
Abstract
Aspirin is one of the most widely used medicines. Although aspirin is commonly utilized for the treatment of several medical conditions, its broadest uptake is for the prevention of recurrent ischemic events in patients with atherosclerotic disease. Its mechanism of action of inhibiting platelet activation via blockade of thromboxane A2 production is unique and is not covered by any other antiplatelet agents. While plain, uncoated, immediate-release aspirin is used in acute settings to help assure rapid absorption, enteric-coated aspirin formulations dominate current chronic use, particularly in North America, including for secondary prevention of cardiovascular events. The unmet needs with current aspirin formulations include a high risk of gastrointestinal (GI) adverse events with plain aspirin, which enteric-coated formulations are not able to overcome, and subject to erratic absorption leading to reduced drug bioavailability. These observations underscore the need for aspirin formulations with a more favorable safety and efficacy profile. Phospholipid-aspirin complex (PL-ASA) is a novel formulation designed to address these needs. It is associated with reduced local acute GI injury compared with plain aspirin, and predictable absorption resulting in more reliable platelet inhibition compared with enteric-coated tablets. This review explores the rationale and pharmacologic profile of PL-ASA intended to address the unmet needs for aspirin therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Although aspirin has been the foundational therapy for the prevention of cardiovascular events for decades, in recent years the role of aspirin in primary prevention or as part of dual antiplatelet therapy has been challenged, largely due to its gastrointestinal (GI) adverse effects. |
Enteric-coated aspirin is the current standard of care, however its clinical performance is limited by erratic absorption and GI injury. |
An unmet need exists for an aspirin formulation that delivers reliable antiplatelet activity while minimizing the risk of GI injury. |
The predictable pharmacokinetic and pharmacodynamic properties of phospholipid-aspirin complex (PL-ASA) compared with enteric-coated tablets, coupled with the reduced risk of acute gastric injury, represent an important advancement to address the unmet needs for aspirin therapy. |
1 Introduction
The first mention of the use of certain natural medicines such as willow bark—later to be found as sources of salicylates—occurred 5000 years ago [1]. In more recent history, salicylate medications have been in use as analgesic, antipyretic, and anti-inflammatory agents since the mid-19th century. In 1897, Felix Hoffman acetylated the phenol group of salicylic acid, and in 1899, as the first commercially viable formulation, this compound was registered as ‘aspirin’ [2]. The dose-dependent inhibition of prostaglandin synthesis by aspirin was first described by Vane in 1971 [3]. Aspirin remains one of the most thoroughly studied and well-characterized drugs in use today [2].
Within the past several decades, aspirin’s antiplatelet effects were recognized and its potential to prevent thrombotic events in patients with cardiovascular (CV) disease or those with multiple risk factors was established in multiple clinical studies. In the late 1990s, the Antithrombotic Trialists Collaboration summarized the clinical evidence and established that doses of aspirin as low as 75–100 mg were sufficient to provide significant protection from CV events [4]. Aspirin was subsequently broadly prescribed in patients with known atherosclerotic cardiovascular disease (ASCVD) to prevent recurrent events, but was also embraced as a strategy to prevent a first event in those without a history of ASCVD. Current guidelines continue to recommend aspirin for patients with ASCVD (secondary prevention) but have retracted previous recommendations for preventive use in people without known ASCVD (primary prevention) based on more recent studies that suggested that the ischemic benefit does not outweigh the risk of bleeding in this setting [5]. Today, aspirin is still a foundational therapy for secondary ASCVD prevention and may also be considered for primary prevention in certain high-risk patient groups [5,6,7,8].
Although aspirin is one of the most widely used medications, the major pitfall of this ubiquitous drug is its associated upper gastrointestinal (GI) adverse effects, ranging from dyspepsia to gastric ulceration. The last innovation in aspirin therapy occurred over 50 years ago with the development of enteric-coated formulations meant to reduce GI adverse effects while still delivering CV protection; however, studies have not consistently shown a protective effect of enteric-coated or buffered aspirin formulations with regard to GI injury [9]. Furthermore, coated aspirins have also been associated with high rates of treatment failure termed ‘aspirin resistance’ due to unpredictable bioavailability secondary to impaired absorption, an issue that may be exaggerated in specific populations such as patients with diabetes or obesity [10]. Therefore, the unmet need remains for new aspirin formulations with reliable bioavailability and reduced risk of gastric effects, for an optimized benefit-to-risk profile.
2 Structure and Mechanism of Action of Aspirin
2.1 Structure
Aspirin is the common name for acetylsalicylic acid (Fig. 1), which, based on its properties, is further classified as a non-steroidal anti-inflammatory drug (NSAID). Following absorption in the GI tract, acetylsalicylic acid rapidly binds to and acetylates a serine residue in its target enzyme family, the cyclooxygenases (COXs). The rapid metabolism of acetylsalicylic acid results in salicylic acid, an active metabolite with residual anti-inflammatory activity [11].

Structure of aspirin.
2.2 Indications and Mechanism of Action
Aspirin is an ‘over-the-counter’ (OTC) medication used for fever and pain relief; however, aspirin is overwhelmingly used in physician-directed CV indications to prevent recurrent CV events. Table 1 shows the indications and uses. Aspirin has been, and continues to be, studied in conditions such as colorectal cancer, pre-eclampsia, and most recently as preventive treatment for thrombotic microangiopathy in hospitalized patients with coronavirus disease 2019 (COVID-19) [12,13,14,15,16].
COX, also known as prostaglandin endoperoxidase synthase, is a membrane-bound glycoprotein with three isoforms (COX-1, -2, and -3) [2]. COX-1 is expressed in various cell types, including endothelial cells and platelets. On COX-1, which produces prostaglandin G2/H2 from arachidonic acid, aspirin acetylates the hydroxyl group of the serine 530 residue in the active binding site of the enzyme. The bulky acetyl group blocks arachidonic acid access to the active binding site of COX-1, leading to irreversible inhibition of the enzyme’s activity. Aspirin is the only COX inhibitor that results in irreversible inhibition.
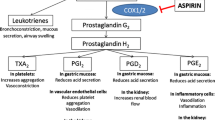
When COX-1 on platelet membranes is acetylated by aspirin, the inhibition lasts for the life of the platelet, i.e. 7–10 days. Blocking COX-1 and the production of prostaglandins also inhibits the eventual production of both thromboxane A2 (TxA2) and prostacyclin (Fig. 2). TxA2 is a platelet activator, thus, aspirin’s utility in prevention of CV events stems from the ability to inhibit platelet aggregation. However, blocking of the production of prostaglandins also produced by COX-1 leads to an increase in the permeability of the gastric barrier, leading to direct, acute injury.

Mechanism of action of aspirin and COX inhibition. Prostaglandins (thromboxane and prostacyclin) are produced from arachidonic acid via the action of the COX-1 and COX-2 enzymes. Aspirin blocks both of these pathways by acetylating the COX enzyme. COX cyclooxygenase, ADP adenosine diphosphate
The clinical benefits of all antiplatelet agents in preventing CV events are directly linked to their ability to deliver effective platelet inhibition. However, it is important to emphasize that aspirin’s mechanism of action is distinct among antiplatelet agents, which is why aspirin is commonly used in combination with platelet P2Y12 receptor inhibitors that work by inhibiting adenosine diphosphate (ADP)-induced platelet activation, but do not inhibit platelet production of thromboxane [17]. Specific platelet function tests have been developed that provide the ability to measure the antiplatelet effects of different classes of antiplatelet agents. Aspirin inhibits the COX-1 enzyme and downregulates the production of thromboxane. Therefore, since the production of thromboxane is COX-1 dependent, measurements of levels of thromboxane are used to extrapolate aspirin effect. Specifically, complete response to aspirin is defined either by demonstration of complete (> 99%) inhibition of thromboxane production or absolute circulating levels < 3.1 ng/mL with the latter having been correlated with recurrent CV events [18]. TxA2 is very unstable and is rapidly converted to thromboxane B2 (TxB2), which is used for measurements of aspirin effect. Additionally, pharmacodynamic (PD) effects can be demonstrated by measuring platelet aggregation, specifically the response of platelet samples (with or without drug) to arachidonic acid as agonist, as measured by light transmittance aggregometry [19].
Aspirin is a potent inhibitor of COX-1 and can potentially achieve complete inhibition with a ‘low dose’ of < 100 mg. However, since about one-seventh of the platelet population is replaced every 24 h, approximately 30% of platelets in circulation will be new and uninhibited within 48 h, which underscores the rationale for daily dosing [20] and why it is essential to have consistent bioavailability and complete platelet response with every aspirin dose. On the other hand, COX-2 is upregulated by inflammatory mediators, resulting in the production of prostaglandins, such as cytokines, which are involved in response to these mediators [21]. Inhibition of COX-2 underlies aspirin’s beneficial effects on pain, fever, and inflammation. In contrast with the COX-1 pathway that only requires low aspirin doses for complete inhibition, much higher doses are required to attain the clinical benefits of COX-2 pathway inhibition [3].
3 Gastrointestinal (GI) Effects
3.1 Molecular and Biophysical Basis of the Barrier Properties of the GI Tract
One of the major drawbacks associated with all NSAIDs, including aspirin, is GI adverse effects, which may span symptoms of gastroesophageal reflux and dyspepsia, to ulceration and GI bleeding [22, 23]. The use of aspirin (and other NSAIDs), due to its vast exposure, is recognized as causing substantial GI effects, which lead to considerable morbidity, patient non-compliance, and increased healthcare costs [24]. Although the overall incidence of GI complications is relatively low, the widespread use of aspirin (and other NSAIDS) results in a ‘considerable clinical problem’.
Aspirin therapy, even in low doses (≤ 325 mg) has long been recognized to frequently result in gastric mucosal damage [25,26,27,28,29], and, specifically, doses as low as 10 mg daily have been observed to reduce gastric mucosal prostaglandin levels [25]. GI effects can manifest in a broad spectrum of clinical symptoms, ranging from dyspepsia to ulceration and bleeding. The clinical significance of ulcers is unquestionable and when they develop they may require GI consultation and specific treatment. Dyspeptic symptoms are much more frequent and usually tend to be self-managed by patients. Frequently, patients take aspirin with food in an effort to minimize bloating and dyspepsia, and may also use antacids or gastric acid-reducing agents. However, patients may also skip doses to avoid dyspepsia and this may be an explanation for the high rates of non-compliance that are reported for aspirin therapy [30]. This is an important concern for physicians because non-adherence with daily aspirin therapy can lead to an increased risk for thrombotic events [30].
Gastric injury is caused by a combination of local and systemic factors. Although the exact contribution of each mechanism is unclear, it appears that local, direct injury by aspirin has an important role in the pathogenesis of gastric damage [31], independent of systemic factors. The gastric mucosa has hydrophobic (non-wettable) properties due to its ability to secrete a surfactant-like phospholipid, which is ‘fortified’ by prostaglandins [32] (Fig. 3). Prostaglandins also inhibit gastric acid secretion [33]. Aspirin and other NSAIDs may reduce surface hydrophobicity by chemically associating with (in a pH-dependent manner) and destabilizing the phospholipid barrier coating the gastric mucosa. Accordingly, NSAIDs can induce changes in the fluidity, permeability, and biomechanical properties of the gastric mucosal cell membranes. The changes make the mucosa less hydrophobic and disrupt the integrity of the protective gel barrier with underlying tissues susceptible to direct injury by gastric acid.

Mechanism of gastric injury. The graphic depicts the gastric epithelial layer, with its normal hydrophobic layer that protects against acid injury. When the phospholipid layer is disrupted, the protective mechanism is compromised, allowing acid diffusion and injury to the epithelial layer. NSAIDs non-steroidal anti-inflammatory drugs
Along with direct injury, there are systemic contributors to gastric damage associated with aspirin (and other NSAID) use. Prostaglandins are important components of the gastric defense mechanism. Systemic prostaglandin depletion by inhibition of the COX enzyme can lead to both qualitative and quantitative decreases in the mucosal barrier function [24]. Prostaglandins also play a role in regulating acid secretion. Therefore, prostaglandin depletion impairs the mucosal barrier and increases acid secretion, which may result in a barrier that is more susceptible to injury and ulceration. Additionally, if a gastric ulcer bleeds, because aspirin interferes with hemostasis (the ability of platelets to aggregate), the bleeding may become a serious event.
3.2 Current Approaches to Mitigate Aspirin Gastric Injury
Aspirin’s direct effects on the gastric epithelial lining together contribute to the risk for proximal GI tract injury and the resultant GI adverse effects. Therefore, the ability to bypass the stomach and deliver aspirin in the intestine is a logical approach to limit this injury. Enteric-coated aspirin was developed with exactly that goal in mind. The coating remains intact in the low pH environment of the stomach and begins degrading and releasing aspirin only after it reaches a pH of 7 in the duodenum. Theoretically, this pH-dependent release mechanism would protect the proximal GI tract from local injury by delivering aspirin directly to the less acidic environment of the duodenum. However, it is not clear from several studies that enteric coating actually protects against such ulceration and bleeding. Multiple reports have confirmed the lack of difference in GI damage between formulations, and some have even suggested that enteric or sustained-release formulations may increase exposure to the drug, thereby increasing adverse effects evidenced by drops in hemoglobin [34,35,36]. Importantly, the delivery mechanism of coated aspirin results in impaired bioavailability and delayed and unpredictable absorption (characterized by the US FDA as ‘erratic’). Several studies have determined that coated aspirin has a very different pharmacokinetic (PK)/PD profile than plain aspirin [37,38,39,40,41,42]. Coated aspirin is not bioequivalent to plain aspirin and therefore different formulations must be considered separately.
Additional strategies used in clinical practice to limit the GI adverse effects include the recommendation to take aspirin with food to minimize dyspepsia, and prescription of proton pump inhibitors (PPI) to limit gastric acid production and reduce ulceration [24]. Although it is unclear whether aspirin taken with food results in fewer dyspeptic symptoms, physicians do routinely recommend taking aspirin with food [43, 44]. It has been shown however that taking coated aspirin with food results in even more erratic absorption, raising the concern for lower bioavailability and reduced clinical effect [39]. Additionally, there is insufficient evidence to support that enteric-coated aspirin reduces the risk of GI injury compared with other formulations [40].
3.3 What is Phospholipid-Aspirin Complex (PL-ASA)? Rationale for Pre-Association with Lipidic Excipients
Phospholipid-aspirin complex (PL-ASA; Vazalore, PLx Pharma, Sparta, NJ, USA) is a novel aspirin formulation with a unique delivery mechanism specifically designed to reduce GI injury while maintaining full aspirin bioavailability. PL-ASA is the first FDA-approved NSAID utilizing the proprietary PlxGuard™ drug delivery platform following considerable scientific investigation over the past 2 decades [23, 33, 45]. PL-ASA capsules contain a pre-associated liquid complex formed by the non-covalent association of aspirin and lipidic excipients. The stability of the non-covalent bond is inversely related to pH, keeping the lipidic excipients and aspirin as a complex when pH is low and allowing separation when pH rises [46]. Therefore, rather than using a mechanical barrier such as a polymer coating, the PlxGuard™ system uses the chemical properties of the complex to ensure that free aspirin is not available to associate with the phospholipids of the proximal GI mucosa, and to ensure targeted delivery of aspirin to the duodenum. This protects the stomach from coming into direct contact with aspirin while also ensuring fast and predictable absorption in the duodenum and preserved bioavailability.
There are obvious similarities between coated aspirin formulations and PL-ASA, but there are also two distinct differences. Both formulations aim to bypass the stomach and deliver aspirin to the intestine, and both leverage the pH gradient to accomplish this goal. The first important difference however is that enteric-coated tablets rely on a static barrier while the PL-ASA capsules rely on a dynamic chemical association (Fig. 4) [47]. This could be an important difference in the context of reflux of duodenal contents back to the low pH of the stomach where the PL-ASA complex could self-assemble again providing bidirectional protection from direct gastric injury, while this is not possible with enteric-coated tablets once the polymer (i.e., static barrier) is dissolved. Spontaneous re-assembly has been demonstrated in in vitro experiments [47]. The second difference relates to the predictability and reliability of aspirin delivery. As previously mentioned, coated formulations have reduced bioavailability and are not bioequivalent to plain aspirin, a difference that may also be extended by food and in patients with certain comorbid conditions (e.g., diabetes, obesity) [10]. PL-ASA on the other hand is bioequivalent to plain aspirin and does not have a clinically relevant food effect, ensuring reliable aspirin absorption and predictable bioavailability [48, 49]. These differences distinguish PL-ASA from existing coated formulations and suggest that this new formulation may be able to address some of the current unmet needs associated with aspirin therapy.

Mechanism of action of PL-ASA. (1) In the stomach, the PL-ASA capsule rapidly dissolves, releasing the liquid lipid-aspirin complex. (2) In the duodenum, the rising pH leads to dissociation of the lipid-aspirin complex. (3) The aspirin is now free for absorption. Data from in vitro experiments, data on file, PLx Pharma. PL-ASA phospholipid-aspirin complex
3.4 Evidence of Less Acute GI Injury with PL-ASA
An endoscopic study using established methodology was performed to determine whether PL-ASA caused less GI injury compared with plain (i.e. non-coated) aspirin. Cryer et al. conducted a randomized, blinded study with an active control looking at rates of endoscopically assessed upper GI injury [50]. The study was performed in 204 healthy subjects (aged 50–74) years following 7 days of 325 mg study drug once daily. Overall, 42.2 % of aspirin-treated subjects developed multiple erosions and/or ulcers compared with 22.2 % of the patients treated with PL-ASA (p = 0.0027). According to a prespecified standardized definition, gastroduodenal ulcers were detected in 17.6% of plain aspirin-treated subjects compared with only 5.1% of subjects treated with PL-ASA (p = 0.0069). Compared with plain aspirin, PL-ASA use was associated with significantly lower rates of acute upper GI injury, as evidenced by lower rates of erosions and ulcers. Based on the 7-day duration of study drug exposure, the authors also commented that the risk for aspirin-induced upper GI injury begins early in the course of treatment. Importantly, this study also demonstrated that compared with plain aspirin, the PLXGuard delivery platform (i.e. pre-association of aspirin and surface-active lipids into a liquid complex) was able to significantly reduce acute aspirin-induced gastric mucosal damage. However, it is not known if this reduction in acute GI injury would translate into less GI bleeding, or how PL-ASA would compare with enteric-coated formulations.
3.5 Lipidic Excipients Do Not Interfere with the Pharmacodynamic Effect
Lipids in general have not been reported to elicit platelet activation and phosphatidylcholine in particular may actually inhibit platelet aggregation [51,52,53]. To confirm the above observations, an in vitro study was performed to determine whether the lipidic excipients used to formulate PL-ASA influence platelet function and/or the PD effects of aspirin [54]. PL-ASA uses a consistent ratio of excipients to aspirin that is maintained across the 81 and 325 mg doses. In the aforementioned experimental study, excipients were tested at double the concentration/ratio as that present in the PL-ASA formulation to increase the likelihood of detecting any potential interference with the antiplatelet effects of aspirin. The experiments were performed with multiple aspirin concentrations ranging from those that exerted only limited inhibition to those exerting maximal inhibition of platelet aggregation. Two methods were used to assess potential PD interactions between the excipients and aspirin. Standard light transmittance aggregometry using arachidonic acid as an agonist was performed using platelet-rich plasma samples containing no excipients or aspirin (control), added excipients, added aspirin at multiple concentrations, or the combination of aspirin and excipients. Flow cytometry is a sensitive method for detecting platelet activation (P-selectin expression), and samples with or without excipients, study drug, and the combination of both (as in the light transmission aggregometry [LTA] experiments above) were assessed. The results demonstrated that the excipients did not have any direct effects on platelets and did not interfere with the PD effects of aspirin.
4 Pharmacokinetics
4.1 Bioavailability of Aspirin Formulations
Following ingestion of plain non-enteric-coated tablets, aspirin is rapidly absorbed, but only about 70% is intact as acetylsalicylic acid due to first-pass hydrolysis in the liver, and thus the portal circulation is where most platelet inhibition has an opportunity to occur [20, 55]. Once in the circulation, aspirin is rapidly hydrolyzed (half-life of about 10 min) to salicylic acid, a more stable metabolite with a longer half-life [56]. Accordingly, bioequivalence studies of aspirin have traditionally relied on the measurement of salicylic acid levels to compare aspirin delivery between different formulations.
In the case of coated formulations, absorption of aspirin is both delayed and erratic [16]. Absorption rates are further delayed and reduced in the presence of food. Multiple studies have shown that coated aspirin formulations are not bioequivalent to plain uncoated aspirin, and, in some patients taking enteric-coated aspirin with food, plasma salicylate levels were undetectable [19, 39,40,41]. The clinical implications of the differing bioavailability of aspirin formulations for patients receiving chronic aspirin therapy remain unclear, however a substantial number suffer breakthrough events, which is typically termed ‘aspirin resistance’, or, more accurately, persistent platelet reactivity despite aspirin treatment [57].
There is considerable discussion around the definition and potential mechanisms underlying aspirin resistance. Reports of aspirin resistance vary widely; the differences may be due to the different definitions employed to characterize the apparent resistance. Of the many proposed causes, the most frequent seems to be simply related to patients either not taking the drug (non-adherence) or taking it but not absorbing it (non-response, low bioavailability) since true ‘genetic’ resistance to aspirin is exceedingly rare. One study looked at subjects who were initially classified as resistant to aspirin [58]. Upon re-challenge with either immediate-release or enteric-coated formulations, no subjects were found to be resistant to immediate-release aspirin, while some subjects still exhibited low responses to enteric-coated aspirin. Importantly, studies have shown that patients with incomplete response to aspirin do experience higher risk for recurrent thrombotic events. In one study, discontinuation of low-dose aspirin was associated with a > 30% increased risk of CV events [59]. The ability of a novel formulation to deliver predictable levels of aspirin and consistently achieve full (> 99%) inhibition of thromboxane production would be helpful in addressing the issue of aspirin resistance.
4.2 PL-ASA Pharmacokinetic Studies
Separate PK clinical studies have shown complete and reliable aspirin absorption with PL-ASA and established bioequivalence to immediate release aspirin.
Bioequivalence of PL-ASA to Plain Immediate-Release Aspirin [48] A bioequivalence study formed part of the FDA approval process with a new drug application (NDA). This study was a randomized, open-label, crossover study comparing PL-ASA with plain immediate-release aspirin (Genuine Bayer® Aspirin, Bayer, Whippany, NJ, USA) at 325 and 650 mg doses. This bioequivalence study demonstrated that PL-ASA is equivalent to immediate-release aspirin from a PK and PD perspective. The 81 mg dose format was included in additional regulatory submissions but was not evaluated in this PK/PD study. Further studies will provide more information about dose-effect relationships.
Healthy volunteers (n = 32) were randomized to treatment with either plain immediate-release aspirin or PL-ASA at one of two doses (325 mg or 650 mg). After completion of the first treatment and a minimum of a 2-week washout period (14–21 days), all but two subjects were crossed over and received two treatments w at the same dose level. Serial PK measurements were taken at baseline, 5, 10, 15, 20, 25, 30, 40, 50, 60, 75, and 90 min post-dose, and 2, 3, 4, 6, 8, 10, and 24 h.
The salicylic acid plasma concentration parameters for both PL-ASA and immediate-release aspirin at each of the dose levels were comparable (Fig. 5). There was a consistent dose-relationship between the two doses (325 mg or 650 mg). Values of the log-transformed PK parameters area under the concentration-time curve from time zero to the last measurable concentration (AUC0-t), AUC from time zero to infinity (AUC0-∞), and peak plasma concentrations (Cmax) for the ratio PL-ASA to immediate-release aspirin were used to determine whether the two formulations were bioequivalent according to FDA guidance standards.

Pharmacokinetic bioequivalence study of PL-ASA and IR (plain) aspirin. Values over time of acetylsalicylic acid and salicylic acid in plasma after a single dose of 325 or 650 mg of PL-ASA. Source: Angiolillo et al. [48]. PL-ASA phospholipid-aspirin complex, IR-ASA immediate-release aspirin
In this study, the 325 mg and 650 mg doses of PL-ASA were both bioequivalent to immediate-release aspirin, with the mean log-transformed salicylic acid values for AUC0-t, AUC0-∞, and Cmax and 90% confidence intervals (CIs) for each were within the 80–125% range required by the FDA (Table 2). These data were the basis of PL-ASA approval as an immediate-release aspirin product.
PK analysis of acetylsalicylic acid levels performed for bioequivalence showed that the 90% CIs for the mean log-transformed acetylsalicylic acid ratio for AUC0-t and AUC0-∞ were within the 80–125% range for both the 325 and 650 mg doses, although the 90% CI for Cmax for both dosing regimens was outside this range.
Slight differences in PK parameters reflected a difference in the rate of absorption between PL-ASA and immediate-release aspirin dosing, likely due to differences in the dissolution rates of the tablets or capsules of the comparators. As noted previously, acetylsalicylic acid is rapidly converted to salicylic acid by hydrolysis and first-pass metabolism. Therefore, the Cmax of acetylsalicylic acid is extremely sensitive to minor variations in disintegration of the formulations, while plasma concentrations of salicylic acid are relatively stable. Overall, this PK study supported the finding of PL-ASA as bioequivalent to immediate-release aspirin.
This study also evaluated bioequivalence with respect to PD parameters related to COX-1 inhibition, namely the inhibition of TxA2 production. The more stable metabolic product of TxA2, TxB2, was measured. TxB2 measurements were taken at baseline and 2, 4, 6, 8, 10 and 24 h; and samples for LTA were taken at 6 and 24 h after study drug administration. Additionally, samples were taken at baseline and at 6 and 24 h after study drug administration for assessment of inhibition of platelet aggregation induced by arachidonic acid and by collagen.
The mean concentration of TxB2 over time following dosing with PL-ASA and immediate-release aspirin were similar. The minimum plasma concentration (Cmin; TxB2) values for both comparators were below 3.1 ng/mL, which has been associated with a decreased occurrence of major adverse CV events in patients taking aspirin for cardioprotection [18]. The ratios of inhibition parameters for PL-ASA and immediate-release (plain) aspirin demonstrate bioequivalence of the two formulations (Table 3).
Light transmittance platelet aggregometry, perfomed at baseline and 6 and 24 h after study drug administration, found > 99% inhibition for both PL-ASA and plain aspirin at both 6 and 24 h, further confirming the PD effect of PL-ASA compared with plain aspirin.
Therefore, this study found that PL-ASA and immediate-release aspirin are bioequivalent from both a PD and PK perspective.
Study of Food Effect on PL-ASA [49] This study was an investigation of the effect of food on the bioavailability of acetylsalicylic acid and its more stable metabolite, salicylic acid, from a single 650 mg dose of PL-ASA in support of the original NDA. At this dose level, food did not affect the bioavailability of salicylic acid; there was a modest effect on acetylsalicylic acid parameters.
A total of 20 healthy volunteers completed this open-label, randomized, two-way crossover, single-center study that compared the PK of salicylic acid after a single 650 mg PL-ASA dose in fed and fasted states. Healthy individuals were randomly assigned to receive the first dose in either the fed or fasted state. Those assigned to the fed group received PL-ASA 30 min after a high-fat meal (i.e., 50% of the caloric content from fat). Subjects assigned to the fasted state received treatment after an overnight (10-h) fasting period. Plasma samples were collected for 24 h after dosing. After a washout period of at least 7 days, individuals were crossed over to the alternate treatment.
Administration of PL-ASA with a high-fat meal resulted in a small decrease in salicylic acid levels and a later peak concentration, but without a significant effect on systemic exposure (AUC). Acetylsalicylic acid levels also showed that food reduced and delayed the peak concentration, but again without impacting the overall exposure (AUC). Overall, these results are consistent with the acknowledgement for regulatory purposes that the rate of absorption of aspirin from the GI tract is dependent on the presence or absence of food, among other factors, but requires no change in dosing with respect to consumption of a meal (US 21 CFR 343.80). This finding is also consistent with the effects of food on aspirin bioavailability as reported in the American Hospital Formulary Service (AHFS) Drug Information publication, which states that food decreases the rate but not the extent of absorption.
Absorption Compared with Enteric Formulation [10] A second PK/PD study evaluated the pharmacologic parameters of immediate-release, enteric-coated, and PL-ASA aspirin formulations in 40 obese patients with diabetes mellitus. The study was a randomized, single-blind, triple-crossover design. Inclusion criteria included age between 21 and 80 years, body mass index of between 30 and 40 kg/m2, and type 2 diabetes mellitus not requiring insulin, while history of CV disease was an exclusion. Subjects received 325 mg of one of the formulations, administered for 3 days in each treatment period, with a 2-week washout period between treatments.
Measurements of drug concentration parameters found that enteric-coated aspirin had lower Cmax and AUCt values and needed longer to reach Cmax compared with the plain aspirin or PL-ASA formulations. The Cmax and AUC0-t values for PL-ASA were similar to that of plain aspirin, but about 2.3- and 4.5-fold higher than those of enteric-coated aspirin (p < 0.0001) (Table 4 and Fig. 6)

Pharmacokinetics of plain aspirin, enteric-coated aspirin, and PL-ASA. Values over time of acetylsalicylic acid in plasma for plain aspirin, PL-ASA, and enteric-coated aspirin. P-values for the comparison at each time point of pharmacokinetic curves were calculated by using the ‘p value from the two-sample Student’s t test. Source: Bhatt et al. [10]. EC enteric-coated. PL-ASA phospholipid-aspirin complex
This study also evaluated the PD properties of the three different aspirin formulations by measuring the time required to achieve complete (> 99%) inhibition of TxB2 production following 3 days of treatment. The main PD finding was that enteric-coated aspirin differed from the other two formulations and required significantly longer time to achieve a complete response (Fig. 7).

Time to complete TxB2 inhibition. PL-ASA and plain aspirin had faster times to >99% inhibition of TxB2 formation compared with enteric-coated aspirin. P-values were calculated using a mixed-effects model. In the event a given patient did not reach the 99% inhibition within 72 h after the initial dose, time to 99% inhibition was imputed as 72 h. Source: Bhatt et al. [10]. PL-ASA phospholipid-aspirin complex, TxB2 thromboxane B2, EC enteric-coated, LS least squares
In addition to the longer time to complete response, the percentage of patients never achieving a complete response was also higher for enteric-coated aspirin compared with the immediate-release and PL-ASA formulations (52.8%, 15.8%, and 8.1%, respectively; p < 0.001 for both comparisons vs. enteric-coated aspirin; p = 0.30 for comparison between plain aspirin and PL-ASA). When looking at absolute serum TxB2 levels after 3 days of therapy, 56% of subjects had > 3.1 ng/mL after taking enteric-coated aspirin, compared with 18% and 11% of subjects after administration of plain aspirin and PL-ASA, respectively (p < 0.0001). As Fig. 8 illustrates, there was also marked interindividual variability in the response to aspirin over the dosing period.

Individual TxB2 inhibition over time. Percentage inhibition of serum TxB2 concentration over baseline after dosing with plain aspirin, PL-ASA, and EC-ASA. Blue lines represent the percentage inhibition in individual patients who reached 99% inhibition by 72 h after the first dose of study drug. Orange lines represent the percentage inhibition in individual patients who did not reach 99% inhibition by 72 h after the first dose of study drug. The horizontal line designates 99% inhibition. Source: Bhatt et al. [10]. PL-ASA phospholipid-aspirin complex, TxB2 thromboxane B2, EC-ASA enteric-coated aspirin
5 Population Pharmacokinetics/Pharmacodynamics
5.1 Weight
Two randomized crossover studies that included obese diabetic patients who received 325 mg of PL-ASA or enteric-coated aspirin each day for 3 days were pooled [60]. The increased sample size of the pooled dataset allowed a dedicated analysis of the potential impact of weight on the PD profile of the two aspirin formulations.
A total of 97 patients were included in the analysis, with 183 samples (EC-ASA, n = 91; PL-ASA, n = 92). Regression analysis showed that weight was inversely associated with the extent of TxB2 inhibition in the aggregate population; the trend line crossed the threshold for complete aspirin response, that is 99% TxB2 inhibition, at 117 kg. The inverse relationship of TxB2 inhibition and weight was present with both formulations, however the association was more pronounced with enteric-coated aspirin, where the trend line crossed the threshold at 95 kg compared with PL-ASA, when that did not occur until a much higher weight of 131 kg (p < 0.001).
Predictable absorption greatly mitigates the impact of weight for the vast majority of patients by driving the risk of incomplete platelet inhibition to the left by about 30 kg (i.e. the problem is evident with patients over 90 kg with enteric-coated aspirin but not until > 120 kg with PL-ASA. This has important epidemiological implications as it affects a very large portion of the target patients [61].
5.2 PL-ASA: Limitations and Future Directions
Studies to date have used PL-ASA mostly at the 325 mg dose. However, the most commonly used dose for secondary prevention of CV events is 81 mg. Although the pharmacologic profile is expected to be the same for both doses, future studies for PL-ASA are needed to confirm the PK and PD profile discussed in this review (NCT04811625). Moreover, the available evidence on the degree of GI injury associated with PL-ASA is limited to short-term treatment, and the GI effects associated with long-term use remains unknown. Additional studies could include registries to explore the impact of this new formulation on gastric symptoms such as dyspepsia, which is a common reason for non-adherence.
6 Conclusions
Aspirin remains the cornerstone therapy for the lifelong prevention of recurrent CV events. While plain, uncoated aspirin may be used in acute settings to help assure rapid absorption, enteric-coated aspirin formulations dominate current chronic use, including for secondary prevention of CV events. The unmet needs with current aspirin formulations include a high risk of GI adverse events with plain aspirin, which enteric-coated formulations are not able to overcome. Enteric-coated formulations are also subject to erratic absorption leading to reduced drug bioavailability. These observations indeed underscore the need for aspirin formulations with a more favorable safety and efficacy profile. PL-ASA is a novel formulation, recently approved by the FDA, designed to address these needs. The combination of predictable aspirin delivery and effective platelet inhibition compared with enteric-coated aspirin with reduced acute direct GI injury compared with plain aspirin makes PL-ASA a potentially attractive treatment option. Future clinical studies are warranted to assess the comparative pharmacologic effects of PL-ASA at the 81 mg dosing regimen with the most commonly utilized enteric-coated formulation at the same dose. An understanding of the long-term safety profile, particularly at the GI level, is also important as current data are limited to acute assessments. Future studies should evaluate whether the advantages of this novel formulation can translate into improved clinical outcomes for the tens of millions of patients who require lifelong aspirin therapy.
References
Steinhubl SR. Historical observations on the discovery of platelets, platelet function testing and the first antiplatelet agent. Curr Drug Targets. 2011;12:1792–804.
Fuster V, Sweeny JM. Aspirin a historical and contemporary therapeutic overview. Circ. 2011;123:768–78.
Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231:232–5.
Antithrombotic Trialists Collaborative. Collaborative metaanalysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high-risk patients. BMJ. 2002;324:71–86.
Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140:e596–646.
Smith SC Jr, Benjamin EJ, Bonow RO, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation. Circulation. 2011;124:2458–73.
Gerhard-Herman MD, Gornik HL, Barrett C, et al. 2016 AHA/ACC guideline on the management of patients with lower extremity peripheral artery disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2017;69:e71-126.
Powers WJ, Rabinstein AA, Ackerson T, et al; on behalf of the American Heart Association Stroke Council. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2019;50(12):e344–e418.
Kedir HM, Sisay EA, Abiye AA. Enteric-coated aspirin and the risk of gastrointestinal side effects: a systematic review. Int J Gen Med. 2021;14:4757–63.
Bhatt DL, Grosser T, Dong JF, et al. Enteric coating and aspirin nonresponsiveness in patients With Type 2 Diabetes Mellitus. J Am Coll Cardiol. 2017;69:603–12.
Randjelović P, Veljković S, Stojiljković N, et al. The beneficial biological properties of salicylic acid. Acta facultatis medicae Naissensis. 2015;32:259–65.
Figueiredo JC, Jacobs EJ, Newton CC, Guinter MA, Cance WG, Campbell PT. Associations of aspirin and non-aspirin non-steroidal anti-inflammatory drugs with colorectal cancer mortality after diagnosis. J Natl Cancer Inst. 2021;113(7):833–40.
Low-dose aspirin use during pregnancy. ACOG Committee Opinion No. 743. American College of Obstetricians and Gynecologists. Obstet Gynecol 2018;132:e44–52
Chow JH, Khanna AK, Kethireddy S, et al. Aspirin use is associated with decreased mechanical ventilation, intensive care unit admission, and in-hospital mortality in hospitalized patients with coronavirus disease 2019. Anesth Analg. 2021;132:930–41.
Vazalore Aspirin Drug Facts. Sparta, NJ: PLx Pharma Inc.; rev 5.17.2021
Aspirin Professional Labeling. 21CFR343Æ80. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=343&showFR=1. Accessed 20 Jul 2021.
Scavone M, Femia EA, Caroppo V, Cattaneo M. Inhibition of the platelet P2Y12 receptor for ADP does not impair the capacity of platelet to synthesize thromboxane A2. EHJ. 2016;37:3347–56.
Frelinger AL 3rd, Li Y, Linden MD, et al. Association of cyclooxygenase-1-dependent and -independent platelet function assays with adverse clinical outcomes in aspirin-treated patients presenting for cardiac catheterization. Circulation. 2009;120:2586–96.
Gum PA, Kottke-Marchant K, Poggio ED, Gurm H, Welsh PA, Brooks L, et al. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am J Cardiol. 2001;88(3):230–5.
Papathanasiou A, Goudevenos J, Tselepis AD. Resistance to aspirin and clopidogrel: possible mechanisms, laboratory investigation, and clinical significance. Hellenic J Cardiol. 2007;48:352–63.
Blobaum AL, Marnett LJ. Structural and functional basis of cyclooxygenase inhibition. J Med Chem. 2007;50:1425–41.
Lichtenberger LM, Barron M, Marathi U. Association of phosphatidylcholine and NSAIDs as a novel strategy to reduce gastrointestinal toxicity. Drugs Today (Barc). 2009;45:877–90.
Li Z, Wang Z, Shen B, Chen C, Ding X, Song H. Effects of aspirin on the gastrointestinal tract: Pros and cons (a Review). Onc Lett. 2020;20:2567–78.
Scheiman J. NSAIDS, Gastrointestinal Injury and Cytoprotection. Gastroent Clin NA. 1996;25:279–98.
Cryer B, Feldman M. Effects of very low dose daily, long-term aspirin therapy on gastric, duodenal, and rectal prostaglandin levels and on mucosal injury in healthy humans. Gastroenterology. 1999;117:17–25.
Yeomans ND, Lanas AI, Talley NJ, et al. Prevalence and incidence of gastroduodenal ulcers during treatment with vascular protective doses of aspirin. Aliment Pharmacol Ther. 2005;22:795–801.
Niv Y, Battler A, Abuksis G, et al. Endoscopy in asymptomatic minidose aspirin consumers. Dig Dis Sci. 2005;50:78–80.
Goldstein JL, Lowry SC, Lanza FL, et al. The impact of low-dose aspirin on endoscopic gastric and duodenal ulcer rates in users of a non-selective nonsteroidal anti-infl ammatory drug or a cyclo-oxygenase-2-selective inhibitor Aliment Pharmacol Ther 2006;23:1489 – 98.
Hernandez-Diaz S, Garcia Rodriguez LA. Cardioprotective aspirin users and their excess risk of upper gastrointestinal complications. BMC Med. 2006;4:22.
Pratt S, Thompson VJ, Elkin EP, Næsdal J, Sörstadius E. The impact of upper gastrointestinal symptoms on nonadherence to, and discontinuation of, low-dose acetylsalicylic acid in patients with cardiovascular risk. Am J Cardiovasc Drugs. 2010;10:281–8.
LaVie CJ, Howden CW, Scheiman J, Tursi J. Upper gastrointestinal toxicity associated with long-term aspirin therapy: consequences and prevention. Curr Prob Cardiol. 2017;42:146–64.
Lichtenberger L. Insight into NSAID-induced membrane alterations, pathogenesis and therapeutics: characterization of interaction of NSAIDS with phosphatidylcholine. Biochim Biophys Acta. 2012;1821:994–1002.
Lichtenberger L. Role of phospholipids in protection of the GI Mucosa. Dig Dis Sci. 2013;58:891–3.
Sostres C, Lanas A. Gastrointestinal effects of aspirin. Nat Rev Gastroenterol Hepatol. 2011;8:385–94.
Davies NM. Sustained release and enteric coated NSAIDs: are they really GI safe? J Pharm Pharm Sci. 1999;2:5–14.
Hirata Y, Kataoka H, Shimura T, et al. Incidence of gastrointestinal bleeding in patients with cardiovascular disease: buffered aspirin versus enteric-coated aspirin. Scand J Gastroenterol. 2011;46:803–9.
Levy G, Hollister LE. Failure of USP disintegration test to assess physiologic availability of enteric coated tablets. NY State J Med. 1964;64:3002–5.
Leonardis JR. Absorption and metabolism of aspirin administered in enteric-coated tablets. JAMA. 1965;193:99–104.
Bogentoft C, Carlsson I, Ekenved G, Magnusson A. Influence of food on the absorption of acetylsalicylic acid from enteric-coated dosage forms. Eur J Clin Pharmacol. 1978;14:351–5.
Bochner F, Williams DB, Morris PM, Siebert DM, Lloyd JV. PK of low-dose oral modified release, soluble and IV aspirin in man, and effects on platelet function. Eur J Clin Pharmacol. 1988;35:287–94.
Haastrup PF. Enteric coating can lead to reduced antiplatelet effect of low-dose acetylsalicylic acid. Basic Clin Pharmacol Toxicol. 2015;116:212–5.
Cox D, Fitzgerald DJ. Lack of bioequivalence among low-dose, enteric coated aspirin preparations. Clin Pharmacol Ther. 2018;103:1047–51.
Cleveland Clinic. Aspirin Therapy in Heart Disease. https://my.clevelandclinic.org/health/drugs/16761-aspirin-therapy-in-heart-disease. Accessed 20 Jun 2021.
Harvard Medical. Answers about aspirin. https://www.health.harvard.edu/heart-health/answers-about-aspirin. Accessed 20 Jun 2021.
Lim YJ, Dial EJ, Lichtenberger LM. Advent of novel phosphatidylcholine-associated nonsteroidal anti-inflammatory drugs with improved gastrointestinal safety. Gut Liver. 2013;7:7–15.
Anand BS, Romero JJ, Sanduja SK, Lichtenberger LM. Phospholipid association reduces the gastric mucosal toxicity of aspirin in human subjects. Aliment Pharmacol Ther. 2008;28:431–42.
PLx Pharma. Data on file.
Angiolillo DJ, Bhatt DL, Lanza F, et al. Pharmacokinetic/pharmacodynamic assessment of a novel, pharmaceutical lipid-aspirin complex: results of a randomized, crossover, bioequivalence study. J Thromb Thrombolysis. 2019;48:554–62.
Angiolillo DJ, Bhatt DL, Lanza F, et al. Bioavailability of aspirin in fasted and fed states of a novel pharmaceutical lipid aspirin complex formulation. J Thromb Thrombolysis. 2020;49:337–43.
Cryer B, Bhatt DL, Lanza FL, Dong JF, Lichtenberger LM, Marathi UK. Low-dose aspirin-induced ulceration is attenuated by aspirin-phosphatidylcholine: a randomized clinical trial. Am J Gastroenterol. 2011;106:272–7.
Kerr JW, Macaulay I, Pirrie R, Bronte-Stewart B. Platelet aggregation by phospholipids and free fatty acids. Lancet. 1965;1:1296–9.
Hornstra G. Dietary fats and arterial thrombosis: effects and mechanism of action. Prog Biochem Pharmacol. 1977;13:326–38.
Kobayashi T, Yamada J, Satoh N, Setaka M, Kwan T. Inhibition of platelet aggregation by synthetic phosphatidylcholines: possible involvement of vesiculation of platelet plasma membranes. Biochim Biophys Acta. 1985;817:307–12.
Schneider DJ, Taatjes-Sommer HS, Prats J, Deliargyris EN. Influence of lipid excipients on platelet function and the pharmacodynamic effects of aspirin. J Cardiovasc Pharmacol. 2021;78(2):297–301.
Rowland M, Riegelman S, Harris PA, Sholkoff SD. Absorption kinetics of aspirin in man following oral administration of an aqueous solution. J Pharm Sci. 1972;61:379–85.
Graham GG, Day RO. Salicylates. In: Parnham M (ed). Compendium of Inflammatory Diseases. Basel: Birkhäuser; 2016. https://doi.org/10.1007/978-3-0348-0620-6_62-3.
Schneider DJ. On defining aspirin resistance. J Am Coll Cardiol. 2005;46:1710–1.
Grosser T, Fries S, Lawson JA, Kapoor SC, Grant GR, FitzGerald GA. Drug resistance and pseudoresistance: an unintended consequence of enteric coating aspirin. Circulation. 2013;127(3):377–85.
Sundstrom J, Hedberg J, Thuresson M, Aarskog P, Johannesen KM, Oldgren J. Low-dose aspirin discontinuation and risk of cardiovascular events. A Swedish Nationwide. Population-Based Cohort Study Circulation. 2017;136:1183–92.
Bhatt DL, Angiolillo DA, Steg PG, et al. Impact of weight on the antiplatelet effects of aspirin-results of a pooled analysis of two randomized crossover studies comparing a liquid aspirin formulation with enteric-coated aspirin. J Am Coll Cardiol. 2020;75:1344.
De Bacquer D, De Backer G, Cokkinos D, Keil U, Montaye M, Ostör E, et al. Overweight and obesity in patients with established coronary heart disease: are we meeting the challenge? Eur Heart J. 2004;25(2):121–8.
Acknowledgements
The authors thank Ms Weihong Fan for her careful reading of the manuscript, and for assistance with study details and statistics.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Funding for PK-PD studies described in this manuscript, for Open Access of this article and for illustrations was provided by PLx Pharma, Inc.
Conflict of interest
Dr. Angiolillo is a member of the PLx Pharma Scientific Advisory Board has received payment as an individual for: a) Consulting fee or honorarium from Abbott, Amgen, Aralez, AstraZeneca, Bayer, Biosensors, Boehringer Ingelheim, Bristol-Myers Squibb, Chiesi, Daiichi-Sankyo, Eli Lilly, Haemonetics, Janssen, Merck, PhaseBio, PLx Pharma, Pfizer, Sanofi, and The Medicines Company; b) Participation in review activities from CeloNova and St. Jude Medical; c) Institutional payments for grants from Amgen, AstraZeneca, Bayer, Biosensors, CeloNova, CSL Behring, Daiichi-Sankyo, Eisai, Eli-Lilly, Gilead, Idorsia, Janssen, Matsutani Chemical Industry Co., Merck, Novartis, Osprey Medical, Renal Guard Solutions and the Scott R. MacKenzie Foundation. Dr. Prats is a consultant to PLxPharma, manufacturer of PL-ASA (Vazalore) Dr. Deliargyris is a member of the PLx Pharma Scientific Advisory Board, no other relevant conflicts. Dr. Schneider has received grant funding from Plx Pharma. Dr. Scheiman is a member of the PLx Pharma Scientific Advisory Board and consultant, Tremeau Pharmaceuticals. Dr. Kimmelstiel is a member of the PLx Pharma Scientific Advisory Board, no other relevant conflicts. Dr. Steg is a member of the PLx Pharma Scientific Advisory Board. He reports research grants from Amarin, Bayer, Sanofi, and Servier; clinical trial conduct (Steering committee, CEC, DSMB) with Amarin, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Idorsia, Novartis, Pfizer, Sanofi, Servier; consulting or speaking activity for Amgen, BMS/Myokardia, Novo-Nordisk, Regeneron ; and is a Senior Associate Editor at Circulation. Dr. Alberts is a member of the PLx Pharma Scientific Advisory Board, no other relevant conflicts. Dr. Rosengart is a member of the PLx Pharma Scientific Advisory Board, no other relevant conflicts. Dr. Mehran reports institutional research grants from Abbott, Abiomed, Applied Therapeutics, Arena, AstraZeneca, Bayer, Biosensors, Boston Scientific, Bristol-Myers Squibb, CardiaWave, CellAegis, CERC, Chiesi, Concept Medical, CSL Behring, DSI, Insel Gruppe AG, Medtronic, Novartis Pharmaceuticals, OrbusNeich, Philips, Transverse Medical, Zoll; personal fees from ACC, Boston Scientific, California Institute for Regenerative Medicine (CIRM), Cine-Med Research, Janssen, WebMD, SCAI; consulting fees paid to the institution from Abbott, Abiomed, AM-Pharma, Alleviant Medical, Bayer, Beth Israel Deaconess, CardiaWave, CeloNova, Chiesi, CSL Behring, Concept Medical, DSI, Duke University, Idorsia Pharmaceuticals, Medtronic, Novartis, Philips; Equity <1% in Applied Therapeutics, Elixir Medical, STEL, CONTROLRAD (spouse); Scientific Advisory Board for AMA, Biosensors (spouse); Faculty CRF (no fee). Dr. Deepak L. Bhatt discloses the following relationships - Advisory Board: Cardax, CellProthera, Cereno Scientific, Elsevier Practice Update Cardiology, Janssen, Level Ex, Medscape Cardiology, MyoKardia, NirvaMed, Novo Nordisk, PhaseBio, PLx Pharma, Regado Biosciences; Board of Directors: Boston VA Research Institute, Society of Cardiovascular Patient Care, TobeSoft; Chair: Inaugural Chair, American Heart Association Quality Oversight Committee; Data Monitoring Committees: Baim Institute for Clinical Research (formerly Harvard Clinical Research Institute, for the PORTICO trial, funded by St. Jude Medical, now Abbott), Cleveland Clinic (including for the ExCEED trial, funded by Edwards), Contego Medical (Chair, PERFORMANCE 2), Duke Clinical Research Institute, Mayo Clinic, Mount Sinai School of Medicine (for the ENVISAGE trial, funded by Daiichi Sankyo), Population Health Research Institute; Honoraria: American College of Cardiology (Senior Associate Editor, Clinical Trials and News, ACC.org; Chair, ACC Accreditation Oversight Committee), Baim Institute for Clinical Research (formerly Harvard Clinical Research Institute; RE-DUAL PCI clinical trial steering committee funded by Boehringer Ingelheim; AEGIS-II executive committee funded by CSL Behring), Belvoir Publications (Editor in Chief, Harvard Heart Letter), Canadian Medical and Surgical Knowledge Translation Research Group (clinical trial steering committees), Duke Clinical Research Institute (clinical trial steering committees, including for the PRONOUNCE trial, funded by Ferring Pharmaceuticals), HMP Global (Editor in Chief, Journal of Invasive Cardiology), Journal of the American College of Cardiology (Guest Editor; Associate Editor), K2P (Co-Chair, interdisciplinary curriculum), Level Ex, Medtelligence/ReachMD (CME steering committees), MJH Life Sciences, Population Health Research Institute (for the COMPASS operations committee, publications committee, steering committee, and USA national co-leader, funded by Bayer), Slack Publications (Chief Medical Editor, Cardiology Today’s Intervention), Society of Cardiovascular Patient Care (Secretary/Treasurer), WebMD (CME steering committees); Other: Clinical Cardiology (Deputy Editor), NCDR-ACTION Registry Steering Committee (Chair), VA CART Research and Publications Committee (Chair); Research Funding: Abbott, Afimmune, Amarin, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Cardax, CellProthera, Cereno Scientific, Chiesi, CSL Behring, Eisai, Ethicon, Ferring Pharmaceuticals, Forest Laboratories, Fractyl, Garmin, HLS Therapeutics, Idorsia, Ironwood, Ischemix, Janssen, Lexicon, Lilly, Medtronic, MyoKardia, NirvaMed, Novartis, Novo Nordisk, Owkin, Pfizer, PhaseBio, PLx Pharma, Regeneron, Roche, Sanofi, Synaptic, The Medicines Company, 89Bio; Royalties: Elsevier (Editor, Cardiovascular Intervention: A Companion to Braunwald’s Heart Disease); Site Co-Investigator: Abbott, Biotronik, Boston Scientific, CSI, St. Jude Medical (now Abbott), Philips, Svelte; Trustee: American College of Cardiology; Unfunded Research: FlowCo, Merck, Takeda.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Angiolillo, D.J., Prats, J., Deliargyris, E.N. et al. Pharmacokinetic and Pharmacodynamic Profile of a Novel Phospholipid Aspirin Formulation. Clin Pharmacokinet 61, 465–479 (2022). https://doi.org/10.1007/s40262-021-01090-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-01090-2