
Abstract
Background and Objectives
Encorafenib is a kinase inhibitor indicated for the treatment of patients with unresectable or metastatic melanoma or metastatic colorectal cancer, respectively, with selected BRAF V600 mutations. A clinical drug–drug interaction (DDI) study was designed to evaluate the effect of encorafenib on rosuvastatin, a sensitive substrate of OATP1B1/3 and breast cancer resistance protein (BCRP), and bupropion, a sensitive CYP2B6 substrate. Coproporphyrin I (CP-I), an endogenous substrate for OATP1B1, was measured in a separate study to deconvolute the mechanism of transporter DDI.
Methods
DDI study participants received a single oral dose of rosuvastatin (10 mg) and bupropion (75 mg) on days − 7, 1, and 14 and continuous doses of encorafenib (450 mg QD) and binimetinib (45 mg BID) starting on day 1. The CP-I data were collected from participants in a phase 3 study who received encorafenib (300 mg QD) and cetuximab (400 mg/m2 initial dose, then 250 mg/m2 QW). Pharmacokinetic and pharmacodynamic analysis was performed using noncompartmental and compartmental methods.
Results
Bupropion exposure was not increased, whereas rosuvastatin Cmax and area under the receiver operating characteristic curve (AUC) increased approximately 2.7 and 1.6-fold, respectively, following repeated doses of encorafenib and binimetinib. Increase in CP-I was minimal, suggesting that the primary effect of encorafenib on rosuvastatin is through BCRP. Categorization of statins on the basis of their metabolic and transporter profile suggests pravastatin would have the least potential for interaction when coadministered with encorafenib.
Conclusion
The results from these clinical studies suggest that encorafenib does not cause clinically relevant CYP2B6 induction or inhibition but is an inhibitor of BCRP and may also inhibit OATP1B1/3 to a lesser extent. Based on these results, it may be necessary to consider switching statins or reducing statin dosage accordingly for coadministration with encorafenib.
Clinical Trial Registration
ClinicalTrials.gov NCT03864042, registered 6 March 2019.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Encorafenib is likely an inhibitor of BCRP but may also inhibit OATP1B1/3 to a lesser extent. Coproporphyrin I (CP-I), a validated endogenous substrate for OATP1B1, was utilized to deconvolute the mechanism of transporter drug–drug interaction (DDI). |
There is a lack of a clinically significant DDI with encorafenib and substrates of CYP2B6. |
Encorafenib in combination with binimetinib was well tolerated when administered with a single oral dose of rosuvastatin and bupropion. |
1 Introduction
Encorafenib is a potent and selective, oral, adenosine triphosphate (ATP)-competitive small-molecule inhibitor of BRAF V600-mutant kinase. Encorafenib in combination with binimetinib or cetuximab is indicated for patients with unresectable or metastatic melanoma or metastatic colorectal cancer, respectively, with selected BRAF V600 mutations. In vitro studies indicate that encorafenib is a reversible inhibitor of CYP2B6 (unbound R1 value of 3.1) [1]. Results from an in vitro hepatocyte assay also show that encorafenib has the potential to be an inducer of CYP2B6 at clinically relevant plasma concentrations per current US Food and Drug Administration and International Conference on Harmonization guidance (R3 values < 0.9) [1,2,3]. In vitro studies indicate that encorafenib is a potent inhibitor of organic anion transporting polypeptide (OATP) 1B1/3 and a weak inhibitor of breast cancer resistance protein (BCRP) based on R values ≥ 1.1 [2]. Substrates for OATP1B1/3 and BCRP transporters, such as HMG-CoA-reductase inhibitors (i.e., statins), may result in increased toxicities (e.g., statin-related rhabdomyolysis) when co-administered with inhibitors for these transporters. Thus, in vivo characterization in a clinical study of drug–drug interaction (DDI) as described here was necessary to provide recommendations when encorafenib is co-administered with substrates for these transporters, as well as for CYP2B6.
The recommended dose of encorafenib is 450 mg once daily in combination with binimetinib (45 mg twice daily) for the treatment of metastatic melanoma and 300 mg once daily in combination with cetuximab (400 mg/m2 initial dose, followed by 250 mg/m2 weekly) for the treatment of metastatic colorectal cancer. Encorafenib pharmacokinetics (PK) has been studied in healthy individuals and patients with solid tumors. Encorafenib is rapidly and nearly completely absorbed (estimated bioavailability at least 86%), with peak concentrations achieved within 2 h. Metabolism is primarily through CYP3A4 (83%) and to a lesser extent by CYP2C19 (16%) and CYP2D6 (1%) [4]. Encorafenib exhibits autoinduction of CYP3A4, resulting in the steady-state exposure being approximately 50% lower compared with exposure following a single dose [5]. For this reason, the effect of encorafenib might be different after a single dose than at steady state, which was accounted for in the DDI study design.
One arm of the phase 1 clinical study C4221003 (NCT03864042) was designed to evaluate the effect of repeat doses of encorafenib on single-dose PK of sensitive substrates for OATP1B1/3, BCRP, and CYP2B6. The study was conducted in patients rather than in healthy volunteers owing to its repeat-dose administration design and the risk of secondary skin neoplasms with selective BRAF inhibitors [6]. No significant DDIs between encorafenib and binimetinib have been observed in clinical studies, and clinically important DDIs involving binimetinib as a perpetrator are unlikely based on in vitro studies and evidence of lack of effect of binimetinib on the PK of midazolam in vivo [7]. Therefore, the recommended dose of encorafenib in combination with binimetinib for melanoma was used in this study.
Rosuvastatin is a known sensitive OATP/BCRP substrate that is not metabolized by CYP3A4 and is therefore commonly used for evaluating the effects on these transporter systems. In addition, rosuvastatin is not known to inhibit or induce any cytochromes (CYPs) or UDP-glucuronosyltransferase (UGT) relevant to encorafenib or bupropion metabolism, respectively [8, 9]. Bupropion was used in this study as it is considered a sensitive substrate of CYP2B6 and no interaction with rosuvastatin would be expected [10, 11]. Hydroxybupropion, the major metabolite of bupropion formed though conversion by CYP2B6 [12], was measured to further characterize the effect of encorafenib on CYP2B6. Both rosuvastatin and bupropion are recommended by the US Food and Drug Administration to be used as probe substrates in clinical drug–drug interaction studies [13]. Although rosuvastatin and bupropion have other minor routes of elimination, changes in their PK, if observed in the DDI study, would likely be attributed to encorafenib’s effect on the major pathways of these probe substrates.
To aid in characterization of the effect of encorafenib on OATP1B1, coproporphyrin I (CP-I), an endogenous substrate used to help predict DDIs related to OATP1B1 inhibition, was measured in PK samples collected from patients treated with encorafenib in a separate phase 3 clinical study (NCT02928224) [14]. Maximum fold change of rich CP-I profiles and a population-based compartmental modeling approach were utilized to help deconvolute the effects on OATP1B1 versus BCRP. On the basis of the results from the DDI study and exploratory CP-I analysis of phase 3 data, the various statins that are currently available were categorized according to potential interaction with encorafenib according to their known metabolic and transporter profile.
2 Methods
2.1 Phase 1 Study Participants
This study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Conference on Harmonization Good Clinical Practice Guidelines. In addition, all local regulatory requirements were followed, particularly those affording greater protection to the safety of trial participants.
Male or female participants were eligible for enrollment if they were ≥ 18 years of age; had histologically confirmed diagnosis of locally advanced, unresectable or metastatic cutaneous melanoma American Joint Committee on Cancer (AJCC) stage IIIB, IIIC, or IV, or other BRAF V600-mutant advanced solid tumors, with evidence of measurable or non-measurable lesions as detected by radiological or photographic methods according to guidelines based on Response Evaluation Criteria in Solid Tumors 1.1; had Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 or 1; and had adequate bone marrow, hepatic, and renal function. Use of contraception was necessary for both males and females of childbearing potential. The type of contraception allowed in this study was determined to be effective and acceptable per Clinical Trial Facilitation Group guidance.
Key exclusion criteria included symptomatic brain metastasis, history of reaction to any of the study medications, symptomatic or untreated leptomeningeal disease, history or current evidence of or current risk factors for retinal vein occlusion (e.g., uncontrolled glaucoma or ocular hypertension, history of hyperviscosity or hypercoagulability syndromes), clinically significant cardiac disease, impaired hepatic function as defined by Child-Pugh class B or C, impaired gastrointestinal function or disease which might have significantly altered the absorption of study drugs (e.g., ulcerative diseases, uncontrolled nausea, vomiting, diarrhea, malabsorption syndrome, and small bowel resection), known hypercoagulability risks other than malignancy (e.g., factor V Leiden syndrome), thromboembolic event except catheter-related venous thrombosis ≤ 12 weeks prior to starting study treatment, discontinuation of prior BRAF and/or MEK inhibitor treatment owing to left ventricular dysfunction, pneumonitis/interstitial lung disease, or retinal vein occlusion.
Participants were also excluded if they had used any herbal medications/supplements or any medications or foods that are moderate or strong inhibitors or inducers of CYP3A4/5, or consumed grapefruit, pomegranates, star fruits, Seville oranges, or products containing the juice, or any substrates, inhibitors, or inducers of CYP2B6, or any substrates or inhibitors of BCRP, OATP1B1, or OATP1B3 within 2 weeks prior to the start of encorafenib/binimetinib treatment on day 1 and through DDI phase (day 28).
2.2 Phase 1 Study Design
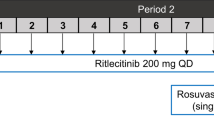
The phase 1 study design is illustrated in Fig. 1. Based on estimates of the within-subject variability for rosuvastatin exposure [18], a total of approximately ten participants were needed to meet the primary study objectives. Participants received a single oral dose of rosuvastatin 10 mg and immediate-release bupropion 75 mg on days − 7, 1, and 14. Encorafenib 450 mg once daily and binimetinib 45 mg twice daily were administered starting on day 1. Blood samples for measurement of plasma concentrations of encorafenib (and its metabolite, LHY746), binimetinib (and its metabolite, AR00426032), rosuvastatin, and bupropion (and its metabolite, hydroxybupropion) were collected at 0, 1, 2, 3, 4, 6, and 8 h post dose on days − 7, 1, and 14. The study drugs were administered in the fasted state, and participants were instructed to refrain from eating for 1 h following encorafenib and binimetinib intake.

Phase 1 study design. PK pharmacokinetics
2.3 Plasma Sample Analysis and PK Parameter Calculation
Human PK plasma samples were analyzed for quantitation of all PK analytes at PPD (Middleton, WI, USA) using validated, sensitive, and specific high-performance liquid chromatography-mass spectrometry/mass spectrometry (HPLC-MS/MS) methods in compliance with laboratory standard operating procedures.
CP-I concentrations were measured retrospectively from remaining PK samples (0–6 h and sparse PK) collected from patients with colorectal cancer who received encorafenib and were analyzed using a validated HPLC-MS/MS. Further details on the study design are reported separately.
PK parameters including, but not limited to, Cmax and AUClast, were calculated for each participant using noncompartmental analysis methods using the WinNonlin software package (Phoenix WinNonlin Professional, version 8.0; Pharsight Corporation, Mountain View, CA, USA) for PK analytes, including rosuvastatin, bupropion, hydroxybupropion, encorafenib and its metabolite LHY746, and binimetinib and its metabolite AR00426032.
2.4 Genotyping
Evaluation of the effect of polymorphisms on the exposure of rosuvastatin or bupropion was an exploratory objective in the study. Polymorphisms for OATP1B1 (SLCO1B1 rs2306283, rs4149056), BCRP (ABCG2 rs2231142, rs72552713), and CYP2B6 (*4,*5,*8,*9,*18,*22,*28) were analyzed on a QuantStudio 12K Flex Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA).
2.5 Safety
Safety was assessed during the DDI phase (day − 7, 1, 14, and 28) and in the post DDI phase every 3–4 weeks until discontinuation of the study drug. Safety monitoring included serious adverse events (SAEs), laboratory profiles (hematology, biochemistry, coagulation, cardiac/muscle enzymes, urinalysis), physical examination (including vital signs, ophthalmic and dermatological examinations), ECOG PS assessment, cardiac profiles (electrocardiogram and multigated acquisition scan or echocardiogram), and concomitant medications or therapies. AEs were classified according to the Medical Dictionary for Regulatory Activities (http://www.meddra.org) classification system, version 22.1, and graded according to the Common Terminology Criteria for Adverse Events, version 4.03.
2.6 Statistical Analysis
For the determination of sample size, the coefficient of variation of the ratio between two rosuvastatin AUClast values for the same participant for rosuvastatin is approximately 47% [18]. Assuming a two-sided significance level of 0.05 and a power of 0.8, approximately ten participants would need to be evaluated to detect a difference of 50% in mean AUClast. Assuming an intrasubject variation of 23% for bupropion AUC0-8, [20] there is an 80% probability with ten participants that a treatment difference will be detected if the true effect size is 33%.
All participants who received at least one dose of any study drug were included in the safety set population. The evaluable PK set included all participants with sufficient concentration data to calculate at least one PK parameter for a probe drug on days − 7, 1, and 14 (days 1 and 14 for binimetinib and encorafenib). Participants who discontinued, missed three or more consecutive doses of encorafenib prior to completion of the last PK sampling on day 14, or required a dose reduction of encorafenib prior to completion of the last PK sampling on day 14 were excluded from the evaluable PK set. In addition, participants who missed any dose of study drugs on any of the PK days or who vomited within 4 h after dosing on any of the PK days were excluded from the evaluable PK set.
An analysis of variance was performed on the natural log transformed Cmax and AUClast of rosuvastatin, bupropion, and hydroxybupropion. The least squares means geometric mean ratio (GMR) and associated 90% confidence interval (CI) for each PK parameter were calculated using the exponentiation of the difference between treatment least squares means from the analyses on the natural log transformed parameters and expressed as a percentage of day 1 relative to day − 7 and day 14 relative to day − 7.
2.7 Analysis of CP-I in Phase 3 Study Participants
CP-I concentrations were measured retrospectively from remaining PK samples (0–6-h profiles and sparse PK) collected from participants with colorectal cancer who received encorafenib in a separate phase 3 study and were analyzed using a validated HPLC-MS/MS. Further details on the study design are reported separately [14, 19]. Two approaches were used to evaluate the potential changes in CP-I concentrations: (1) to determine maximum fold change in the 0–6-h profiles in the safety lead in (SLI) phase participants in the phase 3 clinical study, and (2) to incorporate all of the CP-I patient data from this phase 3 study and use population compartmental modeling for a pooled analysis.
Nonlinear mixed effects models were developed in NONMEM (version 7.5.0) to fit individual participant encorafenib PK and pharmacodynamic (CP-I) profiles. Data formatting, postprocessing, and simulation-based sensitivity analysis were conducted in R Software (v3.6.1). Various PK/PD models were evaluated for their suitability to describe the CP-I concentration–time profiles, including those previously described [15, 17].
Model parameters were then estimated by utilizing encorafenib concentrations predicted on the basis of a previously described population PK model (Pfizer internal data). Encorafenib concentrations were converted to the unbound concentration (nM) on the basis of the molecular weight of encorafenib (540 Da) and the free fraction of drug (0.14) prior to estimation of CP-I model parameters. Final model pharmacodynamic parameters were used for a sensitivity analysis to evaluate the effect of varying the encorafenib in vivo inhibition constant (Ki) against OATP1B1 (range 1–10000 nM).
3 Results
3.1 Phase 1 Study Participants
A total of 12 participants were enrolled in arm 2 and included in the safety set population. All participants received study treatment; however, only ten participants were included in the evaluable PK set for the analysis of the DDI. Two participants had significant dose interruption or reduction, which resulted in their exclusion from the final analysis.
The participants had a mean age of 61.3 years (range 36–82 years) and mean body mass index of 27.92 (range 20.5–34.5); there was an equal number of men and women (Table 1). Within their respective subgroups, most were white, non-smokers, and had an ECOG PS of 0. Of the 12 participants, 10 were stage IV (the others were stage IIIC) and the most frequently reported primary cancer was melanoma, followed by colon, ovarian, pancreatic, and thyroid cancers.
3.2 Pharmacokinetics and Drug–Drug Interactions
Mean (+SD) plasma concentrations of rosuvastatin, bupropion, and hydroxybupropion are presented in Fig. 2. A summary of the plasma PK parameters for rosuvastatin, bupropion, and hydroxybupropion are presented in Table 2, and the statistical comparisons for these analytes are presented in Figs. 3, 4 and 5. The half-life for each analyte could not be calculated for all participants, as some individuals did not have a well-characterized terminal phase (r2 > 0.8).

Mean (+SD) concentration–time profiles for plasma rosuvastatin and bupropion by study day. Solid dots represent the mean analyte concentration at the time point specified. Black lines represent the upper standard deviation of the analyte concentration at each time point. A, C, and E are presented on a linear scale. B, D, and F are presented on a semi-log scale. The lower limit of quantification for the rosuvastatin, bupropion, and hydroxybupropion assays were, 0.1, 0.5, and 1.0 ng/mL, respectively. SD standard deviation

Effect of encorafenib in combination with binimetinib on rosuvastatin. GMRs presented are relative to day − 7. AUClast area under the curve from 0 to the last measurable point; CI confidence interval; Cmax maximum concentration; GMR geometric mean ratio

Effect of encorafenib in combination with binimetinib on bupropion and hydroxybupropion. GMRs presented are relative to day –7. AUClast area under the curve from 0 to the last measurable point; CI confidence interval; Cmax maximum concentration; GMR geometric mean ratio

Observed CP-I concentration data versus time from phase 3 study. Observed CP-I data from both the phase 3 portion and SLI phase. Red dots and lines represent the observed data from the SLI phase and gray dots and lines represent the observed data of the phase 3 portion of the study. CP-I coproporphyrin I, SLI safety lead-in
Encorafenib exposures were consistent with historical studies [5, 21] following administration of encorafenib and binimetinib with geometric mean Cmax values of 6060 and 2940 ng/mL and AUClast values of 25,900 and 13,100 h∙ng/mL on days 1 and 14, respectively. A summary of the plasma PK parameters for encorafenib are presented in Table S1. Mean (+SD) plasma concentrations for encorafenib are presented on linear and semilogarithmic scales in Fig. S1.
3.2.1 Rosuvastatin
Following single oral administrations of 10 mg of rosuvastatin, Cmax of rosuvastatin was characterized by a rapid absorption phase with Tmax of 2 h. Thereafter, plasma concentrations of rosuvastatin declined with half-life of 2–3 h. Rosuvastatin exposures on day − 7 were consistent with historical values, with a mean (CV) of Cmax of 6.98 ng/mL (96.4%) and AUClast of 33.6 h·ng/mL (88.1%) (Fig. 2A, B; Table 2) [22].
Peak exposure to rosuvastatin (Cmax) was approximately 4.3-fold higher on day 1 compared with day − 7 and approximately 2.7-fold higher on day 14 compared with day − 7. Similarly, total exposure to rosuvastatin (AUClast) was approximately 2.8-fold higher on day 1 compared with day − 7 and 1.6-fold higher on day 14 compared with day − 7, with a GMR fold change from 2.79 (90% CI 2.08, 3.74) on day 1 to 1.57 (90% CI 1.17, 2.11) on day 14 (Fig. 3). CIs excluded one in all cases, demonstrating that systemic exposure to rosuvastatin, a sensitive OATP1B1, OATP1B3, and BCRP probe substrate, is significantly higher when rosuvastatin is coadministered with single and repeated doses of encorafenib and binimetinib.
3.2.2 Bupropion
Following single oral administrations of bupropion on days − 7, 1, and 14, mean plasma concentrations of bupropion were characterized by a rapid absorption phase, reaching Tmax within 2 h (Fig. 2C, D). Thereafter, plasma concentrations of bupropion declined with a half-life of 2–3 h. Bupropion exposures on day − 7 were consistent with historical values [23], with mean (CV) Cmax of 94.0 ng/mL (63.9%) and AUClast of 339 h·ng/mL (48.1%; Table 2). Peak and total exposure to bupropion (Cmax and AUClast) was lower on day 1 compared with day − 7, with GMRs ranging from 0.754 to 0.769. A similar decrease in bupropion concentrations was observed on day 14, with GMRs for Cmax and AUClast ranging from 0.736 to 0.755. The decrease in bupropion exposures compared with day − 7 appear to be during the absorption phase whereas bupropion concentration–time profiles for all visits appear to overlap during the elimination phase.
Mean plasma concentrations of hydroxybupropion were characterized by a steady formation phase, with Tmax within 4 h (Fig. 2E, F). Hydroxybupropion Tmax ranged from 3.9 to 7.7 h and terminal phase PK parameters were not estimable. Peak and total exposure to the metabolite hydroxybupropion (Cmax and AUClast) was similar or marginally higher on day 1 compared with day − 7, with GMRs ranging from 0.993 to 1.05 (Table 2). On day 14 the effect of steady-state encorafenib was more pronounced, with GMRs for Cmax and AUClast ranging from 1.42 to 1.48 for hydroxybupropion.
3.3 Genotyping
For the SLCO1B1 gene, which encodes OATP1B1 transporter, there were nine participants in the evaluable PK set with genotypes that resulted in a transporter with normal function. One participant of Asian race had an SLCO1B1 genotype (T521 > C allele), which is characterized as a decreased function phenotype for the OATP1B1 transporter [24]. Evaluation of PK profiles indicated that this participant had a higher rosuvastatin exposure than most participants for both Cmax and AUC on days − 7, 1, and 14 (Fig. S2). A sensitivity analysis excluding the Asian participant with decreased function phenotype was performed, and removing this participant had no impact on the conclusions of the study.
For the ABCG2 gene, which encodes BCRP transporter, one participant was heterozygous for the SNP rs2231142 (Q141K), but all ten participants were predicted to have normal transporter function and therefore no further analysis was conducted.
For CYP2B6, there were two participants who had a genotype that resulted in a phenotype of normal metabolizer (NM), five participants had a genotype that resulted in a phenotype of intermediate metabolizer, and three participants had a phenotype of poor metabolizer (PM) in the evaluable PK set population. Comparison of bupropion and hydroxybupropion median PK profiles for participants in the three groups (normal, intermediate, and poor metabolizers) did not suggest any consistent trend with the known functional relationships for the CYP2B6 genotypes (Fig. S3) [25, 26].
3.4 Safety
During the DDI phase after the first dose of encorafenib plus binimetinib, nine (75.0%) participants experienced all-causality treatment emergent adverse events (TEAEs) of any grade and five (41.7%) experienced all-causality TEAEs of grade ≥ 3. The most frequently reported all-causality TEAEs by primary standard of care were gastrointestinal disorders (66.7%), general disorders and administration site conditions (50.0%), and blood and lymphatic system disorders (41.7%). The all-causality AEs reported in > two participants were nausea (41.7%), anemia (33.3%), diarrhea (25.0%), and fatigue (25.0%). All-causality AEs of grade ≥ 3 were reported in five (41.7%) participants, including anemia, pyrexia, asthenia, increased blood CPK, and decreased ejection fraction, each in one participant.
One participant experienced an SAE of pyrexia, which was considered drug related. Two (16.7%) participants experienced TEAEs leading to study drug discontinuation, retinal detachment, and myalgia, respectively. Both were grade 2 and considered drug related. In addition, two (16.7%) participants had all-causality TEAEs associated with dose reduction, including one participant with a TEAE of grade ≥ 3 (increased blood CPK). Four (33.3%) participants had all-causality TEAEs associated with dose interruption; grade ≥ 3 TEAEs were reported in all four participants.
Overall, encorafenib in combination with binimetinib was generally safe and well tolerated, when administered with a single oral dose of rosuvastatin and bupropion. No new safety findings were observed.
3.5 Exploratory Analysis of CP-I Collected in Phase 3 Study Participants
The clinical CP-I data consisted of 267 concentrations from 68 participants; however, only 11 participants had full profiles collected during an SLI phase of the phase 3 study. The observed CP-I individual profiles are presented in Fig. 5. The observed maximum fold change in CP-I concentrations at C1D1 and C2D1 for participants in the SLI phase were 1.4 and 1.2, respectively.
A population compartmental approach was performed to better characterize the potential impact of encorafenib on CP-I despite the lack of predose CP-I concentration in many participants enrolled in the phase 3 study. A single compartmental model describing CP-I synthesis and degradation rates was able to describe the relatively modest increase in CP-I concentrations over the dosing interval with estimated Ki > 1000 nM, consistent with in vitro results estimating a half maximal inhibitory concentration of approximately 5000 nM. A comparison of the models assessed in this analysis is included in Table S2. The sensitivity analysis was conducted using the final parameters from the single-compartment base model and is presented in Fig. S4. Further details of this compartmental analysis are described separately [27].
4 Discussion
Previously, in vitro studies indicated that encorafenib may act as a reversible inhibitor and potential inducer of CYP2B6 and as a potent inhibitor of OATP1B1/3 and weak inhibitor of BCRP in vivo [2]. In this study, evaluation of the clinical effect of encorafenib in combination with binimetinib on CYP2B6 was conducted using bupropion. Compared with when bupropion was administered on its own, bupropion AUClast was slightly lower (23% and 26%, respectively) following single and repeated doses of encorafenib and binimetinib. Yet, this decrease appeared to be during the absorption phase, whereas bupropion concentration–time profiles for all visits appear to overlap during the elimination phase. Hydroxybupropion AUClast increased by 48% with repeated doses of encorafenib and binimetinib but was not changed following a single dose. This phenomenon of the decreased bupropion absorption when administered with encorafenib could be due to a change in pH of the microenvironment, which could reduce solubility of bupropion.
The results demonstrated both a lack of increase in bupropion exposure and a lack of decrease in hydroxybupropion-to-bupropion ratio after a single dose of encorafenib, indicating that encorafenib does not act as an inhibitor of CYP2B6. Determining the induction effects of CYP2B6 are more difficult as most substrates for CYP2B6 are also substrates for CYP3A4 and there is crosstalk between pregnane X receptor and constitutive androstane receptor in the regulation of these two CYP enzymes. Thus, while bupropion and efavirenz are both recommended clinical CYP2B6 probe substrates for use in DDI studies, caution must be taken when interpreting clinical induction results because of the lack of selectivity of these probes [28]. Here, increases in hydroxybupropion were observed with steady-state encorafenib therapy, but no further decrease in bupropion was noted. By comparison, treatment with carbamazepine, a strong inducer of CYP2B6 and CYP3A4, results in a substantial decrease in bupropion and increase in hydroxybupropion [29]. Taken together, these results suggest that encorafenib is likely not a clinically relevant inducer of CYP2B6.
Evaluation of the clinical effect of encorafenib in combination with binimetinib was also conducted in this study using rosuvastatin, a sensitive probe substrate for OATP1B1, OATP1B3, and BCRP that is not a CYP3A substrate. Rosuvastatin peak and total exposure (Cmax and AUClast) increased approximately 4.3 and 2.8-fold, respectively, in the presence of encorafenib and binimetinib on day 1 and approximately 2.7 and 1.6-fold, respectively, in the presence of encorafenib and binimetinib on day 14. Based on in vitro studies, the increase is attributed to encorafenib but not binimetinib. In addition, the magnitude of the effect was lower at steady state compared with following a single dose, which is a result of lower encorafenib exposure at steady state owing to autoinduction of CYP3A. Based on these results, substrates of OATP1B1, OATB1B3, or BCRP transporters should be coadministered with caution; if coadministered, it is recommended to monitor patients closely for signs and symptoms of increased exposure and consider adjusting the dose of these substrates [4, 30].
Of the potential substrates for these transporters, statins are commonly prescribed and may result in myotoxicity, including myalgia or muscle weakness, with elevations in creatine kinase at high dose levels. Rhabdomyolysis is the most severe AE of statins, which may result in acute renal failure, disseminated intravascular coagulation, or death. The onset of these toxicities after the initiation or intensification of statin treatment has been reported to be around 1 month [31, 32], which suggests that the effects of an interacting drug at steady state are most relevant.
Meta-analyses suggest that the total daily exposure (AUC) rather than Cmax may be most relevant for lowering low-density lipoprotein cholesterol [33]. Likewise, AUClast was proposed for classifying risk of coadministrations with statins [34], which is consistent with the most severe toxicities occurring after repeat dosing. Therefore, the magnitude of change in rosuvastatin exposures, particularly AUClast, observed at steady state is most clinically relevant.
In this study, the observation that the increase in rosuvastatin Cmax was greater than AUClast suggests that the main mechanism is likely intestinal BCRP inhibition rather than OATP1B1 inhibition, considering that concentrations of encorafenib in the gut are likely higher than in the liver, where OATP1B1/3 is highly expressed on hepatocytes [35]. The theoretical maximum fold-change in rosuvastatin exposure based on BCRP alone is 2-fold [36]. The observation of 2.8-fold increase following a single dose, when encorafenib exposures are highest, suggests that inhibition of OATP1B1 may be contributing to the increase in rosuvastatin, and therefore cannot be completely excluded.
CP-I concentrations have been used to help deconvolute the effects on OATP1B1 versus BCRP since clinical studies often have the challenge of using a nonspecific probe substrate (e.g., rosuvastatin) [16]. The plasma CP-I maximum fold-change is approximately 6-fold for a strong inhibitor of OATP1B1 (e.g., rifampin) [37] and approximately 1.8-fold for a mild inhibitor (e.g., JNJ-A) [38]. The observed maximum fold-change in CP-I concentrations from the separate phase 3 study after a single dose in CRC patients receiving encorafenib treatment were consistent with that observed for mild OATP1B1 inhibitors (1.4-fold) [38].
On the basis of the predicted unbound concentrations of encorafenib, the exploratory sensitivity analysis suggests that a Ki between 100 and 1000 nM would be required to achieve peak CP-I concentrations that have been observed following strong OATP1B1 inhibition from compounds, such as rifampin. The Ki from the single compartment model was estimated to be > 1000 nM; taken together, this exploratory analysis suggests that the increase of rosuvastatin exposure following repeat dose administration of encorafenib observed in a dedicated DDI study may be attributed more to inhibition of BCRP than OATP1B1. This result has relevance for prediction of the effects of encorafenib on concomitant medications that are substrates for one or both transporters.
It is well known that all statins are substrates for OATP1B1, while only atorvastatin, lovastatin, and simvastatin are metabolized by CYP3A4 [34]. Experience from PBPK modeling highlights that quantitative prediction of statin DDI is complicated, as is highlighted by limited predictive performance, such as of rosuvastatin [39], simvastatin, or pitavastatin [40]. In general, there is a tendency to underpredict the magnitude for transporter-mediated DDIs, and model parameters may need to be further optimized from their in vitro values to recover the clinical DDI [41]. A qualitative and conservative assessment was performed here in an attempt to categorize risk on the basis of potential of an interaction owing to either transporter or primary metabolic pathway of a particular statin (Table 3).
In larger phase 3 clinical studies, statin use was permitted. There was no evidence of increased muscle related toxicities, and in general, all myalgia was low grade [42]. The highest risk of increased exposure following coadministration with encorafenib is rosuvastatin, fluvastatin, and pitavastatin, given they are not metabolized by CYP3A4 and are substrates for BCRP and OATP1B1. In contrast, lovastatin, atorvastatin, and simvastatin may all be at risk of loss of efficacy owing to CYP3A4 induction observed with encorafenib at steady state. While these three statins are approved over a wide dose range and may be titrated to better manage efficacy, there is still a risk for coadministration if there is an interruption of encorafenib dosing, which could result in a subsequent increase in statin exposure. Pravastatin is least likely to be affected since it does not have CYP3A4 or BCRP contribution, and based on the data presented here, encorafenib has a minimal effect on OATP1B.
5 Conclusions
The results from this phase 1 clinical study and exploratory analysis of CP-I (endogenous substrate of OAT1B1) suggest that encorafenib is likely an inhibitor of BCRP but may also inhibit OATP1B1/3 to a lesser extent. On the basis of these results, coadministration of encorafenib with OATP1B1, OATP1B3, or BCRP substrates can result in increased concentrations of the substrates and may increase the toxicity of these agents. Patients should be monitored closely for signs and symptoms of increased substrate exposure and consider switching statins or lowering the statin dose accordingly when coadministering with encorafenib. The DDI study results also demonstrate a lack of a clinically significant DDI with encorafenib and substrates of CYP2B6.
Overall, encorafenib in combination with binimetinib was well tolerated when administered with a single oral dose of rosuvastatin and bupropion. No new safety findings were observed. In the DDI phase and post-DDI phase, safety data were consistent with the known safety profile of the encorafenib and binimetinib combination.
References
Braftovi (encorafenib) capsules, Full Prescribing Information. <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210496Orig1s000MultidisciplineR.pdf.US. Accessed 3 Aug 2023.
Food and Drug Administration. Immunogenicity testing of therapeutic protein products —developing and validating assays for anti-drug antibody detection Guidance for Industry. 2019. https://www.fda.gov/media/119788/download. Accessed 13 Nov 2023.
European Medicines Agency ICH Guideline M12. Drug Interaction Studies. 2022 https://www.ema.europa.eu/en/documents/scientific-guideline/draft-ich-guideline-m12-drug-interaction-studies-step-2b_en.pdf. Accessed 3 Aug 2023.
BRAFTOVI (encorafenib) capsules, prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/210496s013lbl.pdf. Accessed 3 Aug 2023.
Delord JP, Robert C, Nyakas M, McArthur GA, Kudchakar R, Mahipal A, et al. Phase I dose-escalation and -expansion study of the BRAF inhibitor encorafenib (LGX818) in metastatic BRAF-mutant melanoma. Clin Cancer Res. 2017;23(18):5339–48.
Zimmer L, Hillen U, Livingstone E, Lacouture ME, Busam K, Carvajal RD, et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30(19):2375–83.
MEKTOVI (binimetinib) tablets, Full prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/210498s001lbl.pdf. Accessed 3 Aug 2023.
Neuvonen PJ, Niemi M, Backman JT. Drug interactions with lipid-lowering drugs: mechanisms and clinical relevance. Clin Pharmacol Ther. 2006;80(6):565–81.
Causevic-Ramosevac A, Semiz S. Drug interactions with statins. Acta Pharm. 2013;63(3):277–93.
Han LW, Gao C, Zhang Y, Wang J, Mao Q. Transport of bupropion and its metabolites by the model CHO and HEK293 cell lines. Drug Metab Lett. 2019;13(1):25–36.
Wellbutrin (bupropion hydrochloride) tablets, full prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/018644s043lbl.pdf. Accessed 3 Aug 2023.
Faucette SR, Hawke RL, Lecluyse EL, Shord SS, Yan B, Laethem RM, et al. Validation of bupropion hydroxylation as a selective marker of human cytochrome P450 2B6 catalytic activity. Drug Metab Dispos. 2000;28(10):1222–30.
US Food and Drug Administration. Drug development and drug interactions: table of substrates, inhibitors and inducers. https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers. Accessed 3 Aug 2023.
Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med. 2019;381(17):1632–43.
Barnett S, Ogungbenro K, Menochet K, Shen H, Lai Y, Humphreys WG, et al. Gaining mechanistic insight into coproporphyrin i as endogenous biomarker for OATP1B-mediated drug-drug interactions using population pharmacokinetic modeling and simulation. Clin Pharmacol Ther. 2018;104(3):564–74.
Jones NS, Yoshida K, Salphati L, Kenny JR, Durk MR, Chinn LW. Complex DDI by fenebrutinib and the use of transporter endogenous biomarkers to elucidate the mechanism of DDI. Clin Pharmacol Ther. 2020;107(1):269–77.
Yoshida K, Guo C, Sane R. Quantitative prediction of OATP-Mediated drug-drug interactions with model-based analysis of endogenous biomarker kinetics. CPT Pharmacometrics Syst Pharmacol. 2018;7(8):517–24.
Stopfer P, Giessmann T, Hohl K, Sharma A, Ishiguro N, Taub ME, et al. Pharmacokinetic evaluation of a drug transporter cocktail consisting of digoxin, furosemide, metformin, and rosuvastatin. Clin Pharmacol Ther. 2016;100(3):259–67.
Van Cutsem E, Huijberts S, Grothey A, Yaeger R, Cuyle PJ, Elez E, et al. Binimetinib, encorafenib, and cetuximab triplet therapy for patients with BRAF V600E-mutant metastatic colorectal cancer: safety lead-in results from the phase III BEACON colorectal cancer study. J Clin Oncol. 2019;37(17):1460–9.
Bosilkovska M, Samer CF, Deglon J, Rebsamen M, Staub C, Dayer P, et al. Geneva cocktail for cytochrome p450 and P-glycoprotein activity assessment using dried blood spots. Clin Pharmacol Ther. 2014;96(3):349–59.
Sullivan RJ, Weber J, Patel S, Dummer R, Carlino MS, Tan DSW, et al. A Phase Ib/II study of the BRAF inhibitor encorafenib plus the MEK inhibitor binimetinib in patients with BRAF(V600E/K) -mutant solid tumors. Clin Cancer Res. 2020;26(19):5102–12.
Kanukula R, Salam A, Rodgers A, Kamel B. Pharmacokinetics of rosuvastatin: a systematic review of randomised controlled trials in healthy adults. Clin Pharmacokinet. 2021;60(2):165–75.
Connarn JN, Flowers S, Kelly M, Luo R, Ward KM, Harrington G, et al. Pharmacokinetics and pharmacogenomics of bupropion in three different formulations with different release kinetics in healthy human volunteers. AAPS J. 2017;19(5):1513–22.
Giacomini KM, Balimane PV, Cho SK, Eadon M, Edeki T, Hillgren KM, et al. International Transporter Consortium commentary on clinically important transporter polymorphisms. Clin Pharmacol Ther. 2013;94(1):23–6.
Desta Z, Gammal RS, Gong L, Whirl-Carrillo M, Gaur AH, Sukasem C, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2B6 and efavirenz-containing antiretroviral therapy. Clin Pharmacol Ther. 2019;106(4):726–33.
Eum S, Sayre F, Lee AM, Stingl JC, Bishop JR. Association of CYP2B6 genetic polymorphisms with bupropion and hydroxybupropion exposure: a systematic review and meta-analysis. Pharmacotherapy. 2022;42(1):34–44.
Piscitelli J, Rashid J, Zhang Y, Williams J. Extension to Coproporphyrin I (CP-I) compartmental models to explore unique patterns in cancer patients supporting prediction of OATP1B1 Inhibition. In: 13th Annual American Conference on pharmacometrics. 2022.
Fahmi OA, Shebley M, Palamanda J, Sinz MW, Ramsden D, Einolf HJ, et al. Evaluation of CYP2B6 induction and prediction of clinical drug-drug interactions: considerations from the IQ Consortium Induction Working Group-an industry perspective. Drug Metab Dispos. 2016;44(10):1720–30.
Ketter TA, Jenkins JB, Schroeder DH, Pazzaglia PJ, Marangell LB, George MS, et al. Carbamazepine but not valproate induces bupropion metabolism. J Clin Psychopharmacol. 1995;15(5):327–33.
Encorafenib Summary of Product Characteristics. https://www.ema.europa.eu/en/documents/product-information/braftovi-epar-product-information_en.pdf. Accessed 3 Aug 2023.
Tomaszewski M, Stepien KM, Tomaszewska J, Czuczwar SJ. Statin-induced myopathies. Pharmacol Rep. 2011;63(4):859–66.
Bruckert E, Hayem G, Dejager S, Yau C, Begaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients–the PRIMO study. Cardiovasc Drugs Ther. 2005;19(6):403–14.
Vargo R, Adewale A, Behm MO, Mandema J, Kerbusch T. Prediction of clinical irrelevance of PK differences in atorvastatin using PK/PD models derived from literature-based meta-analyses. Clin Pharmacol Ther. 2014;96(1):101–9.
Kellick KA, Bottorff M, Toth PP. A clinician’s guide to statin drug-drug interactions. J Clin Lipidol. 2014;8(3 Suppl):S30-46.
Mao Q, Unadkat JD. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport–an update. AAPS J. 2015;17(1):65–82.
Elsby R, Martin P, Surry D, Sharma P, Fenner K. Solitary Inhibition of the breast cancer resistance protein efflux transporter results in a clinically significant drug-drug interaction with rosuvastatin by causing up to a 2-fold increase in statin exposure. Drug Metab Dispos. 2016;44(3):398–408.
Lai Y, Mandlekar S, Shen H, Holenarsipur VK, Langish R, Rajanna P, et al. Coproporphyrins in plasma and urine can be appropriate clinical biomarkers to recapitulate drug-drug interactions mediated by organic anion transporting polypeptide inhibition. J Pharmacol Exp Ther. 2016;358(3):397–404.
Kunze A, Ediage EN, Dillen L, Monshouwer M, Snoeys J. clinical investigation of coproporphyrins as sensitive biomarkers to predict mild to strong OATP1B-mediated drug-drug interactions. Clin Pharmacokinet. 2018;57(12):1559–70.
Bowman CM, Ma F, Mao J, Chen Y. Examination of physiologically-based pharmacokinetic models of rosuvastatin. CPT Pharmacometrics Syst Pharmacol. 2021;10(1):5–17.
Duan P, Zhao P, Zhang L. Physiologically based pharmacokinetic (PBPK) modeling of pitavastatin and atorvastatin to predict drug-drug interactions (DDIs). Eur J Drug Metab Pharmacokinet. 2017;42(4):689–705.
Taskar KS, Pilla Reddy V, Burt H, Posada MM, Varma M, Zheng M, et al. Physiologically-based pharmacokinetic models for evaluating membrane transporter mediated drug-drug interactions: current capabilities, case studies, future opportunities, and recommendations. Clin Pharmacol Ther. 2020;107(5):1082–115.
Dummer R, Flaherty KT, Robert C, Arance A, de Groot JWB, Garbe C, et al. COLUMBUS 5-year update: a randomized, open-label, phase iii trial of encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF V600-mutant melanoma. J Clin Oncol. 2022;40(36):4178–88.
Acknowledgements
We thank Kevin Litwiler for his input on design of the study and help with analysis and interpretation of the results. We thank Jonathan Hands and Mark Baird from PPD for noncompartmental analysis. We also thank the patients and their families for their participation in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was sponsored by Pfizer.
Conflict of interest
J.P., M.B.R., L.W., J.G., L.W., Y.Z., K.M., and J.H.W. are employees of Pfizer and hold Pfizer stock. L.D.F. is an employee of Pierre Fabre Medicament.
Availability of Data and Material
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions, and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
Ethics Approval
These studies were conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Conference on Harmonization (ICH) Good Clinical Practice (GCP) Guidelines. In addition, all local regulatory requirements were followed, particularly those affording greater protection to the safety of trial participants. An institutional review board approved the protocols, and all participants gave written informed consent. This trial was registered on ClinicalTrials.gov: NCT03864042.
Consent to Participate
All participants provided written informed consent before undergoing any study procedures.
Author Contributions
JP, MBR, LW, LDF, JG, LW, YZ, KM, and JHW designed and performed the research, analyzed the data, and wrote the manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Piscitelli, J., Reddy, M.B., Wollenberg, L. et al. Clinical Evaluation of the Effect of Encorafenib on Bupropion, Rosuvastatin, and Coproporphyrin I and Considerations for Statin Coadministration. Clin Pharmacokinet 63, 483–496 (2024). https://doi.org/10.1007/s40262-024-01352-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-024-01352-9