
Abstract
Background and Objective
Omecamtiv mecarbil is a novel selective cardiac myosin activator (myotrope) under investigation for the treatment of heart failure with reduced ejection fraction. The objective of this clinical study was to estimate the effect of varying degrees of renal impairment on the pharmacokinetics of omecamtiv mecarbil single dose (50 mg) under fasted conditions.
Methods
This phase I, open-label, non-randomized, parallel-group study evaluated the pharmacokinetics, safety, and tolerability of a single oral dose of omecamtiv mecarbil 50 mg in individuals with normal renal function or mild, moderate, and severe renal impairment, including end-stage renal disease requiring dialysis. Geometric least-squares mean ratios of maximum observed concentration (Cmax) and area under the plasma concentration–time curve (AUC) and 90% confidence intervals were derived for comparisons of renal impairment vs normal renal function. Participants were monitored for adverse events.
Results
Thirty-one participants received treatment and completed the study. Geometric mean exposures were similar for participants with renal impairment (AUC∞ range, 2550–3220 h*ng/mL; Cmax range, 78.9–107 ng/mL) and participants with normal renal function (AUC∞, 2790 h*ng/mL; Cmax, 92.6 ng/mL), with geometric least-squares mean ratios of 85.2–125.9. Exposure was similar on dialysis vs non-dialysis days in participants with end-stage renal disease (AUC0–24, 1650 vs 1700 h*ng/mL; Cmax, 100.0 vs 107.0 ng/mL). Four participants (12.9%) reported four treatment-emergent adverse events. No deaths, treatment-emergent adverse events leading to discontinuation, or serious adverse events occurred.
Conclusions
Omecamtiv mecarbil pharmacokinetics were not meaningfully affected by renal function or hemodialysis, suggesting the same dosing strategy can be used in individuals with normal renal function or renal impairment. Oral administration of omecamtiv mecarbil was not associated with major tolerability findings. This study supports omecamtiv mecarbil for the treatment of heart failure in individuals with or without renal impairment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Omecamtiv mecarbil (OM) is a novel selective cardiac myosin activator under investigation for the treatment of heart failure with reduced ejection fraction |
Results of this open-label, phase I study of a single oral dose of OM in individuals with normal renal function or mild, moderate, and severe renal impairment showed that the pharmacokinetics of OM were not meaningfully affected by renal impairment, and no major tolerability issues with OM were reported |
This study supports OM for the treatment of heart failure in individuals with or without renal impairment without the need for adjustments to the dosing strategy |
1 Introduction
Heart failure (HF) is the final stage for many cardiovascular diseases [1]. Overall, HF affects ~ 64 million people worldwide [2] and is associated with high rates of hospitalization and mortality [3]. Decreased systolic function and impaired cardiac contractility are central factors in the pathogenesis of HF [4].
Omecamtiv mecarbil (OM) is a novel selective cardiac myosin activator that increases cardiac contractility without increasing cardiac myocyte intracellular calcium [5, 6]. In clinical studies, oral and intravenous formulations of OM were associated with improved cardiac function and reduced left ventricular dimensions in patients with HF [7, 8]. The efficacy and safety of oral OM in patients with HF with reduced ejection fraction have been investigated in the phase III GALACTIC-HF study and the ongoing METEORIC-HF study [9, 10]. In these studies, oral OM was administered at starting doses of 25 mg twice daily and was titrated to 25 mg, 37.5 mg, or 50 mg twice daily on the basis of trough plasma concentrations of OM [9].
Renal dysfunction is highly prevalent in patients with chronic HF, with approximately 90% of patients reporting some degree of renal dysfunction (defined as an estimated glomerular filtration rate [eGFR] of < 90 mL/min/1.73 m2) [11]. Omecamtiv mecarbil is extensively metabolized by multiple enzymes, including enzymes from the cytochrome P450 family, with approximately 11% of a single dose recovered as the parent drug in feces and urine. Furthermore, oral OM was demonstrated to have high relative bioavailability in immediate-release (> 90%) and modified-release (> 75%) formulations [12]. However, renal impairment can affect hepatic and intestinal drug metabolism and transport pathways [13], which can alter the pharmacokinetics (PK) of drugs, potentially affecting the dosing or effectiveness of therapies [14]. Thus, pharmacokinetic studies of most drugs intended for long-term use, such as OM, are important to understand, as dosing adjustments may be necessary in patients with renal impairment. The objectives of this study were to evaluate the effect of renal function on the PK, safety, and tolerability of a single oral dose of OM in individuals with normal renal function or with various degrees of renal impairment and to assess the effect of hemodialysis on the PK of OM in individuals with end-stage renal disease (ESRD) requiring dialysis.
2 Methods
2.1 Study Design
This was a phase I, single-dose, open-label, non-randomized, parallel-group study to evaluate the PK, safety, and tolerability of OM in individuals with normal renal function or varying degrees of renal impairment. The study consisted of a screening period lasting up to 21 days followed by a single 8-day treatment period (defined as check-in to discharge) for individuals with normal renal function or mild, moderate, or severe renal impairment (groups 1–4, defined below and in Table 1 of the Electronic Supplementary Material [ESM]) or two treatment periods (period 1, 2 days; period 2, 2 days) separated by a washout period of 7–14 days for individuals with ESRD (group 5; Fig. 1 of the ESM). An end-of-study visit occurred 12–16 days after the single OM dose for groups 1–4 and after the second OM dose (during treatment period 2) for group 5. Participants received a single oral dose of OM 50 mg on day 1 of each treatment period after fasting overnight for ≥ 10 h; fasting continued for ≥ 4 h post-dose. For participants in group 5, OM was administered 3 h before the start of dialysis in treatment period 1 and on a non-dialysis day in treatment period 2. The OM dose was administered as a film-coated oral tablet with a modified-release matrix formulation, which has been previously described [12].
The study was conducted in accordance with ethical guidelines from the Declaration of Helsinki and Council for International Organizations of Medical Sciences, applicable Good Clinical Practice guidelines of the International Council for Harmonization, and applicable local laws and regulations. An Advarra® (Columbia, MD, USA) institutional review board approved the research protocol and study conduct. All study participants provided written informed consent before enrollment in the study and could withdraw from the study at any time. Qualified researchers may request data from Amgen clinical studies; complete details are available at http://www.amgen.com/datasharing.
2.2 Study Participants
Eligibility was determined by medical history, physical examination, vital signs, laboratory values, and cardiac monitoring at screening and check-in. Eligible participants were male or female individuals aged 18–65 years (inclusive) for treatment groups 2 and 3 or aged 18–75 years (inclusive) for treatment groups 1, 4, and 5, with a body mass index between 18.0 and 38.0 kg/m2 (inclusive); only female individuals who were not of childbearing potential were eligible for the study. Non-hypertensive individuals or individuals with treated and stable hypertension were included in renal impairment groups (groups 2–5) if their systolic blood pressure did not exceed 170 mmHg and their diastolic blood pressure did not exceed 100 mm Hg at screening and check-in; individuals with renal impairment were eligible if the dosage and nature of their antihypertensive medication were stable for ≥ 4 weeks before screening and were expected to remain unchanged throughout the study duration.
Eligible participants were enrolled in one of five renal function groups on the basis of US Food and Drug Administration guidance for renal impairment studies [15] and as measured by eGFR and the need for renal replacement therapy (Table 1 of the ESM). The eGFR was calculated using serum creatinine levels and the Modification of Diet in Renal Disease (MDRD) formula [15]: MDRD formula (mL/min/1.73 m2) = 175 × (serum creatinine)−1.154 × (age)−0.203 × (0.742 if female) × (1.212 if African American). Participants in group 1 were considered to have normal renal function (eGFR, ≥ 90 mL/min/1.73 m2 and no history of renal disease); participants in group 2 had mild renal impairment (eGFR, 60–89 mL/min/1.73 m2); participants in group 3 had moderate renal impairment (eGFR, 30–59 mL/min/1.73 m2); participants in group 4 had severe renal impairment (eGFR, 15–29 mL/min/1.73 m2 without dialysis); and participants in group 5 had ESRD requiring dialysis and had been receiving hemodialysis for ≥ 1 month before screening or had an eGFR < 15 mL/min/1.73 m2. Participants in group 1 (normal renal function) were selected so that the mean and distribution of their age, sex, and body mass index matched those of the participants in the renal impairment groups.
Exclusion criteria were related to medical history (e.g., history of uncontrolled or unstable cardiovascular, respiratory, hepatic, gastrointestinal, endocrine, hematopoietic, psychiatric, or neurological disease within 6 months of screening) and laboratory screening tests, including aspartate aminotransferase or alanine aminotransferase levels more than two times the upper limit of normal, and clinically significant hyperkalemia (serum potassium level > 5 mmol/L for groups 1–3 and > 5.5 mmol/L at check-in or within 24 h of the last dialysis session for groups 4–5). Exclusion criteria also included elevated levels of biomarkers associated with coronary events: creatine kinase or creatine kinase muscle/brain levels greater than the upper limit of normal for group 1 or at levels inconsistent with chronic kidney disease (groups 2–5) in the opinion of the investigator at screening, and troponin I levels greater than the upper limit of normal for group 1 or at levels inconsistent with chronic kidney disease (groups 2–5) in the opinion of the investigator at screening or check-in. Participants were excluded if they had previous exposure to the study drug or if they had prior or concomitant use of over-the-counter or prescription drugs that could affect the PK of the study drug. Participants with renal impairment could continue receiving concomitant medication necessary for maintaining their clinical status during the study if they were taking the medication for ≥ 1 month before study drug administration.
2.3 Pharmacokinetic Sampling
Individuals in groups 1–4 remained at the study site and were supervised for approximately 9 days, beginning 2 days before dosing and lasting until collection of the last pharmacokinetic sample at 144 h post-dose. Pooled urine samples for measurement of OM concentrations were collected at 0–12, 12–24, 24–48, 48–72, 72–96, 96–120, and 120–144 h post-dose; urine samples for estimation of creatinine clearance were collected 24 h pre-dose (Table 2 of the ESM). Blood samples collected by venipuncture or cannulation for measurement of plasma OM concentrations were collected pre-dose and at 0.5, 1, 2, 3, 4, 6, 8, 12, 24, 48, 72, 96, 120, and 144 h post-dose. The lower limit of quantification for plasma samples was 1 ng/mL; the lower limit of quantification for urine samples was 50 ng/mL.
Individuals with ESRD (group 5) remained at the study site and were supervised for approximately 3 days in each treatment period, beginning 1 day before dosing and lasting until collection of the last pharmacokinetic samples at 24 h post-dose. Blood samples collected by venipuncture were collected for determining plasma OM concentrations pre-dose and at the same intervals as those used for groups 1–4 on the dialysis day in treatment period 1 and pre-dose and at 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h post-dose on day 1 in treatment period 2 (non-dialysis day; Table 2 of the ESM). During dialysis in treatment period 1, arterial (pre-dialysis) and venous (post-dialysis) plasma samples, as well as dialysate for measurement of OM concentration, were collected at 0.5, 1, 2, 3, and 4 h after the start of dialysis.
2.4 Study Assessments
The primary endpoints used to assess the effect of renal function on the PK of OM were maximum observed plasma concentration (Cmax), area under the plasma concentration–time curve from time zero to infinity (AUC∞), and fraction of dose excreted unchanged in urine (fe); the primary endpoints used to assess the effect of hemodialysis on the PK of OM in individuals with ESRD were Cmax and AUC from time zero to 24 h post-dose (AUC0–24). Additional endpoints included time to reach Cmax (tmax), AUC from time zero to time of last quantifiable concentration, apparent plasma terminal elimination half-life, apparent total body clearance, apparent renal clearance, and apparent volume of distribution.
2.5 Safety Evaluation
Secondary endpoints assessed the safety and tolerability of OM and included the monitoring of adverse events (AEs), clinical laboratory tests, 12-lead electrocardiograms, and vital signs. Monitoring of AEs occurred throughout the study. Clinical chemistry and hematology evaluations were conducted 24 h post-dose; in the case of a suspected coronary event, blood samples for troponin I and/or creatine kinase muscle/brain fraction could be collected at other times. Blood pressure and pulse rate, 12-lead electrocardiograms, and oral body temperature were recorded at screening, checking, scheduled timepoints post-dose, and at the end of the study visit (Table 2 of the ESM).
2.6 Statistical Analysis
The pharmacokinetic parameters Cmax, tmax, apparent plasma terminal elimination half-life, AUC∞ (for groups 1–4; for group 5, period 1 only), fe, and apparent renal clearance were determined using non-compartmental methods. Plasma pharmacokinetic parameter values including arithmetic mean, geometric mean, median, standard deviation, minimum, maximum, and coefficient of variation were calculated for each renal function group.
To determine the effect of renal impairment on the PK of OM, linear regression analyses were performed on the log-transformed Cmax and AUC values as a function of the eGFR and creatinine clearance from pre-dose (− 24 to 0 h); body weight, sex, and age were considered as possible covariates, as these were the characteristics used to match participants in group 1 with the participants in groups 2–5 with renal impairment. For group 5, to determine the effect of hemodialysis on the PK of OM, a repeated-measures analysis was performed.
Geometric least-squares mean ratios (GMRs) and 90% confidence intervals (CIs) were derived for comparisons of renal impairment (groups 2–5) vs normal renal function (group 1) and for the dialysis day (group 5, period 1) vs the non-dialysis day (group 5, period 2) for participants with ESRD. Safety outcomes were summarized using descriptive statistics. All statistical analyses were conducted by SAS Enterprise Guide, Version 7.13 (SAS Institute Inc., Cary, NC, USA).
3 Results
3.1 Participant Disposition and Baseline Characteristics
A total of 31 participants were screened and enrolled in the study; all participants completed the study (Table 1). Each renal function group enrolled six participants, except for group 5 (ESRD requiring dialysis), which had seven participants. Overall, 25 participants (80.6%) were male, mean age was 56.3 years, and mean body mass index was 28.5 kg/m2. The proportion of Black and White participants was 29.0% and 71.0%, respectively.
3.2 Effect of Renal Impairment on PK of OM
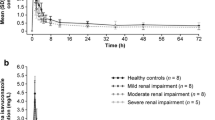
Plasma pharmacokinetic parameters for OM in individuals with normal and impaired renal function are summarized in Table 2, and mean plasma concentration–time profiles are shown in Fig. 1. Plasma concentrations of OM peaked with a median tmax of 3.0 h after a single dose of 50 mg in individuals with normal renal function, compared with a median tmax of 3.5–4.5 h after dosing in participants with mild, moderate, or severe renal impairment or ESRD with dialysis (Table 2, Fig. 1a). Mean apparent plasma terminal elimination half-life of OM was 20.7 h in participants with normal renal function and ranged from 18.8 to 23.3 h in participants with various degrees of renal impairment (Table 2). Total fe was 6.8% in participants with normal renal function and ranged from 4.3 to 8.4% in participants with renal impairment. The proportion of OM unbound to plasma protein ranged from 7.73 to 15.0% and was similar across renal function groups. The regression analysis showed no evidence of a correlation between AUC∞ and Cmax and eGFR or creatinine clearance, with a p-value range for the slopes of 0.70–0.93 (Fig. 2 of the ESM).

Arithmetic mean plasma concentration–time profiles after a single oral administration of omecamtiv mecarbil (OM) 50 mg in individuals with normal renal function (group 1) and individuals with various degrees of renal impairment (groups 2–5) represented in a linear scale. The dashed gray line represents the lower limit of quantitation. Error bars represent standard deviation. ESRD end-stage renal disease
Omecamtiv mecarbil exposure, as estimated by AUC in participants with mild, moderate, or severe renal impairment or ESRD with dialysis was within 9–26% of OM exposure of individuals with normal renal function (Table 3). Omecamtiv mecarbil exposure, as estimated by Cmax, in individuals with mild, moderate, or severe renal impairment or ESRD with dialysis, was within 8–15% of OM exposure of individuals with normal renal function (Table 3).
3.3 Effect of Hemodialysis on PK of OM
Plasma pharmacokinetic parameters for OM in participants with ESRD with or without dialysis are summarized in Table 2. Plasma concentrations peaked with a median tmax of 4.0 h on both the dialysis day and non-dialysis day in participants with ESRD. In addition, the geometric mean (range) AUC from time zero to time of last quantifiable concentration was 416 (355–467) h*ng/mL at the start of the hemodialysis session (arterial samples pre-dialysis) and 369 (316–454) h*ng/mL at the end of the hemodialysis session (venous samples post-dialysis). Arterial and venous samples produced similar plasma concentration–time profiles and therefore similar OM exposures, demonstrating minimal removal of OM by hemodialysis. Of the seven participants with ESRD, one had dialysate samples correctly collected; for the remaining six, owing to an error in how dialysate fluid samples were collected, theoretical dialysate volumes were estimated from the dialysis flow rate and duration of each sampling interval. For the participant with ESRD with correctly collected dialysate fluid samples, 1.20 mg of OM was recovered in the dialysate and the dialysate clearance was 3.55 L/h. For the remaining individuals, estimated amounts of OM in the dialysate were between 1.01 and 1.44 mg, with dialysate clearance values of 2.51–3.55 L/h.
In participants with ESRD requiring hemodialysis, OM exposure as estimated by Cmax was approximately 7% lower on the dialysis day compared with the non-dialysis day (Table 4), with a GMR of 93.2 (90% CI 74.0–117.4). Omecamtiv mecarbil exposure as estimated by AUC0–24 was approximately 3% lower on the dialysis day compared with the non-dialysis day (Table 4), with a GMR of 97.5 (90% CI 83.3–114.2).
3.4 Safety Evaluation
Of the 31 study participants, four (12.9%) reported four treatment-emergent adverse events (TEAEs) during the study; one participant was categorized as having normal renal function (group 1) and three participants had severe renal impairment (group 4). Diarrhea, fatigue, abnormal dreams, and urticaria were reported in one participant each; abnormal dreams, fatigue, and urticaria were considered to be related to OM by study investigators. All TEAEs were mild in severity and resolved by the end of study. No deaths or serious TEAEs were reported. There were no clinically relevant differences in laboratory values, electrocardiogram parameters, or vital signs among treatment groups.
4 Discussion
Given the prevalence of renal impairment among patients with HF that influences the dosing and efficacy of drug therapies, it is important to evaluate the effect of renal impairment on the PK of OM [11, 14]. This phase I trial evaluated the PK, safety, and tolerability of a single dose of oral OM 50 mg in individuals with normal renal function, as well as in individuals with mild, moderate, or severe renal impairment or ESRD requiring dialysis. A 50-mg dose of OM was selected because it is the highest clinical dose evaluated in phase III clinical studies [9, 10]. Results from previous studies of OM demonstrated relatively linear and dose-proportional pharmacokinetic properties [6, 7], suggesting that a single-dose study in participants with renal impairment would be sufficient to accurately describe the PK of OM [15].
The observed half-life of OM was comparable between individuals with various degrees of renal impairment and individuals with normal renal function; it was also comparable to the observed OM half-life previously reported in healthy volunteers (17.1–21.0 h) [6]. Relative OM exposures in individuals with various degrees of renal impairment were not meaningfully different from OM exposures in individuals with normal renal function. The AUC∞ and Cmax values in individuals with impaired renal function were similar to those in individuals with normal renal function, with GMRs for severe renal impairment (group 4) to normal renal function (group 1) of 91.1 (90% CI 60.9–136.5) and 85.2 (90% CI 62.6–115.8), respectively. In addition, there was no evidence of a correlation between the Cmax and AUC∞ and eGFR or creatine clearance.
Omecamtiv mecarbil was generally well tolerated in these populations regardless of exposure level, with no reports of serious TEAEs, no TEAEs leading to discontinuation of the study, and no meaningful changes in laboratory parameters. All AEs were considered mild in severity and resolved by the end of the study.
Patients with ESRD do not experience renal clearance of molecules; therefore, hemodialysis can potentially affect drug elimination in these patients [16], but may restrict the passage of high-molecular-weight species or protein-bound drugs. Pharmacokinetic parameters of OM when administered on dialysis vs non-dialysis days demonstrate that hemodialysis does not limit the clearance of OM in patients with ESRD (GMR [90% CI], 93.2 [74.0–117.4] and 97.5 [83.3–114.2] for Cmax and AUC0–24, respectively). Removal of the drug was further evaluated by comparing the arterial and venous plasma samples collected from individuals with ESRD. The results demonstrate that hemodialysis has a minimal impact on OM concentration–time profiles and OM exposure. Limited amounts of OM (1.01–1.44 mg) were detected in the dialysate; however, because of the estimated dialysate clearance values for six of seven individuals with ESRD, these results should be interpreted with caution. Taken together, these results demonstrate that the PK of OM are minimally affected by renal function, suggesting that individuals with chronic kidney disease, including those undergoing hemodialysis, can use the same dosing strategy as individuals with normal renal function.
While the current study supports the use of OM in patients with ESRD, it is important to consider the findings in the context of HF. The phase III GALACTIC-HF study of oral OM in patients with HF implemented a pharmacokinetic-based dose titration approach to ensure that OM plasma concentrations were within 200–750 ng/mL and to prevent excess exposure [9, 17]. Although patients with eGFR <20 mL/min/1.73 m2 were excluded, the median (interquartile range) eGFR of patients receiving OM was 58.8 [44.3–74.3] mL/min/1.73 m2, suggesting that patients in the study had varying degrees of renal impairment [9]. During the study, plasma concentrations of OM were maintained between 200 and 750 ng/mL in patients with HF and varying eGFR [9, 17]. This observation is generally consistent with findings of the current study, and may support that OM exposure is not substantially affected by renal function in patients with HF.
5 Conclusions
The PK of OM were not significantly affected by renal impairment, and no major tolerability issues with OM were reported. There were no serious AEs or TEAEs leading to study discontinuation. This study supports OM for treatment of HF in individuals with or without renal impairment without the need for adjustments to the dosing strategy.
References
Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011;8:30–41.
Lippi G, Sanchis-Gomar F. Global epidemiology and future trends of heart failure. AME Med J. 2020;5:15.
Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. 2019;139(10):e56–528.
Biering-Sorensen T, Querejeta Roca G, Hegde SM, Shah AM, Claggett B, Mosley TH Jr, et al. Left ventricular ejection time is an independent predictor of incident heart failure in a community-based cohort. Eur J Heart Fail. 2018;20(7):1106–14.
Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, et al. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science. 2011;331(6023):1439–43.
Teerlink JR, Clarke CP, Saikali KG, Lee JH, Chen MM, Escandon RD, et al. Dose-dependent augmentation of cardiac systolic function with the selective cardiac myosin activator, omecamtiv mecarbil: a first-in-man study. Lancet. 2011;378(9792):667–75.
Teerlink JR, Felker GM, McMurray JJV, Ponikowski P, Metra M, Filippatos GS, et al. Acute treatment with omecamtiv mecarbil to increase contractility in acute heart failure: the ATOMIC-AHF study. J Am Coll Cardiol. 2016;67(12):1444–55.
Kaplinsky E, Mallarkey G. Cardiac myosin activators for heart failure therapy: focus on omecamtiv mecarbil. Drugs Context. 2018;7:212518.
Teerlink JR, Diaz R, Felker GM, McMurray JJV, Metra M, Solomon SD, et al. Cardiac myosin activation with omecamtiv mecarbil in systolic heart failure. N Engl J Med. 2020. https://doi.org/10.1056/NEJMoa2025797.
ClinicalTrials.gov. Study to assess the effect of omecamtiv mecarbil on exercise capacity in subjects with heart failure (METEORIC-HF). 2020. https://clinicaltrials.gov/ct2/show/NCT03759392. Accessed 26 June 2020.
Ambrosy AP, Fonarow GC, Butler J, Chioncel O, Greene SJ, Vaduganathan M, et al. The global health and economic burden of hospitalizations for heart failure: lessons learned from hospitalized heart failure registries. J Am Coll Cardiol. 2014;63(12):1123–33.
Palaparthy R, Banfield C, Alvarez P, Yan L, Smith B, Johnson J, et al. Relative bioavailability, food effect, and safety of the single-dose pharmacokinetics of omecamtiv mecarbil following administration of different modified-release formulations in healthy subjects. Int J Clin Pharmacol Ther. 2016;54(3):217–27.
Dreisbach AW, Lertora JJ. The effect of chronic renal failure on drug metabolism and transport. Expert Opin Drug Metab Toxicol. 2008;4(8):1065–74.
Yancy CW, Jessup M, Bozkurt B, Butlet J, Casey DE Jr, Drazner MH, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. J Am Coll Cardiol. 2013;62(16):e147-239.
US Department of Health and Human Services, Food and Drug Administration, and Center for Drug Evaluation and Research. 2020. Guidance for industry: pharmacokinetics in patients with impaired renal function: study design, data analysis, and impact on dosing and labeling. 2010. https://www.fda.gov/media/78573/download. Accessed 14 Aug 2020.
Ahmed JH, Grant AC, Rodger RSC, Murray GR, Elliott HL. Inhibitory effect of uremia on the hepatic clearance and metabolism of nicardipine. Br J Clin Pharmacol. 1991;32(1):57–62.
Teerlink JR, Diaz R, Felker GM, et al. Omecamtiv mecarbil in chronic heart failure with reduced ejection fraction: rationale and design of GALACTIC-HF. JACC Heart Fail. 2020;8(4):329–40.
Acknowledgements
This study was supported by Amgen, Inc. Medical writing and editorial assistance was provided under the direction of the authors by Elizabeth A. Harvie, PhD, CMPP, ELS; Jenna Lewis, MA, ELS; and Emilia Raszkiewicz, BS, MedThink SciCom, with support from Amgen, Inc.
Author information
Authors and Affiliations
Contributions
AT, SD, and EL participated in the conception and planning of the study; all authors participated in the design of the study. All authors participated in the acquisition, analysis, and interpretation of data. All authors critically revised and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Funding
This study was funded by Amgen, Inc.
Conflict of interest
Ashit Trivedi, Rajneet K. Oberoi, Pegah Jafarinasabian, Hanze Zhang, Siddique Abbasi, Sandeep Dutta, and Edward Lee are employees and shareholders of Amgen, Inc. Stephen Flach has no relevant funding or compensation to disclose.
Ethics approval
The study was conducted in accordance with ethical guidelines from the Declaration of Helsinki and Council for International Organizations of Medical Sciences, applicable Good Clinical Practice guidelines of the International Council for Harmonization, and applicable local laws and regulations. An Advarra® (Columbia, MD, USA) institutional review board approved the research protocol and study conduct.
Consent to participate
All study participants provided written informed consent before enrollment in the study and could withdraw from the study at any time.
Consent for publication
Not applicable.
Availability of data and material
Qualified researchers may request data from Amgen clinical studies; complete details are available at http://www.amgen.com/datasharing.
Code availability
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Trivedi, A., Oberoi, R.K., Jafarinasabian, P. et al. Effect of Varying Degrees of Renal Impairment on the Pharmacokinetics of Omecamtiv Mecarbil. Clin Pharmacokinet 60, 1041–1048 (2021). https://doi.org/10.1007/s40262-021-01014-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-01014-0