficacy is lower in opioids that dissociate slowly from the μ-opioid receptors (MOR), such as buprenorphine and carfentanil. There are large intraindividual variations in its pharmacokinetics. Titration of two or more doses every 3 min until satisfactory response (restoration of respiration and not consciousness) to avoid withdrawal is the key to effective treatment. |
Take-home naloxone, enabling use of naloxone by bystanders, is a lifesaving intervention. The nasal route is often preferred. At least two-dose kits should be preprovided to bystanders. It is an interim intervention, and an ambulance should always be called for. |
An initial dose of 0.4–0.8 mg by needle in heroin overdose is well established. Comparable doses (adjusted for bioavailability) of nasal naloxone also works well in these victims. This is documented in controlled clinical studies in the target population. Higher doses may be required in overdoses with the more potent opioid fentanyl, but evidence of need for a great increase of naloxone dose remains uncertain. Controlled clinical trials have not yet been conducted in synthetic opioid overdose cohorts, and safe dosing of naloxone cannot be determined by volunteer or modeling studies only. |
1 Introduction
Naloxone was developed in the 1960s as a pure μ-opioid receptor (MOR) antagonist [1,2,3]. It counteracts the actions of opioids, such as reduced/arrested respiration, reduced level of consciousness, and miosis and constipation. After approval by the Food and Drug Administration (FDA) in 1971 for parenteral use, naloxone entered clinical practice in hospitals and in prehospital medicine. In hospitals, typically low and titrated intravenous naloxone doses were used to counteract opioid-induced postoperative respiratory depression while avoiding provocation of pain. Larger initial doses were required to restore respiration in opioid overdoses. Notably, the effect of naloxone depends not only on the dose the opioid but also on the pharmacological characteristics of the opioid in question, as well as the constitution of the victim [4].
The opioid epidemic, with increasing numbers of deaths from overdose, has sparked new interest in naloxone [5]. However, overdose cases are threatened not only by respiratory failure but also from the risk of airway occlusion including positional asphyxia [6].
Take-home naloxone (THN), that is, preprovision of naloxone for future administration by laypeople who may be present at an overdose scene, was proposed in the late 1990s [7, 8] by harm reduction advocates at clinical addiction arenas across Europe, North America, and Australia. Only parenteral formulations were available at that time, with intramuscular administration being the emerging preferred route, especially using prefilled syringe formulations. However, laypeople were usually not familiar with this procedure. Therefore, the improvised nasal naloxone devices (INNDs) were introduced. A mucosal atomizer device (MAD) was typically attached to a syringe containing 2 ml of a parenteral formulation of 1 mg/ml concentration. This enabled laypeople to administer a simple and prompt interim opioid antagonism to overdose victims via a nasal spray. However, the administration volume of 2 ml by the INNDs far exceeds the recommended volume of 0.1–0.2 ml for nasal use [9], so the systemic dose given was unknown. Surprisingly, no studies of the pharmacokinetics of the INNDs, which are increasingly used for THN, were published [10, 11] until 2019 [12].
There were few publications on the pharmacokinetics of naloxone until 2016. Most studies concerned intravenous administration, alongside a few studies on the population kinetics and pharmacodynamics of naloxone [13,14,15]. From 2012 onwards, the FDA and the National Institute on Drug Abuse (NIDA) in the USA supported the pharmaceutical industry to develop noninjectable alternatives to needle administrations. Among the noninjectable alternatives, the nasal administration was preferred by laypeople [16]. Finally, in 2016, the first plausible study on nasal naloxone pharmacokinetics was published [17]. In the subsequent years, several studies of the pharmacokinetics of intravenous, intramuscular, and intranasal naloxone in healthy volunteers were published, from both academic institutions and the pharmaceutical industry.
The aim of this narrative review is to give an updated presentation of the pharmacokinetics and dynamics of naloxone and its clinical use, especially for the nasal administration of approved products. The safety of the nasal naloxone sprays, considering naloxone’s pharmacokinetics and pharmacodynamics, and the characteristics of various opioids involved in overdoses are also discussed.
2 Methods
This narrative review is based on papers identified by searches primarily in PubMed (Supplementary Information) and the examination of reference lists of papers deemed to be relevant by the authors. Priority was given to studies in humans or human material, with some exceptions. Gray literature (literature that is not formally published in sources such as books or journal articles) in general was not used. Information of gray literature relating to this subject can be found elsewhere [5, 18, 19].
3 History of Naloxone
Naloxone hydrochloride (molecular formula C19H21NO4HCl, CAS number 357-08-4) was synthetized in 1960 by Fishman in the laboratory of Lewenstein. The inventors were granted a USA patent in 1966 [20] claiming that naloxone was a “more potent antagonist to the respiratory effects of potent analgesics than the antagonists hitherto known” [1].
The primary characterization of the pharmacological actions of naloxone in rodents was done by Blumberg and coworkers [21, 22]. Naloxone was shown to be a pure antagonist with no analgesic activity of its own and was not only a far more potent opioid antagonist than nalorphine but was essentially without toxic effects at therapeutic doses [1]. Now that the basis for its clinical use had been established, naloxone became a valuable tool in the exploration of opioid receptors and the endogenous opioid system by Hughes, Kosterlitz, and Terenius, respectively, in the mid-1970s [1].
It was Foldes and coworkers who first studied the efficacy of naloxone in man [2, 3]. Naloxone had no effect on respiration in anesthetized patients and offered significantly greater protection against oxymorphone-induced respiratory depression than nalorphine [2].
After approval by the FDA in 1971, parenteral doses in the range of 0.4–2 mg were used for opioid overdose, while smaller doses (0.04–0.08 mg) were used to counteract postoperative respiratory depression. Titration of naloxone dose was always the key to successful treatment, i.e., to restore respiration without provoking pain postoperatively or withdrawal symptoms in opioid-dependent overdose victims. The clinical significance of naloxone was highlighted by its introduction to the WHO “List of Essential Medicines” in 1983 [23].
With the introduction of take-home naloxone (THN) at the end of the twentieth century, broadening naloxone’s use to become a tool in the hands of laypeople, approved nasal naloxone sprays were developed and introduced, with approvals from 2016 onwards. Guiding this development, the FDA required that any nasal spray should provide similar or higher plasma concentrations than the most common reference administration 0.4 mg intramuscular injection (IM). The FDA supported the use of high naloxone doses in THN to reduce the risk for underdosing at the potential cost of more adverse events such as withdrawal. As a result, an unintended consequence has been higher systemic naloxone exposure from the nasal sprays than from the reference administration [5]. Little consideration has been given thus far to whether this is a disadvantage or an added benefit.
3.1 Miscellaneous Use
Naloxone has been explored for reversal of cardiogenic shock [24], dissociative and eating disorders [25, 26], and prevention of latent sensitization of postsurgical pain [27]. Naloxone has also been added to oral opioid formulations to reduce constipation [28]. A combination of oxycodone and naloxone was reported to reduce drug liking [29, 30].
4 Pharmaceutical Properties
Naloxone (17-allyl-4,5-alpha-epoxy-3,14-dihydroxymorphinan-6-one, Fig. 1) is a synthetic morphinane alkaloid that is morphinone in which the enone double bond has been reduced to a single bond, the hydrogen at position 14 has been replaced by a hydroxy group, and the methyl group attached to the nitrogen has been replaced by an allyl group [31]. Naloxone hydrochloride is a white-to-off-white powder that is soluble in water, acids, and alkali but only slightly soluble in alcohol and insoluble in ether and chloroform. The physiochemical characteristics of naloxone are summarized in Table 1. Naloxone is a racemic drug, the l-enantiomer, levonaloxone, being the active isoform in opioid receptors [33].

The chemical structure of the µ-opioid receptor antagonist naloxone. An asymmetric carbon atom creates a chiral structure, resulting optical isomers of which only the levo-isomer is biologically active in the µ-opioid receptor
5 Mechanism of Action
Naloxone has a high affinity for MOR, to which it competitively binds and antagonizes. The binding affinity of naloxone to MOR is about 1 nM [34,35,36,37]. It is suggested that naloxone needs to block 50% of MOR to reverse the effects of an opioid overdose [38]. Naloxone crosses blood–brain barrier readily and has fast receptor association/dissociation kinetics. Naloxone acts as an inverse agonist causing the rapid removal of any other drugs bound to these receptors.
Naloxone is a pure MOR receptor antagonist and possesses no intrinsic activity. Therefore, it has been difficult to describe its pharmacodynamic effects. Glass et al. [39] studied naloxone and nalmefene in healthy volunteers exposed to fentanyl to establish their potencies and durations of action. They showed a clear dose–response in percent recovery of the slope of the CO2-response indicating a pharmacokinetic explanation for the duration of agonist reversal. Ventilatory depression recurred at a naloxone concentration of 0.28 ng/ml. Naloxone effect depended on the plasma concentration of the agonist at the time of reversal, the duration of agonist infusion, and the dose of the antagonist [39].
Cassel et al. [37] studied the comparative binding kinetics of naloxone and a longer-acting MOR antagonist, alvimopan, in vitro in membrane preparations expressing the cloned human MOR. The in vitro half-life for dissociation of the naloxone from the µ-opioid receptor complex was less than 1 min, while longer-acting antagonists, such as alvimopan, had a slower dissociation rate than naloxone (t1/2= 30–44 min versus 0.82 min, respectively) demonstrating that receptor kinetics modulate the antagonist effects of naloxone.
Yassen et al. (see “Pharmacometric studies” section below) demonstrated a biophase equilibration half-life of buprenorphine of 173 min [15], while Kim et al. reported receptor dissociation half-lives (koff) of 41–68 min for buprenorphine and 6.8 min for fentanyl, respectively [40]. The dissociation half-life of naloxone is between 0.82 [37] and 2.44 min [35].
Despite these earlier studies, robust estimates for the opioid receptor blockade by opioid antagonists were not available until recently. Trøstheim et al. [41] reviewed the existing positron emission tomography data for a detailed analysis of central opioid receptor blockade after opioid antagonism. Positron emission-based techniques can be used to estimate receptor blockade, because antagonist drugs prevent accumulation of the radiotracer in the brain [41]. In two positron emission tomography [42, 43] and two dual-detector studies [44, 45] [11C] carfentanil was used to quantify MOR blockade with intravenous naloxone. Furthermore, timing information was available from a dual detector study with intravenous naloxone [44] and a positron emission study with intranasal naloxone [46]. The authors estimated the time-to-peak blockade from the time series data of [11C] carfentanil activity in the absence and presence of an antagonist in these studies. Blockade estimates were based on the mean signal recorded between 45–65 min after intravenous naloxone administration. From the log-logistic model an median effective dose (ED50) of 0.0023 mg/kg was obtained and an elimination rate constant k = 0.006 was obtained with an average blockade half-life of 110 min [41]. Time-series data from dual-detector studies with [11C] carfentanil [44, 47] showed a maximum reduction in activity from the control condition (i.e., no naloxone) at 23–29 min after administration of intravenous naloxone. Trøstheim et al. [41] showed that 0.1–1.5 mg/kg of intravenous naloxone can completely block all three major opioid receptors.
Pharmacokinetic investigations of concentrated intranasal formulations of naloxone indicate that the drug is rapidly absorbed into the bloodstream, with peak plasma concentrations in plasma reached in 20–30 min [17, 48]. Johansson et al. [46] characterized the µ-opioid receptor occupancy in healthy, young male volunteers after 2 and 4 mg doses of intranasal naloxone by positron emission tomography. Maximum MOR occupancy during the first 60 min after naloxone administration occurred in a dose-dependent manner (54–82% and 71–96% range in occupancy after 2 and 4 mg intranasal naloxone, respectively). [11C]Carfentanil was rapidly displaced from µ-opioid receptors after intranasal naloxone as the time taken to achieve 50% occupancy ranged from 11 to 14 min and from 5 to 13 min after 2 and 4 mg doses, respectively. Receptor occupancy was maintained at these levels during the 60 min observation period. The agonist carfentanil was rapidly (5–14 min) displaced from µ-opioid receptors after 2–4 mg intranasal naloxone dose. Later, after 360 min, a markedly lower µ-opioid receptor occupancy was observed, indicating rapid clearance of naloxone from the brain. The half-life of disappearance of MOR occupancy was estimated to be approximately 100 min. The authors also demonstrated that plasma naloxone concentrations predicted the MOR occupancy in brain, consistent with the early observations by Glass et al. [39]. The dose-occupancy model calculation resulted an average half maximal effective concentration (EC50) estimate in plasma of 0.70 ± 0.12 ng/ml.
The above studies demonstrate that the disposition of naloxone in and out of the brain (effect) compartment and receptor kinetics are fast, and the major limiting factor determining the presence of naloxone at its target site is the elimination half-life from the plasma. Moreover, an opioid’s affinity for the opioid receptor and their kinetics of association and especially dissociation have an impact on the ease of reversal by naloxone [15, 49].
6 Pharmacokinetics
6.1 Metabolism
Although pharmacokinetic studies were sparse until 2015, the metabolism of naloxone was already explored in the seventies. The formation of its major metabolite, naloxone-3-glucuronide (N3G), at that time considered to be by the liver, was rapid. After a radioactive dose, 24–37% appeared in the urine by 6 h [50, 51]. N3G is pharmacologically inactive. Minor naloxone metabolites were also EN-3169 formed by N-dealkylation and EN-2265 formed by reduction [52].
Studies on the formation of N3G in blood were published far later. First, Papathanasiou et al. [53] measured serum concentrations of N3G after a high dose infusion (3.25 mg/kg) study in healthy volunteers. Their major findings were that enzyme saturation did not occur and that N3G kinetics best fitted to a two-compartment model. Tylleskar et al. confirmed [54] the previous claims of a rapid formation of N3G with mean time to maximum concentration (Tmax) of 9.4 min and 17 min after intravenous bolus doses of 1.0 and 0.4 mg IV to healthy volunteers, respectively, far more rapid than, for instance, the corresponding 28 min for midazolam’s major metabolite 1-hydroxy midazolam [55].
6.2 Administration Forms of Naloxone
The oral bioavailability of naloxone is only 1–2%, making this route and the rectal route (bioavailability of 15%) unsuitable for emergency use of naloxone [56]. However, as stated above, oral naloxone in combination with an opioid may reduce constipation without reducing analgesia. Subcutaneous administration of naloxone has been used in the past. Strang et al. examined different potential administration forms without the needle and concluded that of the transmucosal forms, intranasal, buccal and sublingual were the most desirable [16]. However, no buccal formulations have come to the market [57], and major limitations to sublingual administration are uncontrolled swallowing and vomit in the oral cavity. Pharmacokinetic data on subcutaneous and sublingual administration of naloxone can be found in gray literature [18].
Dose-corrected (to 1 mg) peak plasma concentrations (Cmax) and area under the plasma concentration-time curve (AUC) are calculated in the three tables below characterizing the major administration forms of naloxone. This allows a direct comparison of the performance of the respective sprays.
6.3 Intravenous
Intravenous administration is characterized by very rapid onset of action, enabling titration of naloxone every third minute to achieve the desired effect whilst avoiding withdrawal symptoms. Intravenous administration requires skill, and venepuncture also may increase the risk for blood contamination and disease transmission. Intravenous administration is favored in hospital settings where, for postoperative respiratory depression, low initial doses (0.04–0.08 mg) were typically used. Intravenous use in prehospital care has decreased over the years.
Only a few small studies on intravenous administration were published in the pioneer era of naloxone [50, 58,59,60], based on quantification with radioactive immune assays (RIA) and sampling times of 120 min only. Low doses were used (0.04–0.4 mg). The major findings were that naloxone had a short elimination half-life of about 60–120 min, confirmed later by Albeck et al. [61] using higher doses, longer sampling, and quantification of plasma concentrations with high-performance liquid chromatography (HPLC) with electrochemical (ED) detection. A total clearance of about 2000 ml/min and a volume of distribution of 200 l was found in the early studies; however, more reliable figures of volume of distribution of 482 l and total clearance of 3656 ml/min were reported more recently by Tylleskar et al. [62]. Both of these are in the same range as reported earlier by Yassen et al. and Papathanasiou et al. [15, 53] in their population kinetic studies.
Pharmacokinetic data for intravenous administration of naloxone are presented in Table 2. In the era of development of appropriate nasal naloxone sprays, liquid chromatography tandem mass spectrometry (LCMSMS) was used for quantification. Two studies differ from the others by a shorter duration of sampling than 6 h [63, 64], respectively; the former is also under exposure of the volunteers to the opioid remifentanil. As can be seen, 0.4–2 mg doses were studied. The short elimination half-lives (t1/2) found previously were confirmed; central tendencies with arithmetic means were between 70 and 74 min, except for Gufford et al.’s small study (n = 6) that reported a geometric mean of 91 min [64]. Dose-corrected values (to 1 mg) AUCs varied from 3.65 to 5.4 ng × h/ml. This variation is surprising, as all drug is accounted for when given intravenously.
6.3.1 Intravenous Infusion
Intravenous infusion of naloxone may be required in some severe opioid intoxications, that is, large doses in general, and especially with long-acting opioids (methadone and carfentanil) or opioids with strong affinity to, or slow dissociation from, the opioid receptors, such as buprenorphine and carfentanil.
Experiences with naloxone infusions in young children are reported by Lewis et al. [65] and Gourlay and Coulthard [66] after codeine-induced respiratory depression and a near-fatal overdose of nor-methadone, respectively. Infusions rates were about 25 μg/kg/h and were continued for about 10 h. The treatments were uncomplicated.
Waldron et al. reported naloxone infusion in a case of methadone overdose in adults [67]. Bradberry and Raebel described two adult cases with naloxone infusion in methadone and heroin overdoses. An infusion rate of 2.5 μg/kg/h for 12 and 30 h, respectively, successfully restored respiration in both [68].
Goldfrank et al. [69] constructed a dosing nomogram for continuous infusion of naloxone. It was based on determination of a beta-elimination rate constant after 120 min sampling and subsequent reverse phase HPLC-ED quantification in seven overdose patients. The predictions were tested in eight healthy volunteers. Bolus doses of 2 and 4 mg and infusion rates of 1.5 and 3 mg/h were employed, that is, far higher than in the clinical reports above. Based on computer simulations, an infusion rate of half the clinically determined initial bolus dose should be given each hour, in addition, a second half initial dose should be given 15 min after start of infusion.
This study was conducted before the era of synthetic opioids. It is likely that higher infusion rates will be required in intoxications with slowly dissociating opioids, such as buprenorphine, in doses of 0.2–0.4 mg needing naloxone (4 mg/h in healthy volunteers [15]) or carfentanil, also with slow receptor dissociation kinetics [70].
Infusions of naloxone has also been used postoperatively in the past. Rawal et al. infused 5 and 10 µg/kg/h to gallbladder-operated patients under a postoperative epidural with morphine. The lowest infusion rate that maintained analgesia and stimulated respiration in all patients, while patients receiving the higher infusion rate needed additional morphine. A lower infusion rate (3.4 μg/kg/h) after high-dose fentanyl (127 μg/kg) in cardiac surgery allowed early extubation [71].
Apparently, far higher doses were required in the young children with overdose to reverse the opioid effects compared with those given to the adult subjects.
6.4 Extravascular Dosing: Intramuscular Administration
Intramuscular administration usually results in a fairly rapid drug uptake and onset of action and, due to formation of a depot, a longer duration of action than intravenous naloxone. It requires less skill and can be employed by laypeople in take-home naloxone (THN) programs. It also clearly carries a risk for blood contamination. Intramuscular dosing is often favored by many emergency medicine systems (EMS). Typically doses of 0.4–0.8 mg, sometimes up to 2 mg, were used in opioid overdose.
To the best of our knowledge, no study on the pharmacokinetics of intramuscular naloxone was published before 2016 [17]. The first approved device for THN was the Evzio intramuscular autoinjector containing 0.4 mg of naloxone. However, no studies of its pharmacokinetics were published; although, gray literature references had been published previously [5, 19].
A total of five studies of intramuscular administration of naloxone in volunteers, including one on Evzio 2.0 mg intramuscular autoinjector, have more recently been published (Table 3). Doses ranged from 0.4 to 2 mg, and the number of participants from 12 to 34. Central tendencies for Tmax varied from 7.8 to 30 min, and the fastest was under influence of the opioid remifentanil.
The dose-corrected (to 1 mg) area under the curve extrapolated to infinity (AUCinf) after intramuscular naloxone was rather similar, varying from 4.29 to 5.05 ng × h/ml, also under remifentanil exposure. Dose-corrected Cmax varied more, from 2.1 ng/ml of the Evzio 2 mg device to more than 4.5 ng/ml in the two studies by Skulberg et al. Regarding variability, note that all studies showed significant interindividual variability on the central parameters reported.
6.5 Extravascular Dosing: Intranasal Administration
The endothelial lining of the nasal mucosa has characteristics that makes it very open to the external environment. The mucosa is also extensively perfused. Due to the small volume of the nasal cavity (100–150 ml), volumes more than 0.15 ml should be avoided to reduce loss to the pharynx [9]. Mucociliary transport clears up the mucosal surface within 15 min, a critical time-window for drug uptake. Septal abnormalities, nasal trauma, nose-bleeding, and mucus may result in lower nasal absorption of naloxone in people who use opioids (PWUO) [5, 72].
Nasal administration is preferred by laypeople [16]. The risk for blood contamination to the caregiver is small. Nasal uptake may be slower than intramuscular (Fig. 2; Skulberg et al. [73], Tsekouras and Macheras [74]), but this is likely compensated for by the fact that it takes less time to administer nasally than any needle alternative [75].

Time course (left: 0–240 min, right: 0–60 min) of plasma concentrations of intravenous and intramuscular naloxone (0.4 mg) and concentrated intranasal formulations of 1, 2, and 4 mg naloxone. Note the initial closeness of serum concentrations of the regulatory golden standard 0.4 mg intramuscular dose and the 2 mg intranasal dose. Note also that plasma concentrations after 2 mg intranasal naloxone surpass that of 0.4 mg intramuscular naloxone after 6 min and stay above that of intramuscular for the remaining 120 min. Also, note the dramatic overshoot by the 4 mg dose compared with the 0.4 mg intramuscular dose. Redrawn from McDonald et al. [83] with permission
In the studies of Krieter et al. [12] on improvised nasal naloxone devices (INND), nasal spray volumes of 2 ml were used, while Gufford et al. used the mucosal atomizer device (MAD) to administer 0.1 and 0.2 ml. In all other studies, the Aptar spray devices were used to deliver the dose in 0.1 ml (Table 4). Lapidot et al used the dry powder device of Aptar. It should be noted that two approved concentrated sprays, Nalscue (0.9 mg) and Kloxxado (8 mg), are not included in Table 4 as they have only been reported in gray literature, but data can be found in previous publications [5, 19]. Three of the studies on nonapproved formulations [48, 62, 76] were closely related to one of the approved ones [77]. In one study, the volunteers were exposed to the opioid remifentanil [76].
Dose-corrected Cmax for the approved nasal sprays were similar (1.33–1.69), lower than the dry powder formulation of Lapidot et al. (2.3) but far higher than the INNDs (0.6–0.7). The study of Skulberg et al. (2018) under remifentanil had a longer Tmax than the others (28 min versus 15–19 min, respectively). Both Lapidot et al. and Skulberg et al. (2018), the latter with remifentanil, had by far the highest dose-corrected AUCs of about 3.4 ng × h/ml.
Tylleskar et al. was the first to publish a study on absolute bioavailability (54%) of a concentrated nasal naloxone formulation [48]. Nasal bioavailability relative to intramuscular was about 75% in Skulberg et al. (2018) in subjects exposed to the opioid remifentanil. For the others (except Gufford et al., see below) the relative nasal bioavailability was 47–54%.
There was a significant variability in terminal elimination half-life of naloxone in the different studies. Mean/median t1/2 was 61–144 min for all; however, the approved (labeled in bold) varied from 70–132. Thus, the span was larger than for both intravenous and intramuscular naloxone (Tables 2 and 3).
Gufford et al. [64] included only six subjects and had only 240 min sampling for their pharmacokinetic analysis. Whether this or the different composition explain the deviating results for Tmax, dose-corrected Cmax and relative bioavailability remains uncertain.
The approved formulations (Table 4, labeled in bold) have very different compositions [19]; however, they provided fairly similar performance. The relative bioavailabilities were between 46% and 52%, Tmax was between 15 and 30 min, dose-corrected Cmax was from 1.33 to 1.69 ng/ml, and dose-corrected AUC was from 2.13 to 2.42 ng × h/ml. Note that for each of these formulations, the dose-corrected Cmax AUC was independent of dose, indicating a linear dose–concentration relationship (Fig. 3).

Dose–concentration linearity for maximum serum concentration (Tmax) and area under the curve (AUC) after nasal spraying of 0.8–2.0 mg of concentrated nasal naloxone formulations in healthy volunteers (compiled from two different studies [48, 62] with the same formulation). This linearity is the scientific basis for a two-dose administration by laypeople in take-home naloxone (THN)
6.5.1 Improvised Nasal Naloxone Devices
Krieter et al. (Table 4) reported in an important study data on the pharmacokinetics of improvised nasal naloxone devices INNDs [12]. Dose-corrected Cmax and AUCinf were far lower than for the concentrated formulations, and the bioavailability was between 19% and 23%, less than half that of the concentrated ones. This means that the systemically absorbed dose of a standard 2 mg in 2 ml solution administered nasally is about 0.4 mg at best. Note that even lower bioavailabilities for the INNDs were reported in gray literature [5, 19].
Vanky et al. gave 10 μg/kg (0.7 mg in a 70 kg person) by MAD using a 0.4 mg/ml solution (repetitive doses of 0.1 ml). Tmax was 14 min and Cmax of 1.09 ng/ml [78]. Malmros Ollson et al. [79] used a similar procedure with a higher dose (20 μg/kg) in children (6 months to 10 years). Serum concentrations were between 2 and 6 ng/ml at the end of the 20-min sampling period. Dose-corrected plasma concentrations (for body surface area) were far higher in children than previously shown in adults [78].
6.5.2 Variability
Both central tendencies and variability are reported with different methods in the various papers. This reduces the value of comparisons between the studies, especially for the variability. However, it is clear that there may be both significant intraindividual (Fig. 4) and interindividual variability. The latter is clearly shown (Table 4) when comparing central pharmacokinetic data of Narcan 4 mg from Lapidot et al. [80] with the Narcan 4 mg data from Krieter et al. [17], administered with identical liquid sprays. The Narcan spray had Cmax of 5.3 and 8.3 ng/ml, Tmax of 30 and 13 min, and AUCinf of 8.5 and 13.2 ng × h/ml, respectively, in Krieter et al.’s and Lapidot et al.’s studies. While Tmax by Lapidot et al. was 43% lower, the Cmax and AUCinf were about 155% of Krieter et al.’s figures. This variability, even in healthy volunteers, is evident in every aspect and underlines the need for the possibility of repeat dosing in opioid overdose.

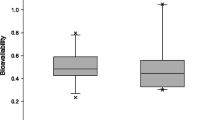
Inter- and intraindividual variability of the absolute bioavailability of intranasal naloxone [AUC(IN)/AUC(IV) × DOSE(IV)/DOSE(IN)] based on AUC0−inf in 22 healthy volunteers. Not only does the absolute nasal bioavailability vary from about 20% to almost 100% between individuals but also is usually reproduced in an individual. Similar findings were observed for the relative intranasal to intramuscular bioavailability in the same subjects [19]
6.5.3 Solution Versus Powder
Middleton et al. [81] reported pharmacokinetic data after intranasal application of crushed buprenorphine/naloxone tablets (two strengths) in recreational users of prescription opioids. Tmax of naloxone was about 18–20 min, dose-corrected Cmax of 0.8–1.2 ng/ml, and AUC 0–72h of 0.8–1.0 ng × ml/h. Bioavailability was 24–30%. Except for bioavailability, these values were similar to the INNDs.
Lapidot et al. compared head-to-head a new dry-powder formulation with the Narcan nasal liquid spray [80], both 4 mg. Although Tmax was about the same for powder and liquid, Cmax was more than 10% higher with powder. The AUCinf ratio was 1.08 in favor of the dry powder, indicating about 8% higher bioavailability than for the comparator Narcan nasal spray. Powder has an advantage in cold climate zones, since it does not freeze (although extremes of cold temperature appear not to damage naloxone [82]).
6.6 Intramuscular Versus Intranasal
Tmax values of intramuscular and intranasal administrations overlapped with large variabilities; consequently, these pharmacokinetic studies cannot be used altogether to depict potential differences in onset of action between the administration forms (Table 3).
As expected from the relative bioavailabilities, both dose-corrected Cmax and AUC were far higher after IM administration. Perhaps more interesting is whether interindividual variability is different between the two administration forms. Contrary to what one might expect (Tables 3 and 4), the variability in the three regulatory studies [17, 77, 83] in Tmax and dose-corrected Cmax (except in Skulberg et al. 2019 [77]) did not differ much between the administration forms, while variability in dose-corrected AUC seemed higher for all after IN.
While the automated intramuscular Evzio device and the disposable single-dose nasal sprayers may not require training for successful use by laypeople [17, 84], this was not the case for INNDs [84]. Indeed, the Evzio device was strikingly superior to INND, both regarding speed of administration (0.9/0.5 min versus 6/2 min, respectively) and success rates (90–100% versus 0–57%), without and with training, respectively.
6.7 Drug Interactions
The effect of nasal decongestants are only described in gray literature [19]. By and large, the effect of oxymetazoline was modest, except for Cmax, which was reduced by about 40%.
Skulberg et al. [63] unexpectedly observed that exposure in healthy volunteers to the opioid remifentanil resulted in an IN/IM bioavailability of 75% [76], which is a significant difference compared with 52% without remifentanil [77]. A follow-up study by Tylleskar et al. [54] determined that the increased nasal bioavailability was likely caused by a reduction of the presystemic metabolism of the swallowed portion of the nasal dose.
7 Pharmacometric Studies
7.1 Modeling Studies
The pharmacokinetics of naloxone has been studied quite extensively by means of noncompartmental analyses and reporting summary statistics. However, there are few actual pharmacometric studies with population pharmacokinetic–pharmacodynamic modeling (Table 5). The population modeling approach utilizes nonlinear mixed effect modeling to determine between-subject and residual unexplained intraindividual variability [85,86,87]. This method enables identifying subject-specific factors that may be valuable when optimizing drug-dosing strategies.
Dowling et al. [88] evaluated the pharmacokinetics of naloxone in an open-label crossover study in six male volunteers. Intravenous (0.8 and 2 mg), intramuscular (0.8 mg), and intranasal (0.8 and 2 mg) administrations were studied. A three-compartment model, with first order absorption [15] for the model for intramuscular and intranasal administration, was used (Table 6). Between-subject variability was included on clearance and central volume, and the effect of lean body weight and weight covariates were incorporated on clearance, and central volume, respectively. The very poor intranasal bioavailability of 4% reported was probably a result of high intranasal volumes as well as low analytical sensitivity [48], rendering little data for accurate determination of nasal AUC.
Recently, Papathanasiou et al. [53] developed a pharmacokinetic model that characterized the pharmacokinetics of naloxone and naloxone-3-glucuronide after high-dose intranasal target-controlled infusion in healthy subjects. The authors administered a total intravenous dose of 3.25 mg/kg in a stepwise approach for 75 min with a step duration of 25 min. A three-compartment model was used to describe naloxone pharmacokinetics and body weight was incorporated to volume of the peripheral compartment (V2). The model adequately described naloxone and naloxone-3-glucuronide pharmacokinetics and the pharmacokinetic parameters (Table 5) from the final model are in line with previous results; although, the reported volume estimates are larger. The results indicate that the naloxone pharmacokinetics remain linear, even after high doses [53].
Algera et al. [89] reviewed human studies on reversal using models that describe and predict the time course of pharmacokinetics and dynamics of opioids and reversal agents. Their analysis provides a detailed overview of opioid-induced respiratory depression through pharmacokinetic–pharmacodynamic modeling.
Yassen et al. [15] studied the pharmacokinetics and pharmacodynamic interaction between buprenorphine and naloxone in 40 healthy volunteers. They used minute ventilation as a biomarker for respiratory depression. Ventilation was measured with a dynamic end-tidal forcing technique using a pneumotachograph, and blood samples were drawn for naloxone and buprenorphine plasma concentration analyses. Two doses of buprenorphine (0.2 mg and 0.4 mg/70 kg) were tested, and half of the dose was administered as a bolus at t = 0 min, followed by a continuous 1-h infusion of the remaining dose. Increasing naloxone doses (1–7 mg) or placebo was given 30 min after buprenorphine bolus, one-half of the dose as a bolus and the remaining half as a 30-min infusion.
Two- and three-compartment models best described the pharmacokinetics of naloxone and buprenorphine, respectively (Table 5). A combined biophase equilibration-receptor association–dissociation pharmacodynamic model was used to evaluate the competitive interaction between buprenorphine and naloxone at the MOR. For buprenorphine, the rate constants of receptor association and dissociation were 0.203 ml/ng/min and 0.0172/min, respectively, and the equilibrium dissociation constant (KD) was 0.18 nmol/l. The half-life (t1/2) of biophase equilibration (tke0) was 173 min. The same tke0 has been previously reported for antinociceptive and respiratory depressant effects of buprenorphine, indicating that disposition kinetics of buprenorphine within the brain tissue was not influenced by naloxone disposition kinetics. Buprenorphine had slow receptor association–dissociation kinetics compared with naloxone, the latter with a half-life of biophase distribution of 6.5 min. The receptor dissociation kinetics were faster compared with the pharmacokinetics in plasma and were not rate-limiting in naloxone kinetics of action at the opioid receptor. Simulation of buprenorphine-induced respiratory depression indicated that the reversal was poorer when naloxone doses over 4 mg/h were used.
Olofsen et al. [13] evaluated the effects of three different naloxone doses (0.025–0.4 mg) on morphine and morphine-6-glucuronide-induced respiratory depression in 24 healthy volunteers using a single-blinded, placebo-controlled, randomized study design. They refrained from plasma sampling to avoid the excitatory effects on breathing and simulated pharmacokinetic data instead. However, the likelihood for bias was considered low. Also, the pharmacokinetic parameter estimates were in accordance with the previous results by Yassen et al. [15] (Table 5). The mechanism-based pharmacokinetic–pharmacodynamic model was applied to estimation of parameters related to biophase distribution and receptor association–dissociation kinetics. Naloxone reversed the morphine-induced respiratory depression faster than that of morphine-6-glucuronide. This was attributed to the receptor kinetics of the opioid used. The authors postulate that an opioid with lower rate factor values (kon and koff) results in more difficult and slowly developing reversal when intravenous bolus dosing is used [90]. Thus, the receptor association-dissociation kinetics of the opioid agonist, not that of the antagonist, dictates the reversal. Furthermore, these findings explain the previous results by Yassen et al. [15] that greater naloxone doses have no effect on the speed of reversal.
Measurements of pupillary size has also been used as a biomarker for assessing the effect of naloxone on the action of an opioid [63, 64, 76, 91]. Pupillary response may serve as an indicator of respiratory depression [92]. Also, miotic pupils outperformed other opioid overdose criteria in guiding prehospital naloxone administration [93].
Gufford et al. [64] used 4 mg oral alfentanil to compare the effect of 2 mg intramuscular and intranasal naloxone in six volunteers. By using oral alfentanil, the pupillary size is determined by both the concomitant alfentanil and the naloxone concentrations. However, the authors only found a modestly higher extent of alfentanil reversal after intramuscular administration than after intranasal.
Skulberg et al. and Tylleskar et al. [63, 76] used target-controlled infusion of remifentanil to enable effect studies of naloxone in 12 volunteers. This procedure gave a steady level of remifentanil in blood. In contrast to Gufford et al.’s procedure [64], the only variable that changed pupillary size was the concomitant blood concentrations of naloxone. In contrast to Gufford et al. [64], Skulberg et al. [76] showed that intramuscular 0.8 mg naloxone increased pupillary size far more than a similar (in mg) intranasal dose. In Tylleskar et al.’s study [63] a full reversal of the opioid effect was achieved within 3 min by 1 mg naloxone intravenously. Moreover, the duration of action of this dose was 118 min (Fig. 5), and the minimum effective blood concentration of naloxone at steady state was 0.5 ng/ml. Finally, the hysteresis plot (Fig. 6) was similar for both arterial and venous concentration of naloxone, indicating that both arterial and venous blood samples can be used in pharmacokinetic/-dynamic modeling.

Time course of pupil diameter in healthy volunteers exposed to remifentanil and naloxone [n = 12, mean (95% confidence interval)]. Remifentanil was started at t = −12 min and stopped at t = 90 min (orange, horizontal line). Miosis was rapidly reversed by a naloxone bolus of 1 mg intravenously given at t = 0. The blue broken line is the regression line [f(x) = − 0.0292x + 6.9924)] and is based on the period 19–89 min. It crosses the nadir baseline (black dotted line) at 118 min. The blood concentration of naloxone at this point was 0.5 ng/ml, which is the minimum effective concentration of naloxone in steady state. Redrawn from Tylleskar et al. [63] with permission

Hysteresis plot of pupil diameter and naloxone concentration during a remifentanil infusion with stable blood concentrations (see legend for Fig. 5). The red line represents arterial samples, and the blue line represent venous samples. The direction of time is illustrated by the arrow. This finding allows for the use of venous concentrations in PK/PD modeling. Redrawn from Tylleskar et al. [63] with permission
Loimer et al. [91] exposed addicted and nonaddicted subjects to intranasal naloxone (1 mg/0.4 ml) to detect opioid dependence. They found a significant decrease in pupillary size at 10 min in addicts. Note that the clinical outcome, a withdrawal rating score, increased significantly simultaneous to the decrease in pupillary size. In another study by the author [94], it was shown that intranasal naloxone was as effective as intravenous naloxone in provoking withdrawal distress in these patients.
As already mentioned, the clinical pharmacology of naloxone does not stand alone; it depends on many factors, not least the characteristics of the agonist it is counteracting [4]. Unfortunately, similar studies as described above are neither available for heroin/6-Monoacetylmorphine (6MAM) nor fentanyl. However, some studies may shed light over these drugs in relation to naloxone. First, it is a common belief that the blood-effect site equilibration time depends heavily on lipid solubility, for instance, in the case of fentanyl. This is not true; the anesthetic opioid alfentanil is far less lipid soluble than fentanyl, but its onset of action is much faster in man [95].The blood-effect site equilibration time for fentanyl (6.3 min) [95] is similar to that of naloxone [13, 15]. Tmax of 6MAM is 1–2 min and 6 min after intravenous and intramuscular injection in man, respectively [96]. Indication of cerebral uptake of 6MAM is only found in a study of rats (Gottås et al. [97]), in which the Tmax of 6MAM in the brain extracellular fluid was reached at 4.3 min, 2.3 min after Tmax in blood. The review of Kiyatkin et al. suggests that both intravenous fentanyl and heroin rapidly reduce oxygen levels in freely moving rats [98]. These data may indicate that the onset time of fentanyl overdose is not dramatically faster than for heroin [99,100,101] for which a real-world median onset time of less than 17 min was observed in a safe injection facility [6]. However, there is also solid experimental evidence that piperidine analogues, such as fentanyl, differ pharmacologically from morphinane analogs, such as morphine and heroin, both when it comes to onset of action and sensitivity of naloxone [102,103,104]. The study of Hill et al. [104] is of particular interest, as assumed equipotent doses of 6MAM, the active metabolite of heroin, and fentanyl was compared in freely moving mice. Onset of respiratory depression with fentanyl was faster than for 6MAM, and naloxone reversed heroin-induced respiratory depression more readily than that of fentanyl.
However, chest wall rigidity from the fentanyl analogues (with a more rapid onset of action than respiratory depression) may be very serious, as it may cause sudden deaths after fentanyl overdose [101]. The muscle rigidity can be antagonized by naloxone [101, 105]; although, naloxone may be inefficient against vocal cord closure [106, 107].
7.2 Simulation of Clinical Scenarios
Skulberg et al. [77] used simulations to demonstrate the time course of blood concentrations after laypeople administration of 1.4 mg naloxone intranasally 10 min before arrival of EMS, who administered 0.4 mg intramuscularly (Fig. 7). Nasal dose administered by bystanders provided higher serum concentrations of naloxone than after the intramuscular administration by EMS for at least 20 min. Moreover, the combination of these two administrations performed better than 0.8 mg intramuscular by EMS at any time. Finally, the interim 1.4 mg nasal naloxone performed as well as EMS 0.4 mg IM, even when given only 2 min before arrival of EMS.

Simulated time courses of mean naloxone concentrations (lines) and standard deviations as shaded areas: 0.8 mg intramuscular (dark gray), 0.4 mg intramuscular (light gray), and 1.4 mg intranasal (blue). Time of administration of injected naloxone is t = 0 min. A Intranasal 1.4 mg administered 10 min prior to injected naloxone (0.4 and 0.8 mg). B Simulation of the shortest time (2.25 min) beneficial to administer the intranasal spray, rather than wait for naloxone to be injected by medical personnel. C Simulation of 1.4 mg intranasal naloxone when intramuscular 0.4 mg naloxone is injected to a patient 10 min before the intranasal dose. Redrawn from Skulberg et al. [77]
In a recent study [108], the blood concentration–time courses of clinically relevant dosing regimens for naloxone currently used to treat opioid overdose were simulated from a sample of 53 volunteers. (Fig. 8). We evaluated dosing against the concentration–time course after a single 0.4 mg intramuscular naloxone dose since, regulatory studies in healthy volunteers, the Cmax (0.9–1.3 ng/ml) and Tmax (10–24 min) of 0.4-mg intramuscular doses are often used as a guidance for nasal naloxone alternatives and are considered indicative of the lowest acceptable blood levels of naloxone [5]. Tylleskar et al. [63] determined a mimum effective naloxone concentration of 0.5 ng/ml under steady-state remifentanil agonism, and this value was used to guide considerations regarding duration of action. Levels in the same range were reported prevously for fentanyl [39] and carfentanil [46]. McDonald et al. [18] suggested that an intramuscular dose resulting in Cmax above that of 0.8 mg (about 3.65 ng/ml, Table 4) could be helpful in guiding dosing to reduce the risk for adverse naloxone effects, in line with the WHO’s recommendation from the heroin era of an upper limit for the initial dose of naloxone [109]. A recent randomized clinical trial on heroin overdose confirmed that 0.8 mg intramuscular dose was highly effective with a low rate of adverse effects, such as withdrawal symptoms [73].

Simulated concentration–time profiles of plasma naloxone after various common clinical dosing schemes in opioid overdose. A pharmacokinetic model based on our own data and 1000 simulations were used (Anesthesiology, manuscript in review) [108]. Two intravenous (0.4 and 2 mg, shown in gray and black, respectively) and three intramuscular doses (0.4, 0.8, and 2.0 mg, shown in light blue, yellow and blue, respectively) were simulated using the final pharmacokinetic model. The lower dotted line suggests the duration of action, represented by an experimentally determined minimum effective concentration of 0.5 ng/ml [76] and the upper dotted line suggests an increased risk of withdrawal, represented by the Cmax (3.2 ng/ml) and 0.8 mg intramuscular dose [18, 109]
The first set of simulations was conducted with the final pharmacokinetic model only to evaluate the model output for naloxone concentration–time profiles for single or repeated bolus dosing using the intravenous, intranasal, or intramuscular routes. Clinically relevant dosing schemes were simulated for 2 h using single and repeated naloxone doses of 0.9, 1.4, 2.0, and 4.0 mg, corresponding to the doses of four commercially available nasal sprays Nalscue, Ventizolve/Respinal, Nyxoid, and Narcan, respectively (Fig. 9). These simulations were conducted to evaluate the probability of achieving clinically effective serum concentrations, as well as project the risk of the intervention provoking adverse events; all the commercially available nasal naloxone formulations, except the 0.9 mg/dose, seemed to perform adequately regarding the clinical effect during the initial 10–15 min. Moreover, all tested IN doses performed better than both intravenous (IV) and intramuscular (IM) dosing for at least 120 min because the concentrations remained above 0.5 ng/ml for 60–90 min longer than after, for instance, the 0.4 mg IM dose. There may, however, be an increased risk of withdrawal with doses above 2 mg given intranasally, even more with repeated dosing, as discussed previously [110,111,112]. However, it should be emphasized that these suggestions are based on simulation results from healthy volunteers only, and further studies are needed to evaluate clinical response. Also based on previous findings, naloxone reversal is dependent on multiple factors, not least pharmacological characteristics of both agonist and antagonist [13, 15] as shown before [70].

Simulated plasma concentration profiles after single bolus doses (0.9, 1.4, 2.0, or 4.0 mg) of intranasal naloxone, alone or with remifentanil dosing (A and B, respectively) using a pharmacokinetic model based on our own data (Anesthesiology, manuscript in review) [108]. Plasma concentration–time course of the regulatory golden standard, an intramuscular 0.4 mg bolus dose, is shown in blue
Moss et al. [36] used a quantitative systems pharmacology model to depict effective intramuscular naloxone doses at high fentanyl blood concentrations. At the most clinically relevant levels of fentanyl (25 ng/ml, see discussion below), 2 mg naloxone intramuscular effectively reduced the receptor occupancy of fentanyl to 50% within 3 min, conforming with reversal of opioid toxicity [38]. However, this dose failed at 75 ng/ml, while 5 and 10 mg reached the desired receptor blockade within 5.5 and 4 min, respectively.
Mann et al. [70] recently published a comprehensive, advanced physiopharmacological modeling of intramuscular naloxone (2 mg) on respiratory depression and cardiac arrest from opioid overdose. Lethal doses of fentanyl and carfentanil were calculated from an autopsy database (about 500 cases). It was concluded that carfentanil had slower receptor dissociation kinetics than fentanyl, which made it more difficult to reverse. The modeling confirmed that a rapid naloxone uptake in the initial phase is important. Naloxone 2 mg intramuscularly prevented more cardiac arrests in fentanyl than carfentanil intoxications (less at the higher doses), and the concentrated intramuscular naloxone (in 0.1 ml) worked better than that in 1.0 ml.
In contrast to the experimental studies of Hill et al. and Kiyatkin [98, 104], Moss et al. and Mann et al. did not include 6MAM, the active metabolite of heroin, as a reference in their modeling [36, 70]. This is unfortunate, not only could we have learned more about differences in response to naloxone, but the abundance of clinical research on effective naloxone dosing in heroin overdose could be translated to fentanyl. Figures for fentanyl were taken from forensic data in both simulation studies. These may be disturbed by postmortem redistribution processes [113]. There may also be a considerable overlap in autopsy blood concentrations and concentrations of those surviving an overdose [113]. Mean autopsy concentrations of fentanyl are usually about 10–15 ng/l [70, 114, 115], far lower than those used by Moss et al. [36]. Finally, it is unclear how Mann et al. converted from autopsy blood concentrations to doses in mg [70]. Modeling is an excellent tool to illustrate relative pharmacological differences between compounds but have shortcomings when it comes to real-life extrapolations, as all models are based on uncertain assumptions as discussed above. Thus, they cannot replace controlled studies in the target population.
8 Special Populations
Unlike most other modern medicines, naloxone’s pharmacokinetics were never studied in special populations, except for oral intake in liver failure.
It is generally recommended to be careful when dosing naloxone to elderly; however, elderly may also be more susceptible to opioids [116]. Care should also be taken in patients with pre-existing cardiovascular and pulmonary disease and those on potentially cardiotoxic drugs. However, it is not known whether these groups should be given lower doses in opioid overdose compared to younger people.
Due to limited data, the risk of using naloxone in pregnant women is not determined. Its use in opioid dependent women may cause withdrawal in both fetus and woman. Breast-feeding can be continued after naloxone exposure.
Children (less than 5 years/20 kg) frequently suffer more severe intoxications than adults. This may be due to differences in drug disposition in children compared to adults [116, 117]. Consequently, naloxone doses of 0.1 mg/kg may be required [116]. However, initial lower doses of naloxone are used in opioid-exposed neonates, as withdrawal may be life-threating in neonates [118].
In subjects with severe hepatic failure, systemic uptake of naloxone after oral administration may increase, especially when large doses (8 mg) are given. Apparently clinically relevant concentrations of naloxone were seen for a couple of hours [119].
9 Clinical Effects of Naloxone
Naloxone has no prominent biological effect in healthy volunteers after administration, even with high doses [53, 120], although Cohen et al reported behavioral changes after accumulative doses up to 4 mg/kg over 3 days [121]. In opioid overdose, naloxone reverses the effects of the opioid overdose triad, that is, respiratory failure, stupor, and pinpoint pupils (miosis). The introduction of take-home naloxone for bystanders/first responders, including by peer drug users as well as by family or by others in the general public, has certainly saved many lives [122, 123] and is now strongly supported by the World Health Organization and by the United Nations, as well as through THN programs at the national level. It is important to note that naloxone also reverses muscle rigidity associated with overdose with fentanyl and its analogues [101, 105, 107], although possibly not vocal cord closure [106]. Interestingly, with a different clinical issue, oral naloxone may also reduce constipation when combined with oral opioid analgesics [28].
9.1 Adverse Effects
Naloxone is a safe drug. In subjects on opioid analgesics for chronic pain or in postoperative patients, care must be taken to avoid provocation of pain. In opioid overdose reversal, the major adverse effect is precipitation of withdrawal symptoms, especially as this may cause hostility and self-discharge with further drug-seeking [124, 125], a pattern which we have termed “behavioural toxicity” [126]. Withdrawal is not considered life threatening in adults [124]; however, the condition may be more severe in the neonates as stated above. Withdrawal symptoms are confusion, restlessness, nausea, and retching/gagging/vomiting. Piloerection, lacrimation, and nasal discharge can be seen, and tachycardia and hyperventilation are not trivial effects. Opioid withdrawal is very unpleasant for the victim and needs to be avoided not only for compassionate reasons but also because it may trigger further drug use [111].
There are many reports of pulmonary complications such as pulmonary edema, aspiration pneumonia and pneumonitis when naloxone has been used. Pulmonary edema has been associated with postoperative naloxone in healthy young people [127, 128]. Linking noncardiac pulmonary edema to naloxone in opioid overdose may be dubious [116], since opioid overdose itself may also be associated with the condition. Nevertheless, recent reports have connected naloxone to pulmonary edema [129,130,131]. Moreover, Farkas et al. reported that naloxone doses exceeding 4.4 mg increased the risk for pulmonary complications after opioid overdose [132]. Kummer et al. [130] also found that similar high doses of naloxone were associated with pulmonary edema, supporting that naloxone by itself may cause pulmonary problems.
Naloxone has also been associated with cardiopulmonary effects. The mechanism may be a surge of adrenaline and noradrenaline with circulatory consequences in subjects under anesthesia [133, 134]. More details on circulatory effects can be found in the review by Rzasa-Lynn and Galinkin [135].
9.2 Adverse Effects in Emergency Medical Setting and Take-Home Naloxone (THN)
Gaddis and Watson [136] reported two cases of naloxone-related violent behavior after intravenous bolus naloxone 2 mg. The interesting conclusion was that naloxone should not be used prehospital in patients with diminished levels of consciousness unless respiratory depression was also present. Buajordet et al. [112] published the first systematic study in 2004 on adverse events from naloxone in opioid overdose in the Oslo ambulance service. The numbers of adverse effects (45%), especially withdrawals, were alarming and related both to dose and to the intravenous administration form. The study resulted in more careful dosing of naloxone in the years to come as shown by our group [110]. In the Bradford Hill analysis of published evidence on take-home naloxone (THN) [122], McDonald and Strang found far lower adverse event rates, such as withdrawals of about 3% and 2% agitation/vomiting. The introduction of intranasal Narcan (4 mg naloxone–HCL) changed the field by increasing naloxone doses in THN. Avetian et al. [137] reported a total of 38% adverse events for the Narcan spray, 14% classified as withdrawal, 12% had irritability/anger and aggressive behavior, and 10% were vomiting/retching. These symptoms are not trivial; vomiting may, for instance, lead to aspiration. The difference between the two studies is striking [122, 137]. The report of Thompson et al. [138] is an interesting response in this context. Initial doses (0.4 and 2 mg, respectively) in a heroin dominated cohort (about 85 %) had similar response to initial dose and requirement of additional doses, but the rate of adverse effects was 2% in the 0.4-mg lower dose and 29% in the 2-mg dose. Also, Moustaqim-Barette et al. [139] reported significant increase in adverse events with number of naloxone ampoules (0.4 mg IM each) in THN programs. Isoardi et al. [140] studied the outcome of a single intramuscular dose of 1.6 mg by the EMS for opioid overdose. They concluded that a rate of severe agitation of 7% and signs of withdrawal in 39% of the victims was acceptable. This conclusion was refuted in an accompanying editorial by Wightman and Lewis [141]. Moe et al. concluded that association of dose with adverse events [142] remains unanswered in this population. Finally, they also believed that withdrawal may impede postoverdose care, as it may induce hostility from the victim toward the health care personnel (although not observed in a recent report [143] but supported in another [144]). Suspected fentanyl exposure, suspected benzodiazepine exposure, and higher doses of naloxone were associated with more frequent withdrawal symptoms [143].
9.3 Recurrent (Rebound) Toxicity
Recurrent or rebound toxicity is return of overdose symptoms after treatment with naloxone. This may be due the short action of naloxone, or a reintoxication to overcome a naloxone blockade. A 12-h observational period is commonly considered. Vilke et al. [145] saw no fatal complications within 12 h in 998 out-of-hospital administrations of naloxone. However, Rudolph et al. [146] reported that 0.13% of 2245 out-of-hospital administrations died from probable recurrent toxicity after discharge on scene. In Wampler et al.’s study [147], no deaths occurred within 48 h in 592 patients who overdosed not transported to the hospital. The safety of leaving an opioid intoxication at the scene was confirmed by Tylleskar et al. [110], in which none of about 980 cases suffered recurrent toxicity. However, about 4% of 201 patients who overdosed included in a randomized controlled trial had another callout requiring naloxone within 12 h [73].
The situation is different in hospitalized patients. Watson et al. [148] reported an overdose recurrence rate of 31 % in hospitalized overdose victims. Boyd et al. [149] found similar rates in victims hospitalized after prehospital naloxone was given. Management of severe opioid overdose may necessitate naloxone infusion to manage recurrent toxicity [150].
10 Clinical Trials
Nasal naloxone has been compared with intravenous [151] or intramuscular naloxone in five randomized controlled studies of opioid overdoses (see Supplementary Information for details). Three of the studies were open-label studies [151,152,153], while two were double-blinded studies [73, 154]. All studies were ethically sound, and all were conducted by academic groups. Despite large dose variations, they showed that intranasal administration was feasible and effective, but the onset of action after intranasal dosing was slower than with intramuscular administration. In the randomized double-blinded study by Skulberg et al., an approved 1.4 mg naloxone nasal spray delivering about 0.77 mg naloxone systemically was compared with 0.8 mg intramuscular dose in patients who overdosed on opioids [73]. The fraction of reversed cases was similar up to 3 min; thereafter, the effect of the nasal administration (1.4 mg) lagged behind the intramuscular dose. At 10 min, the difference was 2.3 min (Fig. 10). However, both administration forms were considered safe with few adverse effects.

A randomized, double-blinded comparison of naloxone given intramuscularly (0.8 mg, blue) or intranasally (1.4 mg, brown) by the ambulance service in opioid overdose (n = 201) Shaded areas represent 95% confidence interval. Kaplan–Meier plot showing the probability of not having reached satisfactory respiration (10 breaths/min). Curves are similar until 3 min, and the restoring of respiration is 2.3 min slower after the intranasal dose at 10 min. In real life, this difference is probably smaller, as intranasal administration is likely faster to complete than intramuscular. These intoxications were also more serious than those in an unselected material in the same catchment area [110]. Redrawn from Skulberg et al. [73]
10.1 Dosing, Safety, and Efficacy
Titration of naloxone increases the extent to which personalization of care can be achieved while ensuring safety in management of opioid overdose. Patients with opioid tolerance frequently have a response to low doses of naloxone that are sufficient to restore breathing without provoking withdrawal [116]. When possible, intravenous administration, while requiring skills in venepuncture and is most often used in the hospital, is probably the best method for titration as onset of action is rapid and easily titratable [116].
In other settings, including EMS, intramuscular administration is a good choice with initial doses of 0.4–0.8 mg [73, 110]. Higher doses increase the risk of provoking withdrawal symptoms [155].
For layperson administration (as with THN), the nasal route is probably the most favorable [16], in particular for operational reasons when in the hands of nonmedical personnel. THN is an interim treatment, and an ambulance should always be called immediately. Some training before being equipped with naloxone is recommended. It has been shown that, after training, laypeople are able to give incremental doses according to response [156].
The experience in several parts of the world with off-label improvised nasal naloxone devices (INND) is interesting and warrants consideration. INND with assumed systemic doses of 0.4 mg at best seemed to work adequately, both before and after the rise of death from fentanyl intoxications [19]. However, after the regulatory approval and introduction of Narcan 4 mg in the USA, the naloxone doses increased significantly [139]. This increase may have been less driven by medical science than by the fact that there was, from the point onwards, no approved naloxone spray for opioid overdose with a unit dose lower than 4 mg, although a 3-mg spray was recently approved for over-the-counter nonprescription use. In contrast, across Europe and in Australia, naloxone nasal spray with doses of 1.4 and 2.0 mg of naloxone hydrocholoride are available. Interestingly, Moe et al. have reported that not only naloxone dosing for heroin overdoses has increased dramatically in the USA but also that naloxone dosing in Europe and Asia was lower than that in the USA, under seeming similar conditions [142]. Unambigious science-based evidence for a medically needed, large increase in naloxone dose for fentanyl overdose is, at the time of writing, still lacking [19, 141, 144], perhaps obscured by the great public and emergency medical concern about the epidemic rise of opioid overdose deaths. Nevertheless, recent modeling studies does support use of higher naloxone doses with the synthetic opioids [36, 70]. In considering the doses of INND, it is likely that all approved formulations, delivering far more naloxone systemically than the INNDs, are much safer products to consider for THN, not least since they are also usually provided in two-spray packages. Therefore, the continuation of a place for INNDs in contemporary take-home naloxone programs is dubious. See Dale [19] for more information on this topic.
10.2 Need for Further Research
There is a lack of comparative knowledge of the receptor kinetics and the receptor interaction with naloxone relating to fentanyl and heroin by means of 6MAM in man. Such knowledge would have made it easier to transfer the vast evidence for heroin overdose treatment to the case of fentanyl and its analogs.
Consideration of the pharmacokinetics in target populations is important. The adequacy and generalizability of pharmacokinetic data from healthy volunteers, on which new naloxone products are approved, both with single and repeated doses, is dubious. The target population may have a history of extensive abuse of drugs by snorting with cocaine-triggered ulceration or may have obstructed nasal passages from a cold or hay fever. Moreover, patients with opioid tolerance frequently have a response to low doses of naloxone that are sufficient to restore breathing without provoking withdrawal [116]. In addition, multiple drugs are often seen in opioid overdoses, not least with tranquilizers that may obscure the overdose picture. The target population may have impaired liver function resulting from chronic hepatitis C. Also, comparison with IV naloxone may not be meaningful since the IV route is often inaccessible in an emergency situation as a result of scarring and damage to surface veins caused by chronic injecting abuse and also collapsed vasculature in the overdose emergency scenario. However, studies of the pharmacokinetics of naloxone in the target population are ethically challenging but not impossible [10].
Also, industry-independent postmarketing studies would add more knowledge to the field, and qualitative follow-up with interviews of victims give a deeper insight into their experiences. Input from ground level experts with first-hand experience is essential [144], also in the planning of clinical trials [73].
Clinical, controlled studies [73, 154] in the target population may compensate for the lack of basic pharmacology and pharmacokinetic studies. Recent publications (conducted in EMS or safe injection facilities) have shown that both blinding and randomization under these difficult clinical circumstances are nevertheless possible. Research ethical challenges were also overcome. There is overwhelming evidence for dosing levels of naloxone in heroin overdose. However, future studies should compare intramuscular and nasal two-step dosing regimens. Moreover, there are no published randomized, controlled studies on two-dose naloxone in fentanyl cohorts. We need to know if a two-step dosing of nasal naloxone not exceeding 2 mg compares well with a two-step intramuscular schedule using 0.8 mg in fentanyl overdose. To enable comparison along studies, respiratory rate and Glasgow coma scale must be reported. The focus should be on criteria for the severity of the overdose, not just on the opioid in question. The latter should be used as an explanatory variable. Finally, new drugs/treatment options, for instance nalmefene, as an alternative to naloxone [4] should be tried against naloxone in target populations, examining speed of onset of benefit as well as duration of action and adverse effects. This is essential in view of the huge intra and interindividual variability, as well as variability, between studies conducted in homogenous cohorts of young healthy volunteers. One may ask, is it more ethical to abstain from controlled studies in this vulnerable minority than to conduct them [10]?
11 Discussion
The use of naloxone by laypeople has sparked a renewed interest in naloxone, not least for noninjectable administration alternatives. This intervention saves lives. Naloxone is an essential medicine due to its specificity to, and lack of any effect on, the MOR. Naloxone’s clinically significant effect is the antagonism of MOR agonists. The overall safety of naloxone in opioid overdose is excellent; however, the dramatic increase in doses observed recently raises concern such as possible cardiopulmonary complications [132].
All treatment with naloxone is empirical [116] and is a balancing act between restoration of respiration and provocation of withdrawal symptoms or premature awakening. All cases, whether iatrogenic in-hospital or self-inflicted overdoses in the community, are unique when it comes to dosing and being dependent on the pharmacological characteristics of actual opioid (the dose), as well as the constitution of the victim [4]. Moreover, there is significant intra- and interindividual variability, and variability between studies even in healthy volunteers in pharmacokinetic studies for all administration forms, which supports the need for individual titration. Consequently, bystanders should have at least a two-dose offer. The aim of naloxone treatment is to achieve adequate ventilation instead of awakening the victim, a consideration that is particularly important as opioid overdoses are often combined with sedatives, such as benzodiazepines, also depressing respiration themselves, on which naloxone has no action [157].
Naloxone is eliminated according to a three-compartment model. Its clearance exceeds the liver perfusion by far; however, the site of extrahepatic biotransformation is unknown. Naloxone has no active metabolites. Bearing in mind the combination of high clearance and a modest volume of distribution, its terminal half-life of 1–2 h is not surprising. However, these data are from young healthy volunteers, and pharmacokinetic studies in the relevant special populations are still disappointingly scarce.
Intravenous administration is the most controllable administration form, allowing for repeated doses, especially when also controlling the victim’s ventilation, e.g., in the hospital. This controllability allows for intravenous infusion in severe intoxications and longer-duration interventions, for instance, by methadone and carfentanil. With ingestion of large doses by children, intravenous infusion is often an important tool.
Intramuscular administration with its relatively rapid uptake is common in prehospital care. The risk for adverse effects with similar intramuscular naloxone doses are lower than for intravenous administration [158]. Duration of action is longer when naloxone is given intramuscularly than intravenously.
The nasal route is increasingly frequently used in take-home naloxone (THN). This route is characterized by somewhat slower onset of action than by the intramuscular route [73]; however, this is partly compensated by the shorter time required to apply as nasal spray. Unlike needle-based alternatives, the risk for blood contamination is small. Also, the duration of action maybe longer (Fig. 9) with nasal naloxone than for the needle alternatives, possibly due to a potential depot function of the nose. Since laypeople may not be able to control ventilation, it is of vital importance that they understand that THN provides interim treatment only and that an ambulance must also be called immediately, otherwise lives may be lost. Laypeople should, in general, have two doses available, and it seems reasonable to assume that dividing the dose administration, even with time interval between the two nasal administrations, may reduce the incidence of provocation of withdrawals in victims. Laypeople have been found to be able to titrate naloxone dose according to effect [159].
Reviews by Algera et al. [89] and Kim et al. [40] show that naloxone reversal is dependent on the several factors: the dose of naloxone administered and its elimination half-life, the koff value of the opioid that requires reversal, naloxone’s potency in dissociating the opioid from the MOR (dependent on KD). Naloxone has a short half-life, and its brain effect diminishes rapidly. Increase in naloxone exposure with alternative dosing modalities (such as using infusions or intranasal dosing) will increase duration of reversal but will not affect the beginning of action. Considering pharmacodynamic determinants, modeling studies have shown that naloxone’s potency differs depending on the opioid that is reversed. Finally, naloxone reversal is slowed down when opioids with high t1/2koff, such as buprenorphine, are present and may even be impossible by a bolus dosing.
Even dosing procedures for naloxone in the hospital are controversial [160]. For THN, widely different opinions exist about the choice of an initial dose by laypersons. The highest dosing for intranasal use is found in the USA by means of Narcan 4 mg (× 2) and Kloxxado 8 mg (×1), compared with 1.4 mg by Ventizolve and 2 mg (× 2) for Nyxoid in Europe and Australia. There are obvious indications that fentanyl overdoses may require higher naloxone doses than for heroin, not least from modeling studies [36, 70], but these should not replace controlled trials. The lack of products with lower nasal doses in the USA may have led to a higher dosing level on an “administrative” basis. The driving force for the higher dosing is a relevant fear of more severe intoxications by the potent synthetic opioids, such as fentanyl, combined with a real fear that laypersons might consider their treatment as a complete treatment (i.e., not as an interim action) and might consequently not call for EMS. These fears are reasonable, the potency of some of these illicitly manufactured drugs may certainly cause more unintended, severe intoxications than usually seen with heroin. However, there are also calls for moderation regarding naloxone dosing [132, 144, 160]. Although the pharmacological properties of fentanyl differ from those of heroin, not least with regard to risk of muscle rigidity, one may question if this explains the dramatic increase in the USA. Notably, not only the dosing of naloxone for fentanyl/ultrapotent opioids increased after 2015 but also the naloxone dose for heroin overdoses increased from 0.8 to 2.0 mg, a strong indication that “administrative” factors are important [142]. While effective doses of naloxone are well known for heroin, similar quality of evidence is lacking for fentanyl. The absence of controlled clinical trials in a synthetic opioid overdose is striking and limits our understanding and ability to respond optimally [142].
There is thus need for more knowledge. However, studies in real patients are very demanding, both logistically and research ethically. Overdoses takes place almost everywhere, and it is likely that relevant studies under controlled conditions can only take place in the ambulance services or at safe injection facilities [73, 154]. When such studies have been conducted, the challenge of informed consent was handled adequately. We urge the FDA and the pharmaceutical companies with products in the overdose reversal field to conduct similar studies in US cohorts and internationally. Finally, it should also be ethically possible to conduct studies on naloxone disposition in which PUWOs are included [10].
12 Conclusions
Naloxone is an essential and safe medicine, which is invaluable in management of opioid overdose. Its effect depends on the pharmacological characteristics of the actual opioid and the constitution of the victim. When possible, naloxone should be given in divided doses according to its effect on respiration to avoid withdrawal, not least since sedatives may also be a part of the overdose. Bystanders should have been preprovided with at least two doses. Bystander treatment is an interim and an ambulance must always be called. Dosing of naloxone is well known in heroin overdose, but a safe initial dose in fentanyl overdose is not yet established, and there is so far no evidence from controlled clinical trials to guide decisions on optimal naloxone dose. This “undone science” with regard to naloxone needs urgent attention so that science can guide evolving clinical practice and important regulatory decisions.
References
Garfield E. The 1982 John Scott Award goes to Jack Fishman and Harold Blumberg for synthesis and investigations on naloxone. Essay Inf Sci. 1983;6:121–30.
Foldes FF, Duncalf D, Kuwabara S. The respiratory, circulatory, and narcotic antagonistic effects of nalorphine, levallorphan, and naloxone in anaesthetized subjects. Can Anaesth Soc J. 1969;16(2):151–61.
Foldes FF, Lunn JN, Moore J, Brown IM. N-Allylnoroxy-morphone: a new potent narcotic antagonist. Am J Med Sci. 1963;245:23–30.
Skolnick P. Treatment of overdose in the synthetic opioid era. Pharmacol Ther. 2022;233: 108019.
Strang J, McDonald R, Campbell G, Degenhardt L, Nielsen S, Ritter A, et al. Take-home naloxone for the emergency interim management of opioid overdose: the public health application of an emergency medicine. Drugs. 2019;79(13):1395–418.
Stam NC, Cogger S, Schumann JL, Weeks A, Roxburgh A, Dietze PM, et al. The onset and severity of acute opioid toxicity in heroin overdose cases: a retrospective cohort study at a supervised injecting facility in Melbourne, Australia. Clin Toxicol. 2022;6:1–8.
Strang J, Darke S, Hall W, Farrell M, Ali R. Heroin overdose: the case for take-home naloxone. BMJ. 1996;312(7044):1435–6.
Strang J. Take-home naloxone and the prevention of deaths from heroin overdose: pursuing strong science, fuller understanding, greater impact. Eur Addict Res. 2022;28(3):161–75.
Dale O, Hjortkjaer R, Kharasch ED. Nasal administration of opioids for pain management in adults. Acta Anaesthesiol Scand. 2002;46(7):759–70.
Dale O. Ethical issues and stakeholders matter. Addiction. 2016;111(4):587–9.
Strang J, McDonald R, Tas B, Day E. Clinical provision of improvised nasal naloxone without experimental testing and without regulatory approval: imaginative shortcut or dangerous bypass of essential safety procedures? Addiction. 2016;111(4):574–82.
Krieter PA, Chiang CN, Gyaw S, McCann DJ. Comparison of the pharmacokinetic properties of naloxone following the use of FDA-approved intranasal and intramuscular devices versus a common improvised nasal naloxone device. J Clin Pharmacol. 2019;59(8):1078–84.
Olofsen E, van Dorp E, Teppema L, Aarts L, Smith TW, Dahan A, et al. Naloxone reversal of morphine- and morphine-6-glucuronide-induced respiratory depression in healthy volunteers: a mechanism-based pharmacokinetic-pharmacodynamic modeling study. Anesthesiology. 2010;112(6):1417–27.
van Dorp E, Yassen A, Sarton E, Romberg R, Olofsen E, Teppema L, et al. Naloxone reversal of buprenorphine-induced respiratory depression. Anesthesiology. 2006;105(1):51–7.
Yassen A, Olofsen E, Romberg R, Sarton E, Teppema L, Danhof M, et al. Mechanism-based PK/PD modeling of the respiratory depressant effect of buprenorphine and fentanyl in healthy volunteers. Clin Pharmacol Ther. 2007;81(1):50–8.
Strang J, McDonald R, Alqurshi A, Royall P, Taylor D, Forbes B. Naloxone without the needle—systematic review of candidate routes for non-injectable naloxone for opioid overdose reversal. Drug Alcohol Depend. 2016;163:16–23.
Krieter P, Chiang N, Gyaw S, Skolnick P, Crystal R, Keegan F, et al. Pharmacokinetic properties and human use characteristics of an FDA-approved intranasal naloxone product for the treatment of opioid overdose. J Clin Pharmacol. 2016;56(10):1243–53.
McDonald R, Danielsson Glende O, Dale O, Strang J. International patent applications for non-injectable naloxone for opioid overdose reversal: exploratory search and retrieve analysis of the Patent Scope database. Drug Alcohol Rev. 2018;37(2):205–15.
Dale O. Pharmacokinetic considerations for community-based dosing of nasal naloxone in opioid overdose in adults. Expert Opin Drug Metab Toxicol. 2022;18(3):203–17.
Lewenstein M, Fishman J, inventors; Moprhine derivate. USA patent 3254088; 1966.
Blumberg H, Dayton HB, George M, Rapaport DN. N-allylnoroxynorphone; a potent narcotic antagonist. Fedn Proc Fedn Am Soc. 1961;20:311.
Blumberg H, Dayton HB, Wolf PS. Counteraction of narcotic antagonist analgesics by the narcotic antagonist naloxone. Proc Soc Exp Biol Med. 1966;123(3):755–8.
WHO. The use of essential drugs. Technical report series. [cited 19th December 2022] 1983. https://www.who.int/publications/i/item/9241206853.
McNicholas LF, Martin WR. New and experimental therapeutic roles for naloxone and related opioid antagonists. Drugs. 1984;27(1):81–93.
Sutar R, Sahu S. Pharmacotherapy for dissociative disorders: a systematic review. Psychiatry Res. 2019;281: 112529.
Valbrun LP, Zvonarev V. The opioid system and food intake: use of opiate antagonists in treatment of binge eating disorder and abnormal eating behavior. J Clin Med Res. 2020;12(2):41–63.
Springborg AD, Jensen EK, Kreilgaard M, Petersen MA, Papathanasiou T, Lund TM, et al. High-dose naloxone: effects by late administration on pain and hyperalgesia following a human heat injury model. A randomized, double-blind, placebo-controlled, crossover trial with an enriched enrollment design. PLoS ONE. 2020;15(11): e0242169.
Morlion BJ, Mueller-Lissner SA, Vellucci R, Leppert W, Coffin BC, Dickerson SL, et al. Oral prolonged-release oxycodone/naloxone for managing pain and opioid-induced constipation: a review of the evidence. Pain Pract. 2018;18(5):647–65.
Colucci SV, Perrino PJ, Shram M, Bartlett C, Wang Y, Harris SC. Abuse potential of intravenous oxycodone/naloxone solution in nondependent recreational drug users. Clin Drug Investig. 2014;34(6):421–9.
Walsh SL, Nuzzo PA, Babalonis S, Casselton V, Lofwall MR. Intranasal buprenorphine alone and in combination with naloxone: abuse liability and reinforcing efficacy in physically dependent opioid abusers. Drug Alcohol Depend. 2016;162:190–8.
Naloxone. PubChem. https://pubchem.ncbi.nlm.nih.gov/compound/Naloxone#section=3D-Conformer. Accessed Oct 26 2022.
Hansch C, Hoekman D, Leo A, Zhang L, Li P. The expanding role of quantitative structure-activity relationships (QSAR) in toxicology. Toxicol Lett. 1995;79:45–53. https://doi.org/10.1016/0378-4274(95)03356-p.
Balyan R, Hahn D, Huang H, Chidambaran V. Pharmacokinetic and pharmacodynamic considerations in developing a response to the opioid epidemic. Expert Opin Drug Metab Toxicol. 2020;16(2):125–41.
Tam SW. (+)-[3H]SKF 10,047, (+)-[3H]ethylketocyclazocine, mu, kappa, delta and phencyclidine binding sites in guinea pig brain membranes. Eur J Pharmacol. 1985;109(1):33–41.
Kanemasa T, Koike K, Takase K, Arai T, Nakamura A, Morioka Y, et al. Pharmacological profile of naldemedine, a peripherally acting μ-opioid receptor antagonist: comparison with naloxone and naloxegol. J Pharmacol Exp Ther. 2020;373(3):438–44.
Moss RB, Pryor MM, Baillie R, Kudrycki K, Friedrich C, Reed M, et al. Higher naloxone dosing in a quantitative systems pharmacology model that predicts naloxone-fentanyl competition at the opioid mu receptor level. PLoS ONE. 2020;15(6): e0234683.
Cassel JA, Daubert JD, DeHaven RN. [(3)H]Alvimopan binding to the micro opioid receptor: comparative binding kinetics of opioid antagonists. Eur J Pharmacol. 2005;520(1–3):29–36.
Melichar JK, Nutt DJ, Malizia AL. Naloxone displacement at opioid receptor sites measured in vivo in the human brain. Eur J Pharmacol. 2003;459(2–3):217–9.
Glass PS, Jhaveri RM, Smith LR. Comparison of potency and duration of action of nalmefene and naloxone. Anesth Analg. 1994;78(3):536–41.
Kim HK, Nelson LS. Reducing the harm of opioid overdose with the safe use of naloxone: a pharmacologic review. Expert Opin Drug Saf. 2015;14(7):1137–46.
Trøstheim M, Eikemo M, Haaker J, Frost JJ, Leknes S. Opioid antagonism in humans: a primer on optimal dose and timing for central mu-opioid receptor blockade. Neuropsychopharmacology. 2023;48(2):299–307.
Frost JJ, Wagner HN Jr, Dannals RF, Ravert HT, Links JM, Wilson AA, et al. Imaging opiate receptors in the human brain by positron tomography. J Comput Assist Tomogr. 1985;9(2):231–6.
Sadzot B, Mayberg HS, Frost JJ. Imaging opiate receptors in the human brain with positron emission tomography. Potential applications for drug addiction research. Acta Psychiatr Belg. 1990;90(1):9–19.
Kim S, Wagner HN Jr, Villemagne VL, Kao PF, Dannals RF, Ravert HT, et al. Longer occupancy of opioid receptors by nalmefene compared to naloxone as measured in vivo by a dual-detector system. J Nucl Med. 1997;38(11):1726–31.
Villemagne VL, Frost JJ, Dannals RF, Lever JR, Tanada S, Natarajan TK, et al. Comparison of [11C]diprenorphine and [11C]carfentanil in vivo binding to opiate receptors in man using a dual detector system. Eur J Pharmacol. 1994;257(1–2):195–7.
Johansson J, Hirvonen J, Lovro Z, Ekblad L, Kaasinen V, Rajasilta O, et al. Intranasal naloxone rapidly occupies brain mu-opioid receptors in human subjects. Neuropsychopharmacology. 2019;44(9):1667–73.
Bice AN, Wagner HN Jr, Frost JJ, Natarajan TK, Lee MC, Wong DF, et al. Simplified detection system for neuroreceptor studies in the human brain. J Nucl Med. 1986;27(2):184–91.
Tylleskar I, Skulberg AK, Nilsen T, Skarra S, Jansook P, Dale O. Pharmacokinetics of a new, nasal formulation of naloxone. Eur J Clin Pharmacol. 2017;73(5):555–62.
Zuurmond WW, Meert TF, Noorduin H. Partial versus full agonists for opioid-mediated analgesia—focus on fentanyl and buprenorphine. Acta Anaesthesiol Belg. 2002;53(3):193–201.
Fishman J, Roffwarg H, Hellman L. Disposition of naloxone-7,8,3H in normal and narcotic-dependent men. J Pharmacol Exp Ther. 1973;187(3):575–80.
Fujimoto JM. Isolation of naloxone-3-glucuronide from human urine. Proc Soc Exp Biol Med. 1970;133(1):317–9.
Weinstein SH, Pfeffer M, Schor JM. Metabolism and pharmacokinetics of naloxone. Adv Biochem Psychopharmacol. 1973;8:525–35.
Papathanasiou T, Springborg AD, Kongstad KT, Staerk D, Moller K, Taylor BK, et al. High-dose naloxone, an experimental tool uncovering latent sensitisation: pharmacokinetics in humans. Br J Anaesth. 2019;123(2):e204–14.
Tylleskar I, Skarra S, Skulberg AK, Dale O. The pharmacokinetic interaction between nasally administered naloxone and the opioid remifentanil in human volunteers. Eur J Clin Pharmacol. 2021;77(12):1901–8.
Dale O, Nilsen T, Loftsson T, Hjorth Tonnesen H, Klepstad P, Kaasa S, et al. Intranasal midazolam: a comparison of two delivery devices in human volunteers. J Pharm Pharmacol. 2006;58(10):1311–8.
Smith K, Hopp M, Mundin G, Bond S, Bailey P, Woodward J, et al. Low absolute bioavailability of oral naloxone in healthy subjects. Int J Clin Pharmacol Ther. 2012;50(5):360–7.
Alqurshi A, Kumar Z, McDonald R, Strang J, Buanz A, Ahmed S, et al. Amorphous formulation and in vitro performance testing of instantly disintegrating buccal tablets for the emergency delivery of naloxone. Mol Pharm. 2016;13(5):1688–98.
Berkowitz BA. The relationship of pharmacokinetics to pharmacological activity: morphine, methadone and naloxone. Clin Pharmacokinet. 1976;1(3):219–30.
Ngai SH, Berkowitz BA, Yang JC, Hempstead J, Spector S. Pharmacokinetics of naloxone in rats and in man: basis for its potency and short duration of action. Anesthesiology. 1976;44(5):398–401.
Berkowitz BA, Ngai SH, Hempstead J, Spector S. Disposition of naloxone: use of a new radioimmunoassay. J Pharmacol Exp Ther. 1975;195(3):499–504.
Albeck H, Woodfield S, Kreek MJ. Quantitative and pharmacokinetic analysis of naloxone in plasma using high-performance liquid chromatography with electrochemical detection and solid-phase extraction. J Chromatogr. 1989;488(2):435–45.
Tylleskar I, Skulberg AK, Nilsen T, Skarra S, Dale O. Naloxone nasal spray—bioavailability and absorption pattern in a phase 1 study. Tidsskr Nor Laegeforen. 2019. https://doi.org/10.4045/tidsskr.19.0162.
Tylleskar I, Skulberg AK, Skarra S, Nilsen T, Dale O. Pharmacodynamics and arteriovenous difference of intravenous naloxone in healthy volunteers exposed to remifentanil. Eur J Clin Pharmacol. 2018;74(12):1547–53.
Gufford BT, Ainslie GR, White JR Jr, Layton ME, Padowski JM, Pollack GM, et al. Comparison of a new intranasal naloxone formulation to intramuscular naloxone: results from hypothesis-generating small clinical studies. Clin Transl Sci. 2017;10(5):380–6.
Lewis JM, Klein-Schwartz W, Benson BE, Oderda GM, Takai S. Continuous naloxone infusion in pediatric narcotic overdose. Am J Dis Child. 1984;138(10):944–6.
Gourlay GK, Coulthard K. The role of naloxone infusions in the treatment of overdoses of long half-life narcotic agonists: application to nor-methadone. Br J Clin Pharmacol. 1983;15(2):269–71.
Waldron VD, Klimt CR, Seibel JE. Methadone overdose treated with naloxone infusion. JAMA. 1973;225(1):53.
Bradberry JC, Raebel MA. Continuous infusion of naloxone in the treatment of narcotic overdose. Drug Intell Clin Pharm. 1981;15(12):945–50.
Goldfrank L, Weisman RS, Errick JK, Lo MW. A dosing nomogram for continuous infusion intravenous naloxone. Ann Emerg Med. 1986;15(5):566–70.
Mann J, Samieegohar M, Chaturbedi A, Zirkle J, Han X, Ahmadi SF, et al. Development of a translational model to assess the impact of opioid overdose and naloxone dosing on respiratory depression and cardiac arrest. Clin Pharmacol Ther. 2022;112(5):1020–32.
Takahashi M, Sugiyama K, Hori M, Chiba S, Kusaka K. Naloxone reversal of opioid anesthesia revisited: clinical evaluation and plasma concentration analysis of continuous naloxone infusion after anesthesia with high-dose fentanyl. J Anesth. 2004;18(1):1–8.
Barton ED, Colwell CB, Wolfe T, Fosnocht D, Gravitz C, Bryan T, et al. Efficacy of intranasal naloxone as a needleless alternative for treatment of opioid overdose in the prehospital setting. J Emerg Med. 2005;29(3):265–71.
Skulberg AK, Tylleskar I, Valberg M, Braarud AC, Dale J, Heyerdahl F, et al. Comparison of intranasal and intramuscular naloxone in opioid overdoses managed by ambulance staff: a double-dummy, randomised, controlled trial. Addiction. 2022;117(6):1658–67.
Tsekouras AA, Macheras P. Re-examining naloxone pharmacokinetics after intranasal and intramuscular administration using the finite absorption time concept. Eur J Drug Metab Pharmacokinet. 2023;48(4):455–62.
Robertson TM, Hendey GW, Stroh G, Shalit M. Intranasal naloxone is a viable alternative to intravenous naloxone for prehospital narcotic overdose. Prehosp Emerg Care. 2009;13(4):512–5.
Skulberg AK, Tylleskar I, Nilsen T, Skarra S, Salvesen O, Sand T, et al. Pharmacokinetics and -dynamics of intramuscular and intranasal naloxone: an explorative study in healthy volunteers. Eur J Clin Pharmacol. 2018;74(7):873–83.
Skulberg AK, Asberg A, Khiabani HZ, Rostad H, Tylleskar I, Dale O. Pharmacokinetics of a novel, approved, 1.4-mg intranasal naloxone formulation for reversal of opioid overdose-a randomized controlled trial. Addiction. 2019;114(5):859–67.
Vanky E, Hellmundt L, Bondesson U, Eksborg S, Lundeberg S. Pharmacokinetics after a single dose of naloxone administered as a nasal spray in healthy volunteers. Acta Anaesthesiol Scand. 2017;61(6):636–40.
Malmros Olsson E, Lönnqvist PA, Stiller CO, Eksborg S, Lundeberg S. Rapid systemic uptake of naloxone after intranasal administration in children. Paediatr Anaesth. 2021;31(6):631–6.
Lapidot T, Bouhajib M, Faulknor J, Khan S, Krayz GT, Abrutzky C, et al. A novel faster-acting, dry powder-based, naloxone intranasal formulation for opioid overdose. Pharm Res. 2022;39(5):963–75.
Middleton LS, Nuzzo PA, Lofwall MR, Moody DE, Walsh SL. The pharmacodynamic and pharmacokinetic profile of intranasal crushed buprenorphine and buprenorphine/naloxone tablets in opioid abusers. Addiction. 2011;106(8):1460–73.
Lai D, Pham AT, Nekkar Rao PP, Beazely MA. The effects of heat and freeze-thaw cycling on naloxone stability. Harm Reduct J. 2019;16(1):17.
McDonald R, Lorch U, Woodward J, Bosse B, Dooner H, Mundin G, et al. Pharmacokinetics of concentrated naloxone nasal spray for opioid overdose reversal: phase I healthy volunteer study. Addiction. 2018;113(3):484–93.
Edwards ET, Edwards ES, Davis E, Mulcare M, Wiklund M, Kelley G. Comparative usability study of a novel auto-injector and an intranasal system for naloxone delivery. Pain Ther. 2015;4(1):89–105.
Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model-based drug development. CPT Pharmacomet Syst Pharmacol. 2012;1: e6.
Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model-based drug development-part 2: introduction to pharmacokinetic modeling methods. CPT Pharmacomet Syst Pharmacol. 2013;2: e38.
Upton RN, Mould DR. Basic concepts in population modeling, simulation, and model-based drug development: part 3-introduction to pharmacodynamic modeling methods. CPT Pharmacomet Syst Pharmacol. 2014;3: e88.
Dowling J, Isbister GK, Kirkpatrick CM, Naidoo D, Graudins A. Population pharmacokinetics of intravenous, intramuscular, and intranasal naloxone in human volunteers. Ther Drug Monit. 2008;30(4):490–6.
Algera MH, Kamp J, van der Schrier R, van Velzen M, Niesters M, Aarts L, et al. Opioid-induced respiratory depression in humans: a review of pharmacokinetic-pharmacodynamic modelling of reversal. Br J Anaesth. 2019;122(6):e168–79.
Olofsen E, Mooren R, van Dorp E, Aarts L, Smith T, den Hartigh J, et al. Arterial and venous pharmacokinetics of morphine-6-glucuronide and impact of sample site on pharmacodynamic parameter estimates. Anesth Analg. 2010;111(3):626–32.
Loimer N, Hofmann P, Chaudhry HR. Nasal administration of naloxone for detection of opiate dependence. J Psychiatr Res. 1992;26(1):39–43.
Ravnborg M, Jensen FM, Jensen NH, Holk IK. Pupillary diameter and ventilatory CO2 sensitivity after epidural morphine and buprenorphine in volunteers. Anesth Analg. 1987;66(9):847–51.
Friedman MS, Manini AF. Validation of criteria to guide prehospital naloxone administration for drug-related altered mental status. J Med Toxicol. 2016;12(3):270–5.
Loimer N, Hofmann P, Chaudhry HR. Nasal administration of naloxone is as effective as the intravenous route in opiate addicts. Int J Addict. 1994;29(6):819–27.
Scott JC, Ponganis KV, Stanski DR. EEG quantitation of narcotic effect: the comparative pharmacodynamics of fentanyl and alfentanil. Anesthesiology. 1985;62(3):234–41.
Rook EJ, Huitema AD, van den Brink W, van Ree JM, Beijnen JH. Pharmacokinetics and pharmacokinetic variability of heroin and its metabolites: review of the literature. Curr Clin Pharmacol. 2006;1(1):109–18.
Gottås A, Øiestad EL, Boix F, Vindenes V, Ripel Å, Thaulow CH, et al. Levels of heroin and its metabolites in blood and brain extracellular fluid after i.v. heroin administration to freely moving rats. Br J Pharmacol. 2013;170(3):546–56.
Kiyatkin EA. Respiratory depression and brain hypoxia induced by opioid drugs: morphine, oxycodone, heroin, and fentanyl. Neuropharmacology. 2019;151:219–26.
Darke S, Duflou J. The toxicology of heroin-related death: estimating survival times. Addiction. 2016;111(9):1607–13.
Poklis J, Poklis A, Wolf C, Mainland M, Hair L, Devers K, et al. Postmortem tissue distribution of acetyl fentanyl, fentanyl and their respective nor-metabolites analyzed by ultrahigh performance liquid chromatography with tandem mass spectrometry. Forensic Sci Int. 2015;257:435–41.
Burns G, DeRienz RT, Baker DD, Casavant M, Spiller HA. Could chest wall rigidity be a factor in rapid death from illicit fentanyl abuse? Clin Toxicol. 2016;54(5):420–3.
Kelly E, Sutcliffe K, Cavallo D, Ramos-Gonzalez N, Alhosan N, Henderson G. The anomalous pharmacology of fentanyl. Br J Pharmacol. 2021;180:797–812.
Sutcliffe KJ, Corey RA, Alhosan N, Cavallo D, Groom S, Santiago M, et al. Interaction with the lipid membrane influences fentanyl pharmacology. Adv Drug Alcohol Res. 2022. https://doi.org/10.3389/adar.2022.10280.
Hill R, Santhakumar R, Dewey W, Kelly E, Henderson G. Fentanyl depression of respiration: comparison with heroin and morphine. Br J Pharmacol. 2020;177(2):254–66.
Coruh B, Tonelli MR, Park DR. Fentanyl-induced chest wall rigidity. Chest. 2013;143(4):1145–6.
Miner NB, Schutzer WE, Zarnegarnia Y, Janowsky A, Torralva R. Fentanyl causes naloxone-resistant vocal cord closure: a platform for testing opioid overdose treatments. Drug Alcohol Depend. 2021;227: 108974.
Torralva R, Janowsky A. Noradrenergic mechanisms in fentanyl-mediated rapid death explain failure of naloxone in the opioid crisis. J Pharmacol Exp Ther. 2019;371(2):453–75.
Saari T, Tylleskär I, Skulberg A, Dale O. Population pharmacokinetic analysis of extravascular naloxone administration in healthy volunteers. Anesthesiology. 2024.
World Health Organization, Management of Substance Abuse Team. Community management of opioid overdose. Geneva: World Health Organization; 2014.
Tylleskar I, Gjersing L, Bjornsen LP, Braarud AC, Heyerdahl F, Dale O, et al. Prehospital naloxone administration—what influences choice of dose and route of administration? BMC Emerg Med. 2020;20(1):71.
Neale J, Strang J. Naloxone—does over-antagonism matter? Evidence of iatrogenic harm after emergency treatment of heroin/opioid overdose. Addiction. 2015;110(10):1644–52.
Buajordet I, Naess AC, Jacobsen D, Brors O. Adverse events after naloxone treatment of episodes of suspected acute opioid overdose. Eur J Emerg Med. 2004;11(1):19–23.
Krotulski AJ, Chapman BP, Marks SJ, Ontiveros ST, Devin-Holcombe K, Fogarty MF, et al. Sentanyl: a comparison of blood fentanyl concentrations and naloxone dosing after non-fatal overdose. Clin Toxicol. 2022;60(2):197–204.
Lee D, Chronister CW, Broussard WA, Utley-Bobak SR, Schultz DL, Vega RS, et al. Illicit fentanyl-related fatalities in Florida: toxicological findings. J Anal Toxicol. 2016;40(8):588–94.
Fogarty MF, Papsun DM, Logan BK. Analysis of fentanyl and 18 novel fentanyl analogs and metabolites by lc-ms-ms, and report of fatalities associated with methoxyacetylfentanyl and cyclopropylfentanyl. J Anal Toxicol. 2018;42(9):592–604.
Boyer EW. Management of opioid analgesic overdose. N Engl J Med. 2012;367(2):146–55.
Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology—drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349(12):1157–67.
Moe-Byrne T, Brown JVE, McGuire W. Naloxone for opioid-exposed newborn infants. Cochrane Database Syst Rev. 2018;10(10):cd003483.
Brennscheidt U, Brunnmüller U, Proppe D, Thomann P, Seiler KU. Pharmacokinetics of tilidine and naloxone in patients with severe hepatic impairment. Arzneimittelforschung. 2007;57(2):106–11.
Cohen MR, Cohen RM, Pickar D, Murphy DL, Bunney WE Jr. Physiological effects of high dose naloxone administration to normal adults. Life Sci. 1982;30(23):2025–31.
Cohen MR, Cohen RM, Pickar D, Weingartner H, Murphy DL, Bunney WE Jr. Behavioural effects after high dose naloxone administration to normal volunteers. Lancet. 1981;2(8255):1110.
McDonald R, Strang J. Are take-home naloxone programmes effective? Systematic review utilizing application of the Bradford Hill criteria. Addiction. 2016;111(7):1177–87.
Razaghizad A, Windle SB, Filion KB, Gore G, Kudrina I, Paraskevopoulos E, et al. The effect of overdose education and naloxone distribution: an umbrella review of systematic reviews. Am J Public Health. 2021;111(8):e1–12.
Clarke SF, Dargan PI, Jones AL. Naloxone in opioid poisoning: walking the tightrope. Emerg Med J. 2005;22(9):612–6.
Wermeling DP. Review of naloxone safety for opioid overdose: practical considerations for new technology and expanded public access. Ther Adv Drug Saf. 2015;6(1):20–31.
Strang J, Neale J, McDonald R, Kalk N. Toxicity: exploring and expanding the concept. Addiction. 2018;113(4):592–4.
Johnson C, Mayer P, Grosz D. Pulmonary edema following naloxone administration in a healthy orthopedic patient. J Clin Anesth. 1995;7(4):356–7.
Prough DS, Roy R, Bumgarner J, Shannon G. Acute pulmonary edema in healthy teenagers following conservative doses of intravenous naloxone. Anesthesiology. 1984;60(5):485–6.
Grout S, Dave M, Lefort R. Naloxone-associated pulmonary edema in a 3-year-old with opioid overdose. J Am Coll Emerg Phys Open. 2022;3(3): e12740.
Kummer RL, Kempainen RR, Olives TD, Leatherman JW, Prekker ME. Naloxone-associated pulmonary edema following recreational opioid overdose. Am J Emerg Med. 2022;53:41–3.
Elkattawy S, Alyacoub R, Ejikeme C, Noori MAM, Remolina C. Naloxone induced pulmonary edema. J Commun Hosp Intern Med Perspect. 2021;11(1):139–42.
Farkas A, Lynch MJ, Westover R, Giles J, Siripong N, Nalatwad A, et al. Pulmonary complications of opioid overdose treated with naloxone. Ann Emerg Med. 2020;75(1):39–48.
Kienbaum P, Thürauf N, Michel MC, Scherbaum N, Gastpar M, Peters J. Profound increase in epinephrine concentration in plasma and cardiovascular stimulation after mu-opioid receptor blockade in opioid-addicted patients during barbiturate-induced anesthesia for acute detoxification. Anesthesiology. 1998;88(5):1154–61.
Tigerstedt I, Tammisto T. Effect of naloxone reversal on CO2 output, oxygen uptake and cardiac index during recovery from fentanyl-supplemented anaesthesia. Acta Anaesthesiol Scand. 1978;22(2):158–66.
Rzasa Lynn R, Galinkin JL. Naloxone dosage for opioid reversal: current evidence and clinical implications. Ther Adv Drug Saf. 2018;9(1):63–88.
Gaddis GM, Watson WA. Naloxone-associated patient violence: an overlooked toxicity? Ann Pharmacother. 1992;26(2):196–8.
Avetian GK, Fiuty P, Mazzella S, Koppa D, Heye V, Hebbar P. Use of naloxone nasal spray 4 mg in the community setting: a survey of use by community organizations. Curr Med Res Opin. 2018;34(4):573–6.
Thompson J, Salter J, Bui P, Herbert L, Mills D, Wagner D, et al. Safety, efficacy, and cost of 0.4-mg versus 2-mg intranasal naloxone for treatment of prehospital opioid overdose. Ann Pharmacother. 2022;56(3):285–9.
Moustaqim-Barrette A, Papamihali K, Williams S, Ferguson M, Moe J, Purssell R, et al. Adverse events related to bystander naloxone administration in cases of suspected opioid overdose in British Columbia: an observational study. PLoS ONE. 2021;16(10): e0259126.
Isoardi KZ, Parker L, Harris K, Rashford S, Isbister GK. Acute opioid withdrawal following intramuscular administration of naloxone 1.6 mg: a prospective out-of-hospital series. Ann Emerg Med. 2022;80(2):120–6.
Wightman RS, Nelson LS. Naloxone dosing in the era of fentanyl: the path widens by traveling down it. Ann Emerg Med. 2022;80(2):127–9.
Moe J, Godwin J, Purssell R, O’Sullivan F, Hau JP, Purssell E, et al. Naloxone dosing in the era of ultra-potent opioid overdoses: a systematic review. CJEM. 2020;22(2):178–86.
Gooley B, Weston B, Colella MR, Farkas A. Outcomes of law enforcement officer administered naloxone. Am J Emerg Med. 2022;62:25–9.
Lemen PM, Garrett DP, Thompson E, Aho M, Vasquez C, Park JN. High-dose naloxone formulations are not as essential as we thought. medRxiv. 2023;6:63.
Vilke GM, Sloane C, Smith AM, Chan TC. Assessment for deaths in out-of-hospital heroin overdose patients treated with naloxone who refuse transport. Acad Emerg Med. 2003;10(8):893–6.
Rudolph SS, Jehu G, Nielsen SL, Nielsen K, Siersma V, Rasmussen LS. Prehospital treatment of opioid overdose in Copenhagen—is it safe to discharge on-scene? Resuscitation. 2011;82(11):1414–8.
Wampler DA, Molina DK, McManus J, Laws P, Manifold CA. No deaths associated with patient refusal of transport after naloxone-reversed opioid overdose. Prehosp Emerg Care. 2011;15(3):320–4.
Watson WA, Steele MT, Muelleman RL, Rush MD. Opioid toxicity recurrence after an initial response to naloxone. J Toxicol Clin Toxicol. 1998;36(1–2):11–7.
Boyd JJ, Kuisma MJ, Alaspaa AO, Vuori E, Repo JV, Randell TT. Recurrent opioid toxicity after pre-hospital care of presumed heroin overdose patients. Acta Anaesthesiol Scand. 2006;50(10):1266–70.
Sutter ME, Gerona RR, Davis MT, Roche BM, Colby DK, Chenoweth JA, et al. Fatal fentanyl: one pill can kill. Acad Emerg Med. 2017;24(1):106–13.
Sabzghabaee AM, Eizadi-Mood N, Yaraghi A, Zandifar S. Naloxone therapy in opioid overdose patients: intranasal or intravenous? A randomized clinical trial. Arch Med Sci. 2014;10(2):309–14.
Kelly AM, Kerr D, Dietze P, Patrick I, Walker T, Koutsogiannis Z. Randomised trial of intranasal versus intramuscular naloxone in prehospital treatment for suspected opioid overdose. Med J Aust. 2005;182(1):24–7.
Kerr D, Kelly AM, Dietze P, Jolley D, Barger B. Randomized controlled trial comparing the effectiveness and safety of intranasal and intramuscular naloxone for the treatment of suspected heroin overdose. Addiction. 2009;104(12):2067–74.
Dietze P, Jauncey M, Salmon A, Mohebbi M, Latimer J, van Beek I, et al. Effect of intranasal vs intramuscular naloxone on opioid overdose: a randomized clinical trial. JAMA Netw Open. 2019;2(11): e1914977.
World Health Organization. WHO guidelines approved by the guidelines. Review committee community management of opioid overdose. Geneva: World Health Organization; 2014.
Madah-Amiri D, Clausen T, Lobmaier P. Rapid widespread distribution of intranasal naloxone for overdose prevention. Drug Alcohol Depend. 2017;173:17–23.
Raman R. High-potency benzodiazepine misuse in opioid-dependent patients: use naloxone with care. Emerg Med J. 2023;40(3):224–7.
Skulberg AK, Tylleskar I, Dale O. Naloxone administration—no balance without titration. Addiction. 2022;117:2750–1.
Madah-Amiri D, Skulberg AK, Braarud AC, Dale O, Heyerdahl F, Lobmaier P, et al. Ambulance-attended opioid overdoses: an examination into overdose locations and the role of a safe injection facility. Subst Abus. 2019;40(3):383–8.
Yugar B, McManus K, Ramdin C, Nelson LS, Parris MA. Systematic review of naloxone dosing and adverse events in the emergency department. J Emerg Med. 2023;65(3):e188–98.
Funding
Open access funding provided by NTNU Norwegian University of Science and Technology (incl St. Olavs Hospital - Trondheim University Hospital).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Teijo I. Saari was supported by State funding for university-level health research to Turku University Hospital (#13821 for T.I.S.). This is a non-commercial, investigator-initiated project and has not received funding from the industry.
Conflict of Interest
Teijo I. Saari has no conflicts of interest to disclose. John Strang declares his employer (King’s College London) has received, connected to his work, research grant support, payment of honoraria, consultancy payments, traveling/accommodation from pharmaceutical and technology companies (including, past 3 years, MundiPharma, Camurus, Accord/Molteni, Pneumowave) and product medication supply from Camurus and from Pneumowave, concerning medicinal or technology products potentially applicable in the treatment of addictions and related problems. This includes exploration of novel naloxone formulations as well as possible wearable devices. NTNU—The Norwegian University of Science and Technology has signed a collaboration and licensing agreement with dne pharma to commercialize a nasal spray formulation. dne pharma has received marketing authorization for a naloxone nasal spray (Ventizolve/Respinal) based on this collaboration. The formulation was invented by Dale, and the agreements ensure potential royalties for him through NTNU, and NTNU’s subsidiary Technology Transfer (TTO). The agreements do not restrict NTNU’s opportunity to publish results from its own research on the product. Dale was Principal Investigator (no personal honorarium) for dne pharma on a study of naloxone spray in volunteers and has been reimbursed for project-related journeys between Oslo and Trondheim. Dale has received honoraria and travel support from dne pharma for presentations on marketing events.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
The datasets analyzed for generating Figs. 3 and 4 are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Authors Contribution
OD conceptualized the manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Saari, T.I., Strang, J. & Dale, O. Clinical Pharmacokinetics and Pharmacodynamics of Naloxone. Clin Pharmacokinet 63, 397–422 (2024). https://doi.org/10.1007/s40262-024-01355-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-024-01355-6