
Abstract
Background and Objectives
Aripiprazole is an atypical antipsychotic drug that is metabolized by cytochrome P450 (CYP) 2D6 and CYP3A4, which mainly form its active metabolite dehydro-aripiprazole. Because of the genetic polymorphism of CYP2D6, plasma concentrations are highly variable between different phenotypes. In this study, phenotype-related physiologically based pharmacokinetic models were developed and evaluated to suggest phenotype-guided dose adjustments.
Methods
Physiologically based pharmacokinetic models for single dose (oral and orodispersible formulation), multiple dose, and steady-state condition were built using trial data from genotyped healthy volunteers. Based on evaluated models, dose adjustments were simulated to compensate for genetically caused differences.
Results
Physiologically based pharmacokinetic models were able to accurately predict the pharmacokinetics of aripiprazole and dehydro-aripiprazole according to CYP2D6 phenotypes, illustrated by a minimal bias and a good precision. For single-dose administration, 92.5% (oral formulation) and 79.3% (orodispersible formulation) of the plasma concentrations of aripiprazole were within the 1.25-fold error range. In addition, physiologically based pharmacokinetic steady-state simulations demonstrate that the daily dose for poor metabolizer should be adjusted, resulting in a maximum recommended dose of 10 mg, but no adjustment is necessary for intermediate and ultra-rapid metabolizers.
Conclusions
In clinical practice, CYP2D6 genotyping in combination with therapeutic drug monitoring should be considered to personalize aripiprazole dosing, especially in CYP2D6 poor metabolizers, to ensure therapy effectiveness and safety.
Similar content being viewed by others

A physiologically based pharmacokinetic (PBPK) model for aripiprazole and dehydro-aripiprazole was successfully developed to predict the pharmacokinetics in genotyped healthy volunteers, taken into account relevant cytochrome P450 (CYP) 2D6-related and CYP3A4-related changes in different CYP2D6 phenotypes. |
Based on CYP2D6 phenotypes, PBPK models for single dose, multiple dose, and steady state were developed and verified to calculate phenotype-guided dose adjustments. |
To ensure therapy effectiveness and safety for each patient, PBPK models indicate a maximum recommended daily dose of 10 mg for CYP2D6 poor metabolizers, but no adjustment is necessary for intermediate and ultra-rapid metabolizers. |
PBPK models show that routine CYP2D6 genotyping of patients with schizophrenia can improve a patient’s safety, especially for CYP2D6 poor metabolizers. |
1 Introduction
Schizophrenia is a chronic and severe mental disorder, affecting 20 million people worldwide [1]. Although medication exists to manage several symptoms of schizophrenia, patients with schizophrenia are two to three times more likely to die earlier than the general population [2]. Therefore, an effective and individualized therapy with antipsychotic agents such as aripiprazole (ARI) is crucial.
Aripiprazole is a an atypical generation antipsychotic commonly indicated for the treatment of schizophrenia and the acute treatment of manic and mixed episodes associated with bipolar I disorder [3]. The drug shows a unique mechanism of action compared with other antipsychotic drugs, displaying partial agonist activity at dopamine D2, D3, and serotonin 5-HT1A receptors, and acts also as a 5-HT2A and 5-HT7 receptor antagonist [4,5,6]. Aripiprazole is mainly metabolized via the hepatic cytochrome P450 (CYP) 2D6 and CYP3A4 by three biotransformation pathways: dehydrogenation, hydroxylation, and N-dealkylation [7]. Based on dehydrogenation, the active metabolite dehydro-aripiprazole (D-ARI) is formed, which accounts for about 40% of the area under the curve (AUC) of the parent drug in plasma at steady state [3, 8]. The pharmacokinetics of ARI is linear and dose proportional within the doses of 5–30 mg [3]. Although ARI is the predominant drug moiety in systematic circulation, both ARI and D-ARI show similar pharmacological properties [3, 9]. The therapeutic reference range of ARI amounts to 10–350 µg/L and for the active moiety (ARI plus D-ARI) 150–500 µg/L, according to the consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology [10].
Considering a therapeutic range with best clinical outcomes and a minimum of side effects, there is evidence for inter-individual variability of plasma concentrations of ARI and D-ARI [8, 11]. The wide pharmacokinetic variability of drugs metabolized by CYP2D6 is largely explained by its genetic polymorphism [12]. CYP2D6 is a highly polymorphic gene with more than 100 allelic variants, which leads to a classification into four different phenotypes [13, 14]: poor metabolizers (PMs), intermediate metabolizers (IMs), normal metabolizers (NMs), and ultra-rapid metabolizers (UMs). Because of the genetic variations in CYP2D6, CYP2D6 PMs carry two no function alleles, leading to a lack of enzyme activity [15]. As a consequence, PMs show higher plasma concentrations and a longer half-life (t1/2) [146 h vs 75 h] for ARI [3, 12]. Based on the wide CYP-mediated metabolism and high inter-individual variability, dose adjustments of ARI should be considered, as has been suggested in the past for other antipsychotics such as risperidone [16] and aripiprazole [3].
The aim of the current study was to develop a mechanistic, whole-body, physiologically based pharmacokinetic (PBPK) model for ARI, D-ARI, and the sum of ARI plus D-ARI, according to the phenotype. Single-dose models for oral and orodispersible formulations as well as multiple-dose models were created and the pharmacokinetics between different phenotypes was analysed. In addition, we investigated the influence of the CYP2D6 phenotype on plasma concentrations of ARI, D-ARI, and the active moiety to support phenotype-guided dose adjustments that compensate for genetic differences. We choose the PBPK approach because this method allows a more mechanistic insight into the pharmacokinetics in comparison to population pharmacokinetic models, which is an empirical approach. To our knowledge, this is the first study examining the effects of CYP2D6 phenotype-related physiological changes on the pharmacokinetics of ARI, D-ARI, and the sum of ARI plus D-ARI, resulting in phenotype-guided dosing strategies using a PBPK approach.
2 Materials and methods
2.1 PBPK Modeling
The modeling and simulation was performed using PK-Sim® and MoBi® Version 8.0 as part of the Open Systems Pharmacology Suite (download: http://www.open-systems-pharmacology.org/). This freely available, whole-body simulation software package allows the description of drugs in humans and several other animal species [17, 18]. Briefly, the software implements a generic structure consisting of 17 different compartments, each representing a single organ or tissue, that are organ-specifically connected to the arterial and venous blood pool [17, 19, 20]. Moreover, each organ is subdivided into subcompartments [20]. Based on physicochemical properties, and specific absorption, distribution, metabolism and excretion data of the drug, the software package estimates a drug-related model parameter to describe the rate and extent of drug disposition in each compartment [19]. For a detailed description, please refer to the user software manual [18].
2.2 Single-Dose Study Data
For PBPK modeling and simulation of ARI, plasma concentration data obtained in six randomized crossover bioequivalence clinical trials (EUDRA-CT: 2012-004241-32, 2012-005274-60, 2012-002016-97, 2012-003196-19, 2013-002478-35, and 2013-004146-42) after single-dose administration to healthy Caucasian volunteers were used. Fifty-five of a total of 97 participants received an oral tablet of 10 mg of Abilify®, while the other 42 participants received an orodispersible formulation of 10 mg of Abilify®. After single-dose administration, blood samples of all 97 participants were collected at the following times after dosing: 1, 2, 4, 6, 8, 10, 12, 24, 48, and 72 h. CYP2D6 genotyping and plasma concentration measurements were conducted by previously reported standard methods [21].
After CYP2D6 genotyping of all 97 healthy volunteers, genotypes were translated to phenotypes, based on the activity score (AS) system [13]. In this system, each allele is assigned to an activity value that is used for the phenotype translation published by Caudle et al. [22]. Here, individuals with an AS of 0 are categorized as PMs, individuals with an AS of 0.25–1 are IMs, individuals with an AS of 1.25–2.25 are NMs, and individuals with an AS > 2.25 are UMs [22]. Demographic data considering different phenotypes of the single-dose study are summarized in Table 1.
2.3 Multiple-Dose Study Data
Furthermore, plasma concentration data of a multiple-dose clinical trial (EUDRA-CT: 2018-000744-26) including 24 healthy volunteers (14 Latin Americans and ten Caucasians) were used for modeling and simulation [23]. Oral 10-mg tablets of ARI were administered during 5 consecutive days and 22 blood samples were collected from each participant. Plasma concentrations of ARI and D-ARI were measured at the following time points: seven samples on day 1 (pre-dose, and 1, 2, 3, 5, 8, and 12 h after dosing); one pre-dose sample on days 2, 3, and 4; seven samples on day 5 (pre-dose, 1, 2, 3, 5, 8, and 12 h after dosing); and one sample on days 6, 7, 9, 11, and 15. CYP2D6 genotyping and subsequent phenotype translation were performed as described above (Sect. 2.2). Demographic data considering CYP2D6 phenotypes of the multiple-dose study are shown in Table 2.
2.4 PBPK Model Development and Verification
For the development of an initial PBPK model, a systematic literature search of previous publications on ARI and D-ARI was conducted. In this context, drug-related and metabolite-related physicochemical data, as well as absorption, distribution, metabolism and excretion-specific and physiologically specific parameters were obtained. For modeling and simulation, ARI, D-ARI, and other inactive metabolites were implemented and all biotransformation pathways of ARI (dehydrogenation, hydroxylation, and N-dealkylation) were considered. Both metabolizing enzymes CYP2D6 and CYP3A4 were integrated and the drug transporter P-glycoprotein (ABCB1) was incorporated in the model. Furthermore, a renal clearance for ARI was assumed to be 0.04 mL/h/kg [24].
Initial PBPK models were verified using plasma concentration data of two independent datasets containing single-dose and multiple-dose clinical trials. First, only single-dose data including data of two different formulations were used: oral tablets and orodispersible tablets. For modeling and simulation of the oral tablet formulation, the release of the drug from the formulation was implemented into PK-Sim® as a Weibull function, with a parabolic dissolution shape of 0.92 and a dissolution time (50% dissolved) of 3.50 minutes. A lag time was not included in the model because the tablets do not have an enteric coating [3]. The orodispersible tablet formulation of Abilify® was integrated into the software as a dissolved formulation. At this point of modeling, individual CYP2D6 phenotypes were not considered. Simulation time was set to 72 h. PBPK models were further evaluated and modeling parameters refined. When single-dose PBPK models showed the best possible fit for both formulations, multiple-dose simulations (simulation time 360 h) were developed and model-building parameters further optimized. Specific hepatic clearance as well as a specific renal clearance of D-ARI were refined and modified according to the predicted values of ARI [25]. Here, specific clearance is defined as the intrinsic clearance normalized to the volume where the process occurs [18]. The catalytic rate constants (kcat), also known as the turnover frequency for both metabolizing enzymes CYP2D6 and CYP3A4 were fitted as close as possible to the mean plasma concentration through parameter optimization. Moreover, modeling parameter partition coefficients for both ARI and D-ARI were calculated using the method described by Poulin and Theil according to the observed data. Model-building parameters are listed in Table 3.
2.5 CYP2D6 Phenotype Implementation
For CYP2D6 phenotype guided modeling, all participants of the single-dose and multiple-dose clinical trials were genotyped and phenotyped (Tables 1, 2). Based on the new subgroups, separate simulations were developed to allow a comparison between the different phenotypes. Previously integrated kcat values for CYP2D6 and CYP3A4 were further refined and set into their proportion of metabolism. Here, a percentage of 40% was set for the dehydrogenation (CYP2D6 and CYP3A4) from ARI to D-ARI and 60% for hydroxylation and N-dealkylation to inactive metabolites.
Next, all kcat values were set into ratio and further evaluated and refined until the best possible fit between observed and estimated plasma concentration–time profiles of ARI and DARI were reached. Best fit was determined using the parameter identification tool of PK-Sim®, based on single-dose modeling as well as on multiple-dose modeling results. For PMs, the kcat values for CYP2D6-mediated dehydrogenation and hydroxylation were set to 0/min in representation of its null metabolizing capacity [12, 13]. For the other phenotypes, CYP2D6 kcat values increased with the number of functional alleles from IMs, NMs, to UMs. For UMs, exhibiting at least three functional alleles, kcat values were set to be 33% higher compared with the kcat values of NMs [13]. In contrast to CYP2D6, kcat values for CYP3A4 were kept constant for all phenotypes, as this enzyme has no clinically relevant polymorphism. An overview of all enzymatic input parameters is shown in Table 4.
2.6 Steady-State PBPK Model
Next, PBPK models were transmitted to steady state. Mean t1/2 are about 75 h and 94 h for ARI and D-ARI, respectively. For simulations, the dose was fixed to 15 mg/day, which is the recommended dose for the management of schizophrenia and bipolar mania in adults, according to the manufacturer’s prescribing information [3]. For steady-state simulations, only oral tablet formulations of Abilify® were performed, as pharmacokinetic studies showed a bioequivalence between oral and orally disintegrating tablets [3]. Furthermore, Abilify® tablets are available in more different strengths (2 mg, 5 mg, 10 mg, 15 mg 20 mg, and 30 mg) than oral disintegrating tablets (10 mg and 15 mg) [3] and allow more clinical-associated simulations in the case of a possible dose adjustment. Overall, modeling and simulation were performed for ARI, D-ARI, and the active moiety, respectively. For the simulation of the active moiety, a new equation has been developed in MoBi®, which was implemented in PK-Sim® (see Eq. 1).
where art is arterial blood, bon is bone, C is concentration in plasma, fQ is blood flow rate, mus is muscle, and skn is skin.
Therapeutic reference ranges were set to 100–350 µg/L for ARI and 150–500 µg/L for the active moiety, according to Hiemke et al. [10]. For predicted plasma concentrations outside the therapeutic range, the daily dose was adjusted until it was within the therapeutic reference range.
2.7 Model Evaluation
The evaluation of the PBPK models was carried out in accordance with the guideline on the reporting of PBPK modeling and simulation [29]. Different pharmacokinetic parameters, such as AUC, maximum plasma concentration of the drug (Cmax), time it takes until the drug reached the Cmax (tmax), trough plasma concentrations as well as the t1/2 were calculated by the software package or performed in R (Version 3.5.2; R Foundation for Statistical Computing, Vienna, Austria) [30].
The quality of the PBPK model evaluation for single dose and multiple dose were analysed using goodness-of-fit plots and weighted residuals vs observed plasma concentrations. To describe the accuracy and precision of the models, prediction error (PE), mean prediction error (MPE), and mean absolute prediction error (MAPE) were calculated according to Eqs. 2–4:
3 Results
3.1 Single-Dose Modeling
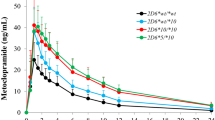
Single-dose PBPK models accurately described plasma concentrations for a population of healthy volunteers after 10-mg single-dose administration of both oral and orodispersible tablet formulations of ARI (Figs. 1, 2). Aripiprazole is rapidly absorbed and shows a predicted Cmax between 52.1 and 55.5 µg/L for oral tablet formulations and between 50.0 and 55.0 µg/L for orodispersible tablet formulations, depending on the CYP2D6 phenotype. Predicted Cmax occurred within 3 h after dosing and predicted mean t1/2 of ARI amounts to 68.6 h, which are both in accordance with the information provided by the prescribing information of the manufacturer [3]. In PMs, predicted and observed Cmax and tmax of ARI tended to be higher and the elimination much slower than in the other phenotypes (Figs. 1, 2).

Plasma concentration–time curves (mean and standard deviation) of aripiprazole (filled circles) and dehydro-aripiprazole (open circles) after the administration of an oral single 10-mg dose of aripiprazole in healthy adults according to their cytochrome P450 2D6 phenotype. Solid colored lines indicate the mean and the colored area indicates the ± standard deviation of the physiologically based pharmacokinetic model. Circles indicate observed plasma concentrations. a Normal metabolizer (n = 28). b Intermediate metabolizer (n = 21). c Poor metabolizer (n = 1). d Ultra-rapid metabolizer (n = 5)

Plasma concentration–time curves (mean and standard deviation) of aripiprazole (filled circles) and dehydro-aripiprazole (open circles) after the administration of an orodispersible single 10-mg dose of aripiprazole in healthy adults according to their cytochrome P450 2D6 phenotype. Solid colored lines indicate the mean and the colored area indicates the ± standard deviation of the physiologically based pharmacokinetic model. Circles indicate observed plasma concentrations. a Normal metabolizer (n = 27). b Intermediate metabolizer (n = 13). c Poor metabolizer (n = 2)
Physiologically based pharmacokinetic models for oral and orodispersible tablets of ARI can accurately predict the influence of the CYP2D6 phenotype, as can be seen by the respective MPE and MAPE values (Table 5). For oral tablet formulations, models show a minimal bias (MPE) with a range of − 2.80 to 4.36% for ARI and − 17.1 to 20.2% for D-ARI and a good precision (MAPE) with a range of 4.26–18.9% for ARI and 11.6–22.3% for D-ARI in their prediction. In accordance with this, simulations for the orodispersible tablet formulation also show a high accuracy illustrated by a minimal bias (MPE) with a range of − 30.0 to 19.4% for ARI and D-ARI as well as a good precision (MAPE) with a range of 5.53–30.0% for ARI and D-ARI, respectively. Moreover, 92.5% (oral tablet) and 79.3% (orodispersible tablet) of the ARI plasma concentrations and 67.5% (oral tablet) and 69.0% (orodispersible tablet) of the D-ARI plasma concentrations were in the 1.25-fold error range in the goodness-of-fit plots (Fig. 3a, c). It is noticeable that the simulations overestimate the plasma concentrations of ARI in PMs, especially in the terminal phase. The overestimation of ARI can be seen in both types of formulations, suggesting that the clearance of ARI is under-predicted. This in turn leads also to an underestimation of the plasma concentrations of the active metabolite D-ARI resulting in respective values for bias and precision.

a, c Goodness-of-fit plot for the model prediction of aripiprazole (filled circles) and dehydro-aripiprazole (open circles) plasma concentrations in healthy adults. b, d Relative residuals vs observed aripiprazole and dehydro-aripiprazole concentrations for healthy adult volunteers over the entire concentration range. The solid black line indicates the line of identity, dotted lines indicate the ±30% range, dashed lines indicate ± 50%, and filled (aripiprazole) and open (dehydro-aripiprazole) circles indicate observed data. Black circles indicate normal metabolizers, blue circles indicate intermediate metabolizers, red circles indicate poor metabolizers, and green circles indicate ultra-rapid metabolizers. a, b oral formulation. c, d orodispersible formulation
3.2 Multiple-Dose Modeling
Multiple-dose PBPK models were able to predict the pharmacokinetics of ARI and its active metabolite in healthy volunteers, especially in NMs and IMs (Fig. 4). Simulations for both phenotypes showed minimal bias illustrated by an overall MPE of 21.0% (NMs), 23.6% (IMs), and a good precision represented by an overall MAPE of 31.9% (NMs) and 33.9% (IMs) of ARI. At trough concentrations (24, 48, 72, and 96 h after the first dose), models showed a MAPE range for ARI of 1.12–11.9% for NMs, 7.71–14.3% for IMs, and 34.8–97.6% for UMs as well as a range for D-ARI of 8.34–22.3% for NMs, 1.98–38.5% for IMs, and 12.3–28.2% for UMs. An overestimation of the plasma concentrations of the parent drug can be detected in UMs (n = 2), resulting in respective values for bias and precision compared to NMs (n = 14) and IMs (n = 8). However, 63.6% of all UMs observed plasma concentrations of the two participants were within the predicted standard deviation. For PMs, only predicted plasma concentration–time curves are presented, as no healthy volunteer enrolled in the multiple-dose clinical trial was genotyped as CYP2D6 PM.

Plasma concentration–time curve profiles (mean and standard deviation) of aripiprazole (filled circles) and dehydro-aripiprazole (open circles) after the administration of multiple oral 10-mg tablets of aripiprazole in healthy adults according to their cytochrome P450 2D6 phenotype. Solid colored lines indicate the mean and the colored area indicates the ± standard deviation of the physiologically based pharmacokinetic model. Circles indicate observed plasma concentrations. a Normal metabolizer (n = 14). b Intermediate metabolizer (n = 8). c Poor metabolizer. For poor metabolizers, only the physiologically based pharmacokinetic simulation is shown. d Ultra-rapid metabolizer (n = 2)
3.3 Steady-State Modeling
Steady-state simulations were performed for ARI and the active moiety and compared to the respective therapeutic reference ranges [10]. With respect to the simulations of ARI, all predicted mean trough plasma concentrations for NMs, IMs, PMs, and UMs were in the therapeutic reference range of 100–350 µg/L (Fig. 5). Here, mean predicted Css,min amounts to 164 µg/L (NMs), 174 µg/L (IMs), 215 µg/L (PMs), and 153 µg/L (UMs) and its corresponding Css,max amounts to 224 µg/L (NM), 235 µg/L (IMs), 277 µg/L (PMs), and 214 µg/L (UMs). However, predicted upper standard deviation at Css,max for both IMs (362 µg/L) and PMs (424 µg/L) were outside the therapeutic reference range. This is in accordance with the steady-state simulations of the active moiety (Fig. 6). Here, mean predicted Css,max of the active moiety amounts to 332 µg/L for IM resulting in an upper value of the standard deviation of 505 µg/L. In PMs, mean predicted Css,max of the active moiety tended to be higher than IMs and amounts to 372 µg/L and its corresponding upper value of the standard deviation to 566 µg/L. Thus, predicted upper values of the standard deviation of IMs and PMs are above the therapeutic reference range. In contrast to the upper limit of the standard deviation, all predicted mean trough plasma concentrations of the active moiety are within the therapeutic reference range.

Predicted concentration–time profiles of aripiprazole after the administration of multiple oral 15-mg daily doses of aripiprazole according to their cytochrome P450 2D6 phenotype. The solid lines indicate the mean plasma concentrations, the dashed lines indicate the ± standard deviation. The colored boxes represent the therapeutic reference range between 100 and 350 µg/L. a Normal metabolizer. b Intermediate metabolizer. c Poor metabolizer. d Ultra-rapid metabolizer

Predicted concentration–time profiles of the active moiety (aripiprazole plus dehydro-aripiprazole) after the administration of multiple oral 15-mg daily doses of aripiprazole according to their cytochrome P450 2D6 phenotype. The solid lines indicate the mean plasma concentrations, the dashed lines indicate the ± standard deviation. The colored boxes represent the therapeutic reference range between 150 and 500 µg/L. a Normal metabolizer. b Intermediate metabolizer. c Poor metabolizer. d Ultra-rapid metabolizer
3.4 Dose Adjustment
Based on steady-state simulations, dose adjustments for all phenotypes were considered. For IMs and UMs, no adjustment was necessary. Regarding PMs, the daily dose was reduced from 15 to 10 mg (Fig. 7). As a consequence, mean predicted Css,min of ARI decreased from 215 to 172 µg/L and mean Css,max from 277 to 222 µg/L. This trend was also evident in the simulations of the active moiety. Mean predicted Css,min decreased from 308 to 249 µg/L and Css,max decreased from 372 to 301 µg/L. The dose reduction also leads to a decrease of the upper predicted standard deviation of Css,max by − 22.6%, which means a value of 438 µg/L. As a result of the dose adjustment for PMs, both predicted means and standard deviations do not exceed the upper limit of the therapeutic reference range.

Predicted concentration–time profiles after aripiprazole dose adjustment to 10 mg/day in cytochrome P450 2D6 poor metabolizers. The solid lines indicate the mean plasma concentrations and the dashed lines indicate the ± standard deviation. The colored boxes represent the therapeutic reference range between 150 and 500 µg/L. a Aripiprazole. b Active moiety
4 Discussion
In the present study, we analyzed the influence of CYP2D6 polymorphism on the pharmacokinetics of ARI and D-ARI by PBPK modeling and simulation. For the first time, various PBPK models for single-dose and multiple-dose predictions of the plasma concentrations of ARI, D-ARI, and the active moiety were developed and evaluated, according to the CYP2D6 phenotype. Although a previous PBPK study reported a relationship between the CYP2D6 genetic polymorphism and the pharmacokinetics of ARI [31], to the best of our knowledge no other PBPK studies integrated CYP2D6-related information to optimize dosing strategies according to phenotypes.
The developed PBPK models successfully predict the pharmacokinetics of ARI and D-ARI according to the different CYP2D6 phenotypes, as demonstrated by minimal bias and good precision after single-dose administration in both formulations (Table 5). Overall, the models accurately predicted absorption, distribution, and elimination phases, showing precise AUC ratio values (Table 5), a Cmax 3 h after dosing, and a mean ARI t1/2 of 68.6 h, which is in accordance with the prescribing information provided by the manufacturer and previous investigations [3, 32].
In CYP2D6 PMs, predicted Cmax, tmax, and t1/2 of ARI tended to be higher and the elimination much slower compared with NMs, IMs, and UMs (Figs. 1, 2). The main reason for the variation in pharmacokinetic parameters of PMs is the loss of catalytic enzyme activity of CYP2D6 based on two no function CYP2D6 alleles [12, 33, 34]. Consequently, PMs do not convert ARI into its active metabolite D-ARI through CYP2D6, but only through CYP3A4. Therefore, Cmax and tmax of ARI increase and t1/2 extends compared with the other phenotypes. Regarding the clearance and AUC within PMs, single-dose PBPK models suggest an under-prediction of the clearance of ARI, resulting in a greater predicted AUCtend than observed (ratio 1.19 oral and 1.23 orodispersible formulation; Table 5). In contrast, clearance of the active metabolite was over-predicted with an AUCtend ratio of 0.92 for the oral formulation and 0.77 for the orodispersible formulation (Table 5). For model application in a clinical environment, more data from CYP2D6 PMs are needed to verify and further revise the determination of hepatic and renal clearance. In addition, information on a patient’s hepatic and/or renal impairment is required for dose individualization. Multiple-dose PBPK models for UMs showed an overestimation in the plasma concentrations of ARI (Fig. 4), whereas single-dose PBPK models showed a slight underestimation in the terminal phase (Figs. 1, 2). One reason for this variation is the low number of participants in the multiple-dose study. Here, only two participants were genotyped as UMs (Table 2). In addition to the limited plasma concentration data, one of the participants was identified as Caucasian, the other one as Latin American, resulting in a large observed inter-individual variability of ARI and D-ARI between both participants. For modeling and simulation of UMs, an European population has been created according to the International Commission on Radiological Protection guideline [35], as Latin Americans are not part of the modeling software package [18]. Although a European population was developed, almost 63.6% of the observed plasma concentrations were within the standard deviation of the prediction. To further optimize the PBPK model for UMs, more data are needed to consider more plasma concentrations for modeling and simulation. In this context, not only data from healthy volunteers should be used, but also data from patients with schizophrenia.
In the present study, our steady-state simulations of ARI and the sum of ARI plus D-ARI showed higher predicted plasma concentrations of PMs compared with the other three phenotypes (Figs. 5, 6). Because of the complex mechanisms of action of ARI at different receptors (see Sect. 1), the clinical consequences of higher plasma concentrations might be difficult to predict, especially in PMs. As previously reported, the most frequent adverse events leading to discontinuation were psychosis and agitation [36]. Furthermore, different case reports of exacerbation of psychosis related to initiation of therapy have been described [37]. Based on the reported adverse drug reaction profiles of ARI, it is possible that an increased plasma concentration actually triggers psychotic symptoms, especially in PMs [38]. The increase in predicted steady-state plasma concentrations indicates that a dose reduction for PMs should be considered (Figs. 5, 6). This dose adjustment would achieve similar plasma concentrations as NMs, IMs, and UMs and reduces the risk of suffering under potential adverse events. Our PBPK simulations for CYP2D6 PMs permit us to recommend a reduction from 15 to 10 mg/day, representing a decrease of 33.3% (Fig. 7). These findings are in accordance with previous investigations by Hendset et al. reporting a dose reduction of 30–40% for PMs [38]. In contrast to that, the manufacturer’s prescribing information referring to the US Food and Drug Administration label [3] as well as the Dutch Pharmacogenetics Working Group recommend half of the usual dose for known CYP2D6 PMs. Here, our PBPK simulations show that an overall dose reduction of 50% should be avoided and a more individualized dose adjustment is advantageous. As the maximum daily dose of ARI amounts to 30 mg [3], a potential dose adjustment of 50% would lead to a daily dose of 15 mg, which will cause plasma concentrations above the therapeutic reference range (Fig. 7). Regarding the predicted plasma concentrations and the available oral dosage forms and strengths (2 mg, 5 mg, 10 mg, 15 mg, 20 mg, and 30 mg of Abilify®), an administration of not more than 10 mg/day should be recommended in PMs to ensure the best and safest therapy for each patient with schizophrenia.
Potential limitations should be considered when interpreting our results of PBPK modeling and simulation. First, study data from healthy volunteers were used rather than patients with schizophrenia, which may be limiting when applying the results in clinical practice. The applicability needs to be evaluated, including pharmacokinetic data of patients with schizophrenia as well as elderly patients. Furthermore, more data of non-Caucasian individuals such as African or Asian individuals should be taken into account to analyze the impact of different ethnicities and to further optimize PBPK models. It is apparent that not only genetic but also environmental, physiological, pathological, and pharmacological aspects such as co-medication or co-morbidity should be considered to personalize dosing of ARI in a clinical environment. In this context, other variables such as age and renal or hepatic impairment as well as acute infections or inflammations should be taken into consideration when interpreting modeling and simulation results for clinical application. Nonetheless, PBPK models presented in this work allow us to optimize dosing strategies for CYP2D6 PMs. Within CYP2D6 IMs and UMs, no dose adjustments have to be taken into account. It is apparent that not only genetic polymorphisms but also concomitant strong CYP2D6 and/or CYP3A4 inhibitors should be considered. In this regard, dose adjustments are necessary according to the prescribing information of the manufacturer [3]. Furthermore, food–drug interactions and other variables such as co-morbidity should be considered to finally personalize dosing in the clinical environment [39]. Nonetheless, based on predicted and observed high inter-individual variability of plasma concentrations between different CYP2D6 phenotypes, therapeutic drug monitoring in combination with genotyping should be considered to ensure the best clinical outcome and a minimum of adverse drug reactions. For this purpose, analytical methods for plasma concentration measurements of ARI and D-ARI within therapeutic drug monitoring are already developed and ready for use. Nonetheless, the implementation of pharmacogenetic analysis and therapeutic drug monitoring is not common in psychiatry, but would be very beneficial for patients to ensure the effectiveness and drug safety of ARI therapy.
5 Conclusions
The present PBPK study demonstrates a CYP2D6-related influence of the genetic polymorphisms on the plasma concentrations and pharmacokinetics of ARI and D-ARI according to different phenotypes. For CYP2D6 PMs, PBPK models indicate a dose reduction to achieve steady-state plasma concentrations comparable to those of the other phenotypes. Here, PBPK models suggest a maximum daily dose for patients with schizophrenia of 10 mg. However, IMs and UMs do not need a dose adjustment. Based on the highly recommended dose adjustment for CYP2D6 PMs, close therapeutic drug monitoring of ARI and D-ARI in combination with CYP2D6 genotyping should be considered to ensure a personalized and safe therapy for each patient. Therefore, efforts must be made to convince and involve psychiatrists more often, to make use of personalized treatment strategies through pharmacogenetics or therapeutic drug monitoring to ensure the efficacy, safety, and adherence of the patient.
References
James SL, Abate D, Abate KH, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392(10159):1789–858.
Laursen TM, Nordentoft M, Mortensen PB. Excess early mortality in schizophrenia. Annu Rev Clin Psychol. 2014;10:425–48.
Abilify® (aripiprazole): U.S. full prescribing information. Available from: https://www.otsuka-us.com/media/static/Abilify-PI.pdf. Accessed 10 Oct 2020.
de Bartolomeis A, Tomasetti C, Iasevoli F. Update on the mechanism of action of aripiprazole: translational insights into antipsychotic strategies beyond dopamine receptor antagonism. CNS Drugs. 2015;29(9):773–99.
Lawler C, Prioleau C, Lewis M, et al. Interactions of the novel antipsychotic aripiprazole (OPC-14597) with dopamine and serotonin receptor subtypes. Neuropsychopharmacology. 1999;20(6):612–27.
Stip E, Tourjman V. Aripiprazole in schizophrenia and schizoaffective disorder: a review. Clin Ther. 2010;32(Suppl. 1):S3-20.
McGavin JK, Goa KL. Aripiprazole. CNS Drugs. 2002;16(11):779–86 (discussion 787–788).
Kirschbaum KM, Müller MJ, Malevani J, et al. Serum levels of aripiprazole and dehydroaripiprazole, clinical response and side effects. World J Biol Psychiatry. 2008;9(3):212–8.
Wood MD, Scott C, Clarke K, et al. Aripiprazole and its human metabolite are partial agonists at the human dopamine D2 receptor, but the rodent metabolite displays antagonist properties. Eur J Pharmacol. 2006;546(1–3):88–94.
Hiemke C, Bergemann N, Clement HW, Conca A, Deckert J, Domschke K, Eckermann G, Egberts K, Gerlach M, Greiner C, Gründer G, Haen E, Havemann-Reinecke U, Hefner G, Helmer R, Janssen G, Jaquenoud E, Laux G, Messer T, Mössner R, Müller MJ, Paulzen M, Pfuhlmann B, Riederer P, Saria A, Schoppek B, Schoretsanitis G, Schwarz M, Gracia MS, Stegmann B, Steimer W, Stingl JC, Uhr M, Ulrich S, Unterecker S, Waschgler R, Zernig G, Zurek G, Baumann P. Consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology: Update 2017. Pharmacopsychiatry. 2018;51(1-02):9–62. https://doi.org/10.1055/s-0043-116492.
Molden E, Lunde H, Lunder N, Refsum H. Pharmacokinetic variability of aripiprazole and the active metabolite dehydroaripiprazole in psychiatric patients. Ther Drug Monit. 2006;28(6):744–9.
Zhou S-F. Polymorphism of human cytochrome P450 2D6 and its clinical significance: part I. Clin Pharmacokinet. 2009;48(11):689–723.
Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharmacol Ther. 2008;83(2):234–42.
Gaedigk A, Sangkuhl K, Whirl-Carrillo M, Klein T, Leeder JS. Prediction of CYP2D6 phenotype from genotype across world populations. Genet Med. 2017;19(1):69–76.
Ingelman-Sundberg M, Oscarson M, McLellan RA. Polymorphic human cytochrome P450 enzymes: an opportunity for individualized drug treatment. Trends Pharmacol Sci. 1999;20(8):342–9.
Kneller LA, Abad-Santos F, Hempel G. Physiologically based pharmacokinetic modelling to describe the pharmacokinetics of risperidone and 9-hydroxyrisperidone according to cytochrome P450 2D6 phenotypes. Clin Pharmacokinet. 2020;59(1):51–65.
Willmann S, Lippert J, Sevestre M, Solodenko J, Fois F, Schmitt W. PK-Sim®: a physiologically based pharmacokinetic ‘whole-body’ model. Biosilico. 2003;1(4):121–4.
Open Systems Pharmacology Suite. Open system pharmacology manual. Available from: https://docs.open-systems-pharmacology.org/. Accessed 19 Sept 2020.
Willmann S, Höhn K, Edginton A, et al. Development of a physiology-based whole-body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J Pharmacokinet Pharmacodyn. 2007;34(3):401–31.
Eissing T, Kuepfer L, Becker C, et al. A computational systems biology software platform for multiscale modeling and simulation: integrating whole-body physiology, disease biology, and molecular reaction networks. Front Physiol. 2011;2:4.
Belmonte C, Ochoa D, Román M, et al. Influence of CYP2D6, CYP3A4, CYP3A5 and ABCB1 polymorphisms on pharmacokinetics and safety of aripiprazole in healthy volunteers. Basic Clin Pharmacol Toxicol. 2018;122(6):596–605.
Caudle KE, Sangkuhl K, Whirl-Carrillo M, et al. Standardizing CYP 2D6 genotype to phenotype translation: consensus recommendations from the Clinical Pharmacogenetics Implementation Consortium and Dutch Pharmacogenetics Working Group. Clin Transl Sci. 2020;13(1):116–24.
Koller D, Saiz-Rodríguez M, Zubiaur P, et al. The effects of aripiprazole and olanzapine on pupillary light reflex and its relationship with pharmacogenetics in a randomized multiple-dose trial. Br J Clin Pharmacol. 2020;86(10):2051–62.
Mallikaarjun S, Shoaf SE, Boulton DW, Bramer SL. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet. 2008;47(8):533–42.
Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–82.
Poulin P, Theil F-P. Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J Pharm Sci. 2002;91(1):129–56.
Ursu O, Holmes J, Knockel J, et al. DrugCentral: online drug compendium. Nucleic Acids Res. 2017;45(D1):D932–9.
Takács-Novák K, Urac M, Horváth P, Völgyi G, Anderson BD, Avdeef A. Equilibrium solubility measurement of compounds with low dissolution rate by Higuchi’s facilitated dissolution method: a validation study. Eur J Pharm Sci. 2017;106:133–41.
European Medicines Agency. Guideline on the reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation. Amsterdam. 2018. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-reporting-physiologically-based-pharmacokinetic-pbpk-modelling-simulation_en.pdf.
R Core Team (2020). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/
Vieira MDLT, Kim M-J, Apparaju S, et al. PBPK model describes the effects of comedication and genetic polymorphism on systemic exposure of drugs that undergo multiple clearance pathways. Clin Pharmacol Ther. 2014;95(5):550–7.
Mallikaarjun S, Kane JM, Bricmont P, et al. Pharmacokinetics, tolerability and safety of aripiprazole once-monthly in adult schizophrenia: an open-label, parallel-arm, multiple-dose study. Schizophr Res. 2013;150(1):281–8.
Sachse C, Brockmöller J, Bauer S, Roots I. Cytochrome P450 2D6 variants in a Caucasian population: allele frequencies and phenotypic consequences. Am J Hum Genet. 1997;60(2):284–95.
Mizutani T. PM frequencies of major CYPs in Asians and Caucasians. Drug Metab Rev. 2003;35(2–3):99–106.
Valentin J. Basic anatomical and physiological data for use in radiological protection: reference values: a report of age- and gender-related differences in the anatomical and physiological characteristics of reference individuals. Ann ICRP. 2002;32(3–4):5–265.
Marder SR, McQuade RD, Stock E, et al. Aripiprazole in the treatment of schizophrenia: safety and tolerability in short-term, placebo-controlled trials. Schizophr Res. 2003;61(2–3):123–36.
Ramaswamy S, Vijay D, William M, Sattar SP, Praveen F, Petty F. Aripiprazole possibly worsens psychosis. Int Clin Psychopharmacol. 2004;19(1):45–8.
Hendset M, Hermann M, Lunde H, Refsum H, Molden E. Impact of the CYP2D6 genotype on steady-state serum concentrations of aripiprazole and dehydroaripiprazole. Eur J Clin Pharmacol. 2007;63(12):1147–51.
Spina E, de Leon J. Clinical applications of CYP genotyping in psychiatry. J Neural Transm (Vienna). 2015;122(1):5–28.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
Open Access funding enabled and organized by Projekt DEAL. Dora Koller and the multiple-dose phase I trial (EUDRA-CT: 2018-000744-26) were co-financed by the H2020 Marie Sklodowska-Curie Innovative Training Network 721236 grant.
Conflict of interest
Lisa Alina Kneller, Pablo Zubiaur, Dora Koller, Francisco Abad-Santos, and Georg Hempel have no conflicts of interest that are directly relevant to the content of this article.
Ethics approval
All clinical trials were duly authorized by the Spanish Drugs Agency (AEMPS) and the Ethics Committee of Hospital Universitario de La Princesa, Madrid, Spain. They were carried out under the guidelines of Good Clinical Practices, complying with current Spanish legislation on clinical research in humans and with the Declaration of Helsinki. EUDRA-CT (https://eudract.ema.europa.eu/) for single-dose clinical trials: 2012-004241-32, 2012-005274-60, 2012-002016-97, 2012-003196-19, 2013-002478-35, and 2013-002478-35. EUDRA-CT for multiple-dose phase I trial: 2018-000744-26 (ethics committee registry number: 3411)
Consent for participation
All subjects provided their informed consent for the present research.
Consent for publication
All authors provided their informed consent for publication.
Availability of data and material
Data are available from the corresponding author upon reasonable request.
Code availability
Codes are available from the corresponding author upon reasonable request.
Author contributions
Writing of the manuscript: LAK. Writing of the manuscript, review and editing: PZ, DK, FA-S, GH. Modeling and simulation: LAK, GH. Designed research: LAK, FA-S, GH. Determination of drug concentration: PZ, DK. Pharmacogenetics: PZ, DK.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kneller, L.A., Zubiaur, P., Koller, D. et al. Influence of CYP2D6 Phenotypes on the Pharmacokinetics of Aripiprazole and Dehydro-Aripiprazole Using a Physiologically Based Pharmacokinetic Approach. Clin Pharmacokinet 60, 1569–1582 (2021). https://doi.org/10.1007/s40262-021-01041-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-01041-x