
Abstract
Pulmonary arterial hypertension (PAH) is a progressive disease of the lung vascular system, which leads to right-sided heart failure and ultimately death if untreated. Treatments to regulate the pulmonary vascular pressure target the prostacyclin, nitric oxide, and endothelin (ET) pathways. Macitentan, an oral, once-daily, dual ETA and ETB receptor antagonist with high affinity and sustained receptor binding is the first ET receptor antagonist to show significant reduction of the risk of morbidity and mortality in PAH patients in a large-scale phase III study with a long-term outcome. Here we present a review of the available clinical pharmacokinetic, pharmacodynamic, pharmacokinetic/pharmacodynamic relationship, and drug–drug interaction data of macitentan in healthy subjects, patients with PAH, and in special populations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Macitentan is a novel dual endothelin receptor antagonist. |
Macitentan requires once-daily dosing (active metabolite present) and has a low propensity to elicit drug–drug interactions. |
Macitentan is not a substrate of active drug transporters, possibly leading to a better liver safety profile. |
1 Introduction
Pulmonary arterial hypertension (PAH) is a chronic disease characterized by an increase in pulmonary vascular resistance, which leads to right ventricular failure and ultimately death if not treated [1, 2]. PAH is a rare condition affecting 15–50 people per million population. Discussion of the classification, diagnosis, and assessment of PAH at consensus meetings has resulted in recommendations that were incorporated into international guidelines and were recently updated during the 5th World Symposium on Pulmonary Hypertension in 2013 in Nice, France [3, 4]. In short, PAH is the first of five general categories of pulmonary hypertension. It can be idiopathic or heritable, but can also be the result of drug or toxin use or associated with other diseases such as mixed connective tissue disease, HIV infection, portal hypertension, congenital heart disease, schistosomiasis, and chronic hemolytic anemia [3–9].
The basis of the increase in pulmonary vascular resistance resides in a combination of factors such as endothelial dysfunction, increased contractility of small pulmonary arteries, proliferation and remodeling of endothelial and smooth muscle cells, and in situ thrombosis [10–12].
The three identified mechanisms that are mainly involved in regulation of the pulmonary vascular pressure are the prostacyclin, nitric oxide (NO), and endothelin (ET) pathways. Both the prostacyclin and the NO pathway act directly on the vascular bed [11, 13].
ET-1, part of a family of closely related 21-amino-acid polypeptides, works through a different mechanism promoting endothelial dysfunction and vascular remodeling [11]. ET-1 achieves its effects by activation of specific G-protein-coupled cell surface receptors. Two subtypes of ET-1 have been identified, termed ETA and ETB receptors. ETA receptors are predominantly located in vascular smooth muscle cells and cardiomyocytes and mediate contraction. ETB receptors are also present in smooth muscle cells in which they have a similar function as ETA, but are mainly located in vascular endothelial cells where they mediate vascular dilatation through NO release and regulate ET-1 uptake and production [14–21]. Under normal conditions, ET-1 production and clearance of ET-1 from the vascular bed is balanced, but in diseases such as PAH, the balance is disrupted and leads to an increase in circulating levels of ET-1 and detrimental effects [22–24]. It has been observed that in chronic pathological situations, ETB receptors are down-regulated on endothelial cells and up-regulated on smooth muscle cells and fibroblasts [25–28]. While it could be hypothesized that the beneficial effects of ETB on the endothelial cells could be reduced by blockade of ETB, selective ETA blockade would leave the ETB receptors that are up-regulated on smooth muscle cells functional. Specific blockade of ETA has been shown to activate the renin–angiotensin system, potentially resulting in edema [29, 30]. Therefore, sustained blockade of both ET receptors may be a better strategy to obtain optimal efficacy and safety [26, 28, 31–33].
ET receptor antagonists (ERAs) have been proven to be effective in the treatment of PAH [34–38]. Treatment with approved ERAs has been shown to confer improvements in a number of important clinical endpoints, including exercise capacity, modified New York Heart Association (NYHA) functional class, and pulmonary hemodynamics [34–38]. Both ETA/ETB receptor antagonists and selective ETA receptor antagonists are currently licensed for the treatment of PAH [39, 40]. Traditionally, approval was granted based on short-term studies (12–16 weeks) with low patient numbers, in which an improvement in exercise capacity was shown, based on the distance walked in 6 min [41, 42]. However, current guidelines for clinical research on PAH encourage the conduct of long-term outcome studies with morbidity/mortality endpoints [43–45]. Besides ERAs, other specific pharmacotherapies such as treatment with prostanoids, phosphodiesterase type-5 (PDE-5) inhibitors, or a stimulator of soluble guanylate cyclase are used. Guidelines have recently been revised to reflect current strategies with regard to targeted and combination therapy [1, 36].
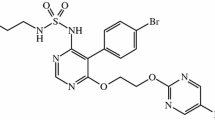
Macitentan (Fig. 1), N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N′-propylsulfamide, is an oral, once-daily, potent dual ETA/ETB receptor antagonist.

Proposed metabolic pathways of macitentan in humans and animals
Macitentan is metabolized to a major and pharmacologically active metabolite, ACT-132577. In functional assays, both macitentan and ACT-132577 behave as dual receptor antagonists. ACT-132577 is approximately fivefold less potent than macitentan on ETA receptors and presents an ETA/ETB inhibitory potency ratio of 16, versus 50 for its parent compound [46]. Free median 50 % inhibition concentration (IC50) values for ETA and ETB were within the range of free concentrations observed in humans treated with macitentan at the therapeutic dose of 10 mg, suggesting that ETB blockade is achieved. In vivo, ET-1 plasma concentrations were increased, which is typical of treatment with a dual ERA and not a selective ERA [46].
This sulfamide was the result of a targeted medicinal chemistry program to identify a novel, potent ERA with high oral efficacy [47]. In preclinical models of hypertension and PAH, macitentan showed a dose-dependent decrease in mean arterial blood pressure without affecting the heart rate [46, 47]. This decrease was even observed when macitentan was given on top of the ERA bosentan, whereas this decrease was not observed when bosentan was given on top of macitentan [48]. Tissue targeting was achieved through an increase in acid dissociation constant (pKa) and log D values resulting in a higher affinity of macitentan for lipophilic structures [46]. In vitro, macitentan displayed insurmountable antagonism to ET receptors, due to its slower apparent receptor association rate when compared with other ERAs [49]. Therefore, macitentan could lead to a more effective blockage blockade of ET-1 than other ERAs [49].
Macitentan was granted approval for the treatment of PAH in several countries including the USA, Canada, Europe, and Switzerland and marketed as Opsumit® at a dosing regimen of 10 mg once daily [50–52] following the results of SERAPHIN (Study with an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome), a landmark phase III study. SERAPHIN was a long-term, randomized, placebo-controlled study in PAH patients. The primary endpoint used was a clearly defined morbidity/mortality endpoint allowing evaluation of the efficacy and safety of macitentan [53, 54]. Two dose levels (3 and 10 mg) were studied; macitentan reduced the risk of a morbidity/mortality event vs. placebo (10 mg dose, risk reduction 45 %, p < 0.001; 3 mg dose, risk reduction 30 %, p = 0.01) and was well-tolerated at both dose levels [53]. The number of adverse events (AEs) reported and of patients discontinuing treatment due to AEs was similar across all treatment groups and the incidence of macitentan-induced liver injury was comparable to that induced by placebo [53]. A thorough discussion of the study and its limitations is provided by Pulido et al. [53] as well as Dingemanse et al. [54].
2 Drug Formulations
In early clinical phase I and II studies, macitentan was administered in hard gelatin capsules [55]. A film-coated tablet of macitentan 10 mg, which is easier to ingest than the capsule, was developed for later phase I and III studies and subsequently marketed as Opsumit®. The in vitro dissolution rate of a macitentan 10 mg capsule was slightly higher than that of the tablet formulation, while dissolution characteristics of an uncoated tablet formulation did not differ from the coated formulation [55]. In healthy male subjects, the extent of absorption of macitentan given as a tablet was slightly lower than the capsule formulation, leading to the lower of the 90 % confidence limits of the geometric mean ratio for maximum plasma concentration (C max) to fall outside the commonly referenced range of bioequivalence of 0.80–1.25. Geometric mean ratios and 90 % confidence intervals (CIs) of total exposure expressed as area under the plasma concentration–time curve (AUC) from time zero to infinity (AUC∞) and AUC from time zero to last timepoint t of the measurable plasma concentrations (AUCt) were completely within the referenced bioequivalence range [55]. As the change in C max observed in the tablet formulation was minor and total exposure, which is a more relevant measure for drugs that are used long-term, was not different from that of the capsule formulation, the observation was considered to be not clinically relevant. Therefore, no dose adjustment was needed for the tablet formulation.
3 Bioanalysis of Macitentan and Metabolites
In the course of its clinical development, in a dedicated study investigating the absorption, distribution, metabolism, and elimination (ADME) of macitentan, a pharmacologically inactive metabolite, ACT-373898, was discovered [56]. As the inactive metabolite had been characterized adequately in animals (see Sect. 4.3) and did not appear in high enough concentrations to raise concern in human studies [56], ACT-373898 was not routinely measured in healthy subjects and only assessed in subjects with hepatic and renal impairment.
Macitentan, its active metabolite ACT-132577, and its pharmacologically inactive metabolite ACT-373898 (Fig. 1) were determined in human plasma using a validated liquid chromatography–tandem mass spectrometry (LC-MS/MS) method. After thawing, human plasma samples were vortexed and centrifuged, and a mixture of acetonitrile/ethanol was added followed by addition of the deuterated standard. After protein precipitation, plasma samples were filtrated through a protein precipitation plate and the filtrate was injected onto the high-performance liquid chromatography (HPLC) column. The analysis of macitentan and ACT-132577 was performed by separation with reverse-phase chromatography followed by detection with triple-stage quadrupole tandem mass spectrometry (MS/MS) in positive ion mode. The assay was linear in the concentration range 1–2000 ng/mL for both analytes. The performance of the method was monitored using quality control samples at concentrations of 3, 100, and 1500 ng/mL [55, 57]. In the study with renally impaired subjects, ACT-373898 was measured separately and was detected using triple-state quadrupole MS/MS in negative ion mode [57].
4 Pharmacokinetics in Healthy Adults
4.1 Absorption
4.1.1 Single-Dose Administration
The entry-into-human study was performed in healthy male subjects with a single-center, double-blind, randomized, placebo-controlled study design [58]. Doses of macitentan 0.2, 1, 5, 25, 100, 300, and 600 mg or placebo were investigated under fasted conditions in groups of eight subjects (six on active, two on placebo) each. The pharmacokinetic parameters after single-dose administration are shown in Table 1. After oral administration, macitentan was slowly absorbed with median time to C max (t max) varying from 8 to 30 h for the different dose groups. Thereafter, macitentan plasma concentrations decreased slowly. The apparent elimination half-life (t ½) could only be reliably estimated for the 300 and 600 mg dose groups and was approximately 16 h. The C max of macitentan increased less than dose proportionally as indicated by a value for the dose-proportionality coefficient β of 0.83 (95 % CI 0.79–0.87) [58].
Formation of the pharmacologically active metabolite ACT-1323577 was slow with C max values attained at the earliest 30 h after dosing (Table 1). In comparison to the parent compound, plasma concentrations of the metabolite were higher and elimination was slower, as demonstrated by mean t ½ values varying from 40 to 66 h (Table 1). Total exposure (AUC∞) to the metabolite was 2.7-fold (95 % CI 2.5–2.9) greater than to macitentan at a dose of 600 mg [58]. Data from other studies in which a single dose of 10 mg was administered to healthy subjects showed similar pharmacokinetic results [57].
4.1.2 Effect of Food on Absorption
The effect of food on the pharmacokinetics of single-dose macitentan and ACT-132577 was investigated in healthy subjects [59]. In an exploratory, open-label, randomized, two-period, crossover study, subjects received macitentan 10 mg once under fasted conditions and, after a washout period, once after consumption of a high-fat, high-calorie breakfast as recommended in US FDA and European Medicines Agency (EMA) guidelines [60, 61]. Exposure to both macitentan and its active metabolite expressed in C max and AUC was unchanged in the presence of food. This was evidenced by the point estimates for C max and AUC and their 90 % CIs falling entirely within the referenced bioequivalence range of 0.80–1.25 [99]. Therefore, macitentan can be administered with and without food [59, 62, 63].
4.1.3 Multiple-Dose Administration
The multiple-ascending dose study was performed in healthy male subjects in a single-center, double-blind, randomized, placebo-controlled study design [64]. Oral doses of macitentan 1, 3, 10, and 30 mg or placebo, administered once daily under fasted conditions for 10 days, were investigated in groups of eight subjects (six on active, two on placebo) each. Steady-state conditions for macitentan and ACT-132577 were reached by days 3 and 7, respectively [64]. The pharmacokinetic parameters after multiple-dose administration are shown in Table 2. After multiple-dose administration, macitentan reached C max at approximately 8 h and was eliminated slowly as indicated by its t ½ varying from 14 to 19 h [64]. Accumulation was limited when comparing AUC during a dosing interval (AUCτ) on day 10 with that of day 1 (Table 2). Both AUCτ and C max were dose-proportional over the dose range tested.
Similar to the entry-into-human study, ACT-132577 was slowly formed and eliminated with t max and t ½ values of 8 and 48 h, respectively (Table 2). Due to its slow formation and slow elimination, ACT-132577 accumulated significantly as indicated by an accumulation index of 7–10 for the different doses (Table 2) [64].
4.1.4 Absolute Bioavailability
No absolute bioavailability study has been performed. However, oral bioavailability was estimated using Simcyp Population-Based ADME Simulator. Using a physiologically based pharmacokinetic (PBPK) model approach combined with genetic, physiological, and demographic variables derived from population databases, an oral bioavailability of 74 % was estimated for macitentan [65].
4.2 Distribution, Protein Binding, and Blood Partitioning
Using Professional WinNonlin® (Pharsight Corp., Mountain View, CA, USA) a one-compartment model with a lag time and first-order input and elimination was fit to describe the steady-state plasma concentration vs. time data of macitentan and ACT-132577 after oral administration. The model indicated an apparent oral volume of distribution at steady state (V ss/F) of approximately 50 and 40 L for macitentan and ACT-132577, respectively. Macitentan and ACT-132577 distributed well into tissues as indicated by V ss/F exceeding volume of total body water [63].
Macitentan and its active metabolite are highly bound to plasma proteins (>99 %), primarily to albumin and to a smaller extent to α1-acid glycoprotein [56, 63].
In the ADME study, concentrations of total radioactivity in plasma were greater than in whole blood [56]. The blood/plasma ratio of the total radioactivity concentration of 0.55 was close to the theoretical blood-to-plasma distribution ratio (l-hematocrit = 0.57), indicating that macitentan and its metabolites poorly bind to or penetrate into blood cells. This was also in accordance with in vitro data on blood cell/plasma partitioning, which showed no notable uptake of either macitentan or ACT-132577 into human erythrocytes [56].
4.3 Metabolism and Elimination
The disposition and metabolism of macitentan were investigated in a dedicated ADME study in which six male subjects received a single oral dose of 14C-labeled macitentan with a target radioactivity of 2.78 MBq (970 µ Sievert) [56].
Excretion of the radioactivity involved both renal and fecal routes, with urine being the most important elimination route. In humans, there were three, four, and five entities detected in plasma, urine, and feces, respectively (Fig. 1). Besides macitentan and M6 (ACT-132577), a pharmacologically inactive metabolite (M5, ACT-373898) was observed in plasma. Macitentan is metabolized in humans by four independent pathways: (1) oxidative depropylation of the sulfamide group to form the pharmacologically active metabolite M6; formation of M6 is mainly catalyzed by cytochrome P450 (CYP) 3A4, with minor contributions of CYP2C8, CYP2C9, and CYP2C19; (2) oxidative cleavage of the ethylene glycol catalyzed by CYP2C9 to form the alcohol M4, which is subsequently oxidized to the corresponding acid M5 and then hydrolyzed to m/z 324; (3) oxidative depropylation of one of the distal carbon atoms to form M7, mainly catalyzed by CYP2C9, with minor contributions of CYP2C8 and CYP2C19; and (4) chemical hydrolysis of macitentan and ACT-132577 to form M3 (ACT-080803). The most abundant entities in urine and feces were M5 and M3, respectively. The metabolites identified in humans had also been detected in animal studies and the latter provided sufficient coverage for the human situation (Fig. 1). Therefore, no unique human metabolite was identified in this study. In addition, no metabolite structure was identified, which would be indicative of a safety concern. Based on in vitro and in vivo toxicology studies, there was no evidence of genotoxicity with macitentan [56].
5 Pharmacokinetics in Special Populations
5.1 Pharmacokinetics in Healthy Asian Subjects
The pharmacokinetics of a drug could be influenced by ethnicity due to intrinsic differences such as genetic polymorphisms in metabolic pathways, differences in dose–response relationships, and differences in bioavailability, but also due to extrinsic differences such as body composition and food habits [66]. Therefore, pharmacokinetic studies were conducted to investigate the effect of ethnicity on the pharmacokinetics of macitentan.
After single-dose administration of macitentan 10 mg to ten healthy male and female Caucasian and ten healthy male and female Japanese subjects, the geometric mean ratio and its 90 % CI of C max of macitentan and ACT-132577 fell entirely within the referenced bioequivalence range of 0.80–1.25. The t ½ was slightly shorter in Japanese subjects, resulting in a 15 % decrease in AUC∞ [67]. As this decrease in exposure was small and exposures were within the ranges observed in other phase I studies, this difference was considered to be of no clinical relevance [67].
The pharmacokinetics of macitentan and ACT-132577 were further investigated in multiple-ascending dose studies in 30 healthy Korean subjects [68] and 24 Japanese subjects [100]. Doses of macitentan 3, 10, and 30 mg or placebo were studied in groups of ten Korean subjects (eight on active, two on placebo) and eight Japanese subjects (six on active, two on placebo) each. The pharmacokinetic results were similar to those observed in the multiple-ascending dose study in Caucasians.
Overall, based on these studies, no dose adjustment would be needed for subjects of different ethnicities [68].
5.2 Effect of Sex and Age on Pharmacokinetics
The effect of sex and age on the pharmacokinetics of macitentan and ACT-132577 was investigated in the context of the study in healthy Japanese subjects, subjects with renal and hepatic impairment, and pharmacokinetic assessments of patients in phase II and III [57, 67, 69]. Results indicate that, in general, females show a small increase in t ½ of up to 22 %, leading to an increase in exposure of up to 25 % to macitentan and ACT-132577. This finding is likely to be caused by a difference in body size and body fat between females and males [57, 67, 69]. Sub-analysis of pharmacokinetic data of macitentan and ACT-132577 from the SERAPHIN study showed no clear effect of age (Actelion Pharmaceuticals Ltd, data on file). In summary, as exposures were not or only marginally different and exposures were in line with population ranges, no dose adjustment of macitentan for sex and age is required [62, 63].
5.3 Pharmacokinetics in Subjects with Hepatic Impairment
The pharmacokinetics of macitentan, ACT-132577, and ACT-373898 were studied after administration of a single dose of macitentan 10 mg to subjects with mild, moderate, and severe hepatic impairment due to liver cirrhosis in a single-center, open-label design. Hepatic impairment was determined according to Child-Pugh grade based on screening laboratory test results for serum bilirubin, serum albumin, prothrombin time, and the stage of hepatic encephalopathy, with or without ascites. The study investigated sequential groups of eight subjects with mild, moderate, and severe hepatic impairment. A control group of eight healthy subjects was matched to and studied together with the severe hepatically impaired subjects [57]. Plasma concentrations were generally lower in subjects with hepatic impairment than in healthy subjects, which is reflected in geometric mean ratios varying between 0.54 and 1.13 for C max and AUC∞ (Table 3). No correlation between degree of hepatic impairment and pharmacokinetics could be discerned. In fact, subjects with severe hepatic impairment were very similar to the matched healthy subjects, as indicated by geometric mean ratios varying between 0.74 and 1.03 for the different analytes (Table 3) [57]. The unbound macitentan and ACT-132577 fraction in plasma was determined in all study groups and ranged between 0.09–0.26 % and 0.52–1.09 %, respectively. No pattern related to the level of hepatic impairment in plasma protein binding of macitentan and ACT‐132577 was detected. Levels of unbound ACT-373898 increased with severity and were 2.39 ± 0.26 % (healthy subjects), 2.11 ± 0.52 % (mild hepatic impairment), 3.05 ± 0.71 % (moderate hepatic impairment), and 3.97 ± 0.95 % (severe hepatic impairment) [57].
No dose adjustments based on pharmacokinetics would be needed. However, no clinical data are available in PAH patients with moderate or severe hepatic impairment [63]. Macitentan, as have other ERAs, has been associated with hepatotoxicity. Therefore, macitentan should not be initiated in PAH patients with severe hepatic impairment [54, 63].
5.4 Pharmacokinetics in Subjects with Renal Impairment
The pharmacokinetics of a single dose of macitentan 10 mg were investigated in eight subjects with severe renal function impairment (SRFI) and eight healthy subjects matched for age, sex, and weight, in a single-center, open-label design. Subjects with SRFI were defined as having a creatinine clearance of 15–29 mL/min using the Cockroft-Gault formula. Healthy subjects had a creatinine clearance >70 mL/min for subjects aged 50–60 years and >80 mL/min for subjects <50 years [57].
The plasma concentration–time profiles of macitentan and ACT-132577 were similar in healthy subjects and those with SRFI. However, clearance of macitentan and ACT-132577 was slower in subjects with SRFI as indicated by a slightly longer t ½. This resulted in increases in exposures demonstrated by a geometric mean ratio (90 % CI) of 1.11 (0.80–1.54) and 1.24 (0.83–1.85) for C max and AUC∞, respectively, of macitentan and 1.39 (1.07–1.81) and 1.58 (1.24–2.01) for C max and AUC∞, respectively, of ACT-132577, which were not considered clinically relevant. As ACT-373898 is predominantly eliminated by the kidneys, its exposure increased markedly in SRFI subjects as indicated by a geometric mean ratio (90 % CI) of 4.24 (3.10–5.80) and 7.31 (5.42–9.86) for C max and AUC∞, respectively [57]. Due to the low concentrations of circulating ACT-373898, its lack of activity on the ET receptor, and sufficient margin established in toxicology studies, the increase is not clinically relevant and does not necessitate dose adjustment.
5.5 Pharmacokinetics in Patients with Essential Hypertension
Trough plasma samples were collected at week 4 and 8 in a multicenter, double-blind, randomized, placebo- and active-controlled, dose-ranging study to evaluate the efficacy and safety of macitentan in subjects with mild-to-moderate essential hypertension. Dose levels that were investigated were macitentan 0.3, 1, 3, and 10 mg once daily for up to 8 weeks [70]. Trough concentrations (C trough) were consistent after week 4 and 8 of treatment. Pharmacokinetic analysis showed that exposure in terms of C trough of both macitentan and ACT-132557 was dose proportional over the dose range tested [70].
5.6 Pharmacokinetics in Patients with Pulmonary Arterial Hypertension
In the pivotal phase III SERAPHIN study in patients with PAH, plasma samples were taken at end of treatment (EOT) in all 742 patients and at month 6 in 187 patients participating in a sub-study [69]. Macitentan steady-state C trough values ranged from below limit of quantification to 801 ng/mL. Arithmetic means (±standard deviation) showed clear differences between the two doses (month 6: 92 ± 53 and 291 ± 155 ng/mL, for macitentan 3 and 10 mg, respectively; EOT: 76 ± 61 and 208 ± 139 ng/mL, for macitentan 3 and 10 mg, respectively) [69]. A cross-study comparison showed that C trough values of macitentan and ACT-132577 were higher in patients with PAH than in healthy subjects. However, differences in exposure in terms of C max and AUC between PAH patients and healthy subjects were minor. This was based on a pharmacokinetic profile over 24 h at steady-state that was obtained from 20 patients treated with macitentan 10 mg in the open-label extension study of SERAPHIN. Results indicated that exposure to macitentan and ACT-132577 was 1.2- and 1.3-fold, respectively, greater in PAH patients than in healthy subjects and was not influenced by the severity of the disease [62, 63].
6 Pharmacodynamics
6.1 Pharmacodynamics in Healthy Subjects
6.1.1 ET-1 Concentrations in Healthy Subjects
In the single- and multiple-ascending dose studies, ET-1 concentrations were measured in plasma at regular intervals using a commercially available radioimmunoassay [58, 64]. In these studies ET-1 concentrations increased with dose. Statistical significance compared with placebo was observed at doses of 25 mg and up for single-dose administration and 10 mg and up for multiple-dose administration (Fig. 2). ET-1 concentrations did not significantly increase at 30 mg compared with 10 mg of macitentan given as multiple doses, suggesting that its maximum had been reached. Based on these results, a 10 mg dose was selected for clinical development.

Arithmetic mean (±standard deviation) plasma endothelin-1 concentrations in healthy subjects after single-dose (left panel) and multiple-dose (right panel) administration of macitentan (n = 6 per dose level) or placebo (n = 14 left panel, n = 8 right panel). AUC τ area under the concentration–time curve during a dosing interval, AUC 48 area under the concentration–time curve from time zero to 48 h, ET endothelin, *p < 0.05 vs. placebo
6.1.2 Effect on Cardiac Repolarization
The effect of macitentan on cardiac repolarization was studied in a thorough corrected QT (QTc) interval (TQT) study following regulatory guidance [71, 72]. The study was performed as a double-blind, randomized, placebo- and positive-controlled, four-way crossover study. Treatment arms consisted of moxifloxacin 400 mg, macitentan 10 mg (therapeutic dose), macitentan 30 mg (supra-therapeutic dose), and placebo for 9 days [73]. The upper bound of the 90 % CI of the primary endpoint, which was the baseline-adjusted, placebo-corrected QT interval using the Fridericia correction method (∆∆QTcF), was below 10 ms at all timepoints, whereas moxifloxacin showed a clear prolongation of QTcF. No correlation between pharmacokinetic parameters and ECG variables and no treatment-related morphological ECG abnormalities were detected [73]. The data indicate that macitentan and ACT-132577 do not affect the QTc interval and that, therefore, no specific ECG monitoring is needed for clinical use.
6.1.3 Bile Salts
Serum bile salts were measured in the single- and multiple-ascending dose studies at regular intervals using a commercially available enzymatic assay [58, 64].
No significant differences between the macitentan and placebo groups were detected, therefore suggesting that macitentan does not interfere with hepatic bile salt transport, unlike some other ERAs such as bosentan [58, 64, 74]. Hepatic disposition of macitentan is driven by passive diffusion with no role of active uptake transporters such as organic anion-transporting polypeptide (OATP). As macitentan is highly bound to plasma proteins, hepatocellular drug concentrations are too low to inhibit transport proteins involved in bile salt trafficking [75]. Indeed, macitentan appears to have a favorable hepatic safety profile with liver enzyme elevations similar to placebo, as observed in clinical trials [54].
6.2 Pharmacodynamics in Patients
6.2.1 ET-1 Concentrations in Patients
ET-1 measured at trough in a study with patients with essential hypertension showed a clear dose-related effect at doses of macitentan 3 and 10 mg once daily (Actelion Pharmaceuticals Ltd, data on file) [70].
6.3 Pharmacokinetic/Pharmacodynamic Relationships
Pharmacokinetic/pharmacodynamic analysis in PAH patients was performed to investigate possible relationships between pharmacokinetics (trough samples collected at EOT and month 6) and hemodynamic parameters, 6 min walk distance (6MWD), laboratory parameters, and AEs leading to study drug discontinuation [69]. Different approaches [linear, log linear, and maximum effect (E max) models] were assessed to establish the best fit. The results indicated that the pharmacodynamic variables pulmonary vascular resistance, mean pulmonary artery pressure, total pulmonary resistance, and cardiac output correlated with plasma concentration. No relationship was established between plasma concentrations and mean right arterial pressure or mixed venous oxygen saturation [69]. No correlations could be established between plasma concentrations and laboratory variables or the probability of AEs leading to discontinuation [69].
7 Drug–Drug Interactions
7.1 Drug–Drug Interaction Potential
Weiss et al. investigated the interaction potential of macitentan on critical drug-metabolizing enzymes and drug transporters in vitro [76]. Macitentan was found to moderately inhibit breast cancer resistance protein (BCRP), CYP3A4, CYP2C19, and clearly inhibited P-glycoprotein (P-gp) [76]. However, further in vitro and in vivo studies, performed at clinically relevant concentrations, showed that macitentan is not a substrate of P-gp [62]. Hepatic uptake is mostly driven by passive diffusion and is not dependent on OATP transport, unlike other ERAs such as bosentan [75, 77, 78]. Macitentan showed a potential to inhibit OATPB1 and OATPB3; however, taking into consideration the high protein binding and low clinical drug concentrations, it is unlikely that treatment with macitentan would affect the pharmacokinetics of coadministered drugs dependent on OATP transport [76, 78]. The limited drug interaction potential of macitentan was demonstrated in a number of in vivo drug–drug interaction studies summarized in the next sections. The effects of other drugs on the pharmacokinetics of macitentan and ACT-132577 are shown in Fig. 3.

Effects of other drugs on macitentan and ACT-132577 pharmacokinetics. Black circle macitentan, Cross ACT-132577
7.2 Warfarin
Warfarin is an anticoagulant commonly administered to patients with PAH [1, 4, 79, 80]. It consists of a racemic mixture of R- and S-enantiomers. The elimination of warfarin is almost entirely by metabolism, after which it is excreted into the urine. Warfarin is metabolized by CYP enzymes (CYP2C9, CYP2C19, CYP2C8, CYP2C18, CYP1A2, and CYP3A4) to inactive hydroxylated metabolites and by reductases to reduced metabolites with minimal anticoagulation activity. The S-enantiomer is primarily metabolized by CYP2C9, and less by CYP2C19 and CYP3A4, whereas the R-enantiomer is mainly metabolized by CYP1A2, with a smaller contribution of CYP3A4 [81]. As warfarin has a narrow therapeutic index, which requires careful monitoring, it was important to investigate the potential effect of macitentan on warfarin pharmacokinetics and pharmacodynamics in a drug–drug interaction study. In this study, 12 healthy male subjects were exposed to two treatments: (A) a single dose of 25 mg; and (B) a loading dose of macitentan 30 mg on day 1 followed by 10 mg once daily for 8 days, with a single 25 mg dose of warfarin on day 4 in a randomized, open-label, single-center, two-way crossover design [82]. A loading dose strategy was chosen to ensure that steady-state conditions of macitentan and ACT-132577 would be attained earlier than day 7 (i.e., day 3). The pharmacokinetic parameters t max, C max, AUC∞, and t ½ of R- and S-warfarin were studied. Pharmacodynamics were investigated by determination of AUC from time zero to 144 h (AUC144) and t max of international normalized ratio (INR) and factor VII activity [83].
The mean plasma concentration–time profiles of R- and S-warfarin in the presence and absence of macitentan were similar and geometric mean ratios and 90 % CI of C max and AUC∞ fell entirely in the reference bioequivalence range of 0.80–1.25. No effect of macitentan on INR and factor VII activity was detected [82].
7.3 Sildenafil
Sildenafil, a PDE-5 inhibitor used for the treatment of PAH, is metabolized predominantly via CYP3A4 (major route) and CYP2C9 (minor route). The major circulating active metabolite results from N-desmethylation of sildenafil and is, itself, further metabolized [84, 85]. To investigate the potential mutual interaction between macitentan and sildenafil, 12 healthy male subjects received (A) a loading dose of macitentan 30 mg on day 1 followed by 10 mg once daily for 3 days; (B) sildenafil 20 mg three times a day for 3 days and a single 20 mg dose on day 4; and (C) both treatments A and B concomitantly in an open-label, randomized, three-way crossover study. On day 4 of each treatment the pharmacokinetics of macitentan, ACT-132577, sildenafil, and N-desmethylsildenafil were determined [86]. In the presence of sildenafil, macitentan pharmacokinetics were not affected, as shown in Fig. 3. Exposure to ACT-132577, on average, decreased by approximately 17 and 15 % for C max and AUCτ, respectively, which was not clinically relevant. In the presence of macitentan, plasma concentrations of sildenafil were higher than during treatment with sildenafil alone, resulting in increased C max and AUCτ values. The respective geometric mean ratios (90 % CI) were 1.26 (1.07–1.48) and 1.15 (0.94–1.41). The pharmacokinetics of N-desmethylsildenafil were not affected by macitentan [86].
7.4 Ketoconazole
Ketoconazole, a broad-spectrum antifungal agent, is a strong inhibitor of CYP3A4 and was used as a sensitive tool for assessing this effect on the pharmacokinetics of macitentan [87, 88]. In a two-period, randomized, open-label crossover study, ten healthy subjects received (A) a single oral dose of macitentan 10 mg; and (B) ketoconazole 400 mg once daily for 4 days, coadministration of macitentan 10 mg with ketoconazole 400 mg on day 5, followed by continued administration of ketoconazole 400 mg once daily for 19 additional days [89]. In the presence of ketoconazole, exposure to macitentan increased approximately twofold, while exposure to ACT-132577 was reduced by 26 % (Fig. 3). As macitentan is prescribed for long-term use, pharmacokinetic modeling was performed to predict the interaction between ketoconazole and macitentan under steady-state conditions. Results of the modeling indicated that exposure to macitentan in the presence of ketoconazole would be threefold that without macitentan, which is an exposure well-tolerated in the multiple-ascending dose study [64]. Exposure to ACT-132577 would be reduced by 25 % [89]. Results indicate that macitentan, in principal, could be administered together with strong CYP3A4 inhibitors as the increased exposure is well-tolerated by subjects. However, due to the lack of long-term safety data in subjects and patients exposed to higher concentrations of macitentan, caution should be exercised using concomitant treatment with strong CYP3A4 inhibitors [62, 63].
7.5 Cyclosporine
Cyclosporine (ciclosporin), which is an inhibitor of OATP, Pg-p, and BCRP as well as a weak inhibitor of CYP3A4, is a co-medication used in PAH patients. Drug–drug interactions between cyclosporine and other ERAs that are substrates for OATP have been reported [90, 91]. The effect of cyclosporine on macitentan pharmacokinetics was investigated in a single-center, open-label, one-sequence, crossover study design [78, 92, 93]. Ten healthy subjects received a loading dose of macitentan 30 mg on day 1 followed by macitentan 10 mg once daily for 17 days. On days 6–17, subjects received concomitant cyclosporine 100 mg in a twice-daily dosing regimen. Based on dosing recommendations for cyclosporine, both macitentan and cyclosporine were administered after a meal. In the presence of cyclosporine, macitentan C trough values were increased on average by 38 %; however, the increase in exposure during a dosing interval was limited (i.e., 10 %) (Fig. 3). In this study, the pharmacokinetics of ACT-373898 were also studied, which were not affected by concomitant cyclosporine [78]. The data indicate that macitentan and its metabolites are not substrates of the drug transporters OATP, P-gp, and BCRP. Overall, macitentan can be used concomitantly with cyclosporine with no need for dose adjustment.
7.6 Rifampin
Rifampin (rifampicin), an antibacterial mainly used for the treatment of tuberculosis, is a strong inducer of CYP3A4 [87, 94]. In a single-center, open-label, one-sequence, crossover study design ten healthy subjects received a loading dose of macitentan 30 mg on day 1 followed by macitentan 10 mg once daily for 12 days. On day 6, subjects received concomitant rifampin 600 mg once daily until day 12 [78]. Macitentan concentrations decreased markedly, i.e., AUCτ decreased by approximately 80 % (Fig. 3). Daily exposure to ACT-132577 in the presence of rifampin did not change (geometric mean ratio of 1.00; 90 % CI 0.89–1.12) [78]. Although ACT-132577 contributes to the overall pharmacological effect, the approximately fourfold reduction in exposure to macitentan in the presence of a strong CYP3A4 inducer could impact the overall efficacy of macitentan. Therefore, concomitant treatment should be avoided [62, 63, 78].
8 Discussion
Macitentan was approved at a dose of 10 mg after having undergone one of the largest phase III studies conducted in PAH and used a composite morbidity/mortality primary endpoint, consisting of clinically highly relevant components. The pharmacokinetics of macitentan can be characterized by slow absorption of macitentan and slow formation of ACT-132577 with t ½ values of 16 and 48 h, respectively, therefore justifying a once-daily dosing regimen. Formation of ACT-132577 was mainly catalyzed by CYP3A4 with minor contributions of CYP2C8, CYP2C9, and CYP2C19. Over the dose range tested in multiple-dose regimens the pharmacokinetics of macitentan and ACT-132577 were dose proportional and not influenced by meals, sex, age, or ethnicity. Due to elimination both by liver and kidney, no dose adjustment based on pharmacokinetics appeared necessary in patients with hepatic or renal impairment. It is remarkable that the pharmacokinetics of macitentan were not influenced by hepatic impairment, given the observation that macitentan is extensively metabolized before being excreted and that unchanged macitentan was not detected in urine. A possible explanation could be that the major metabolites identified in urine and feces are hydrolysis products and, therefore, only in part dependent on metabolism in the liver. Further, it is known that in hepatically impaired patients hepatic levels of CYP enzymes are diminished but this does not result in a decrease of CYP activity [95]. In fact, many patients with chronic liver disease (without cirrhosis) can metabolize drugs at a normal rate [95–98]. A third hypothesis could be that urine and/or feces samples were not sufficiently stabilized to prevent hydrolysis of macitentan and ACT-132577, therefore underestimating the amount of unchanged macitentan and ACT-132577 excreted.
It must be noted that no clinical data of PAH patients with severe or moderate hepatic impairment are available. As the class of ERAs is associated with hepatotoxicity, macitentan should not be used in patients with severe hepatic impairment.
The pharmacokinetic characteristics of macitentan and ACT-132577 were consistent in healthy subjects and patients with essential hypertension or PAH. This allows utilization of population pharmacokinetic and pharmacokinetic/pharmacodynamic analysis methods to predict concentrations in other patient populations, in particular children, for whom no data exist at this time. Also, the observed data can be useful for assessing the potential for other drug–drug interactions. At the approved clinical dose of macitentan of 10 mg, its drug–drug interaction potential is considered low. Macitentan pharmacokinetics were not affected by commonly co-prescribed medications in PAH patients such as warfarin and sildenafil. Unlike other ERAs such a bosentan, macitentan and ACT-132577 are not substrates of OATP and can be administered concomitantly with cyclosporine without need for dose adjustment. The results of the study with cyclosporine, which inhibits other drug transporters besides OATP, indicate that the pharmacokinetics of macitentan would not be affected by inhibitors of OATP, P-gp or BCRP. Concomitant treatment with a strong inhibitor or inducer of CYP3A4 affected the pharmacokinetics markedly. Therefore, this combination should be avoided. Using the clinical data of the study with sildenafil, a sensitive substrate of CYP3A4, it is unlikely that macitentan would affect other CYP3A4 substrates including new oral anticoagulants (e.g., rivoraxaban) or hormonal contraceptives. These drugs are frequently used by PAH patients and, therefore, an absence of such an interaction would provide benefit to the patient. The use of modeling could support the absence of drug–drug interactions.
The potential of macitentan to have an improved liver safety profile is supported by a lack of effect on concentrations of bile salts and demonstrated in the pharmacokinetic/pharmacodynamic analysis of the SERAPHIN study in which no correlations between macitentan concentrations and increases in ALT, AST, and γ-glutamyl transferase were detected. ET-1 data in healthy subjects and patients with essential hypertension supported a dose of 10 mg as being efficacious, which was confirmed by the SERAPHIN results. Results of the TQT study indicated that macitentan, both at the therapeutic dose of 10 mg and at a supratherapeutic dose of 30 mg, does not have a pro-arrhythmic potential. The safety of macitentan has been reviewed and reported in several other publications [53, 54, 75]. In clinical studies, macitentan had a good safety and tolerability profile. As with other ERAs, macitentan carries a black box warning because of risk of embryo-fetal toxicity and a decrease in hemoglobin. Also, due to an assumed pharmacologic class effect, macitentan could have an adverse effect on spermatogenesis.
Data from the long-term phase III SERAPHIN study indicate a favorable profile with respect to liver enzyme elevations and edema/fluid retention.
In conclusion, the studies discussed in this review support the use of macitentan as an efficacious and well-tolerated treatment for patients with PAH with possible benefits compared with treatment with other registered ERAs.
References
Galie N, Corris PA, Frost A, Girgis RE, Granton J, Jing ZC, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62:D60–72.
D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115:343–9.
Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D42–50.
Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2009;34:1219–63.
Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J. 2007;30:104–9.
Degano B, Sitbon O, Simonneau G. Pulmonary arterial hypertension and HIV infection. Semin Respir Crit Care Med. 2009;30:440–7.
Sitbon O, Lascoux-Combe C, Delfraissy JF, Yeni PG, Raffi F, De Zuttere D, et al. Prevalence of HIV-related pulmonary arterial hypertension in the current antiretroviral therapy era. Am J Respir Crit Care Med. 2008;177:108–13.
Gashouta MA, Humbert M, Hassoun PM. Update in systemic sclerosis-associated pulmonary arterial hypertension. Presse Med. 2014;43:e293–304.
Ahmed S, Palevsky HI. Pulmonary arterial hypertension related to connective tissue disease: a review. Rheum Dis Clin North Am. 2014;40:103–24.
Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circ Res. 2014;115:115–30.
Chester AH, Yacoub MH. The role of endothelin-1 in pulmonary arterial hypertension. Glob Cardiol Sci Pract. 2014;2014:62–78.
Preston IR. Properly diagnosing pulmonary arterial hypertension. Am J Cardiol. 2013;111:2C–9C.
Patel BB, Feng Y, Cheng-Lai A. Pulmonary arterial hypertension: a review in pharmacotherapy. Cardiol Rev 2015;23(1):33–51.
Arai H, Hori S, Aramori I, Ohkubo H, Nakanishi S. Cloning and expression of a cDNA encoding an endothelin receptor. Nature. 1990;348:730–2.
Brunner F, Bras-Silva C, Cerdeira AS, Leite-Moreira AF. Cardiovascular endothelins: essential regulators of cardiovascular homeostasis. Pharmacol Ther. 2006;111:508–31.
Martin ER, Brenner BM, Ballermann BJ. Heterogeneity of cell surface endothelin receptors. J Biol Chem. 1990;265:14044–9.
Takayanagi R, Kitazumi K, Takasaki C, Ohnaka K, Aimoto S, Tasaka K, et al. Presence of non-selective type of endothelin receptor on vascular endothelium and its linkage to vasodilation. FEBS Lett. 1991;282:103–6.
Farhat N, Matouk CC, Mamarbachi AM, Marsden PA, Allen BG, Thorin E. Activation of ETB receptors regulates the abundance of ET-1 mRNA in vascular endothelial cells. Br J Pharmacol. 2008;153:1420–31.
Verhaar MC, Strachan FE, Newby DE, Cruden NL, Koomans HA, Rabelink TJ, et al. Endothelin-A receptor antagonist-mediated vasodilatation is attenuated by inhibition of nitric oxide synthesis and by endothelin-B receptor blockade. Circulation. 1998;97:752–6.
Cardillo C, Kilcoyne CM, Waclawiw M, Cannon RO 3rd, Panza JA. Role of endothelin in the increased vascular tone of patients with essential hypertension. Hypertension. 1999;33:753–8.
Bauer M, Wilkens H, Langer F, Schneider SO, Lausberg H, Schafers HJ. Selective upregulation of endothelin B receptor gene expression in severe pulmonary hypertension. Circulation. 2002;105:1034–6.
Stewart DJ, Levy RD, Cernacek P, Langleben D. Increased plasma endothelin-1 in pulmonary hypertension: marker or mediator of disease? Ann Intern Med. 1991;114:464–9.
Yamane K, Miyauchi T, Suzuki N, Yuhara T, Akama T, Suzuki H, et al. Significance of plasma endothelin-1 levels in patients with systemic sclerosis. J Rheumatol. 1992;19:1566–71.
Shao D, Park JE, Wort SJ. The role of endothelin-1 in the pathogenesis of pulmonary arterial hypertension. Pharmacol Res. 2011;63:504–11.
Thorin E, Clozel M. The cardiovascular physiology and pharmacology of endothelin-1. Adv Pharmacol. 2010;60:1–26.
Dupuis J, Goresky CA, Fournier A. Pulmonary clearance of circulating endothelin-1 in dogs in vivo: exclusive role of ETB receptors. J Appl Physiol. 1996;81:1510–5.
Black SM, Mata-Greenwood E, Dettman RW, Ovadia B, Fitzgerald RK, Reinhartz O, et al. Emergence of smooth muscle cell endothelin B-mediated vasoconstriction in lambs with experimental congenital heart disease and increased pulmonary blood flow. Circulation. 2003;108:1646–54.
Trow TK, Taichman DB. Endothelin receptor blockade in the management of pulmonary arterial hypertension: selective and dual antagonism. Respir Med. 2009;103:951–62.
Schirger JA, Chen HH, Jougasaki M, Lisy O, Boerrigter G, Cataliotti A, et al. Endothelin A receptor antagonism in experimental congestive heart failure results in augmentation of the renin–angiotensin system and sustained sodium retention. Circulation. 2004;109:249–54.
Ohnishi M, Wada A, Tsutamoto T, Fukai D, Kinoshita M. Comparison of the acute effects of a selective endothelin ETA and a mixed ETA/ETB receptor antagonist in heart failure. Cardiovasc Res. 1998;39:617–24.
Iglarz M, Clozel M. Mechanisms of ET-1-induced endothelial dysfunction. J Cardiovasc Pharmacol. 2007;50:621–8.
Dupuis J. Endothelin: setting the scene in PAH. Eur Respir Rev. 2007;16:3–7.
Sauvageau S, Thorin E, Caron A, Dupuis J. Endothelin-1-induced pulmonary vasoreactivity is regulated by ET(A) and ET(B) receptor interactions. J Vasc Res. 2007;44:375–81.
Anderson JR, Nawarskas JJ. Pharmacotherapeutic management of pulmonary arterial hypertension. Cardiol Rev. 2010;18:148–62.
Badesch DB, Abman SH, Simonneau G, Rubin LJ, McLaughlin VV. Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest. 2007;131:1917–28.
Taichman DB, Ornelas J, Chung L, Klinger JR, Lewis S, Mandel J, et al. Pharmacologic therapy for pulmonary arterial hypertension in adults: CHEST guideline and expert panel report. Chest. 2014;146:449–75.
Zamanian RT, Kudelko KT, Sung YK, de Jesus Perez V, Liu J, Spiekerkoetter E. Current clinical management of pulmonary arterial hypertension. Circ Res. 2014;115:131–47.
Boniface S, Reynaud-Gaubert M. Endothelin receptor antagonists—their role in pulmonary medicine. Rev Mal Respir. 2011;28:e94–107.
Dingemanse J, van Giersbergen PL. Clinical pharmacology of bosentan, a dual endothelin receptor antagonist. Clin Pharmacokinet. 2004;43:1089–115.
Vatter H, Seifert V. Ambrisentan, a non-peptide endothelin receptor antagonist. Cardiovasc Drug Rev. 2006;24:63–76.
Channick RN, Simonneau G, Sitbon O, Robbins IM, Frost A, Tapson VF, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. 2001;358:1119–23.
Galie N, Badesch D, Oudiz R, Simonneau G, McGoon MD, Keogh AM, et al. Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2005;46:529–35.
McGlinchey N, Peacock AJ. Endpoints in PAH clinical trials in the era of combination therapy: how do we decide whether something is working without going bankrupt? Drug Discov Today. 2014;19:1236–40.
Gaine S, Simonneau G. The need to move from 6-minute walk distance to outcome trials in pulmonary arterial hypertension. Eur Respir Rev. 2013;22:487–94.
Hassoun PM, Nikkho S, Rosenzweig EB, Moreschi G, Lawrence J, Teeter J, et al. Updating clinical endpoint definitions. Pulm Circ. 2013;3:206–16.
Iglarz M, Binkert C, Morrison K, Fischli W, Gatfield J, Treiber A, et al. Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist. J Pharmacol Exp Ther. 2008;327:736–45.
Bolli MH, Boss C, Binkert C, Buchmann S, Bur D, Hess P, et al. The discovery of N-[5-(4-bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N′-propylsulfamide (macitentan), an orally active, potent dual endothelin receptor antagonist. J Med Chem. 2012;55:7849–61.
Iglarz M, Bossu A, Wanner D, Bortolamiol C, Rey M, Hess P, et al. Comparison of pharmacological activity of macitentan and bosentan in preclinical models of systemic and pulmonary hypertension. Life Sci. 2014;118(2):333–9.
Gatfield J, Mueller Grandjean C, Sasse T, Clozel M, Nayler O. Slow receptor dissociation kinetics differentiate macitentan from other endothelin receptor antagonists in pulmonary arterial smooth muscle cells. PLoS One. 2012;7:e47662.
Food and Drug Administration. FDA approves Opsumit to treat pulmonary arterial hypertension [press release]. October 18, 2013. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm371362.htm. Accessed Oct 2014.
Actelion Pharmaceuticals Ltd. Actelion receives Health Canada approval of Opsumit (macitentan) for the long-term treatment of pulmonary arterial hypertension. http://www1.actelion.com/en/investors/news-archive/index.page?newsId=1742459. Accessed Apr 2015.
SwissMedic, Schweizerisches Heilmittelinstitut. Overview of newly registered human drugs. https://www.swissmedic.ch/zulassungen/00153/00189/00200/01986/index.html?lang=de. Accessed Oct 2014.
Pulido T, Adzerikho I, Channick RN, Delcroix M, Galie N, Ghofrani HA, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809–18.
Dingemanse J, Sidharta PN, Maddrey WC, Rubin LJ, Mickail H. Efficacy, safety and clinical pharmacology of macitentan in comparison to other endothelin receptor antagonists in the treatment of pulmonary arterial hypertension. Expert Opin Drug Saf. 2014;13:391–405.
Kummer O, Haschke M, Hammann F, Bodmer M, Bruderer S, Regnault Y, et al. Comparison of the dissolution and pharmacokinetic profiles of two galenical formulations of the endothelin receptor antagonist macitentan. Eur J Pharm Sci. 2009;38:384–8.
Bruderer S, Hopfgartner G, Seiberling M, Wank J, Sidharta PN, Treiber A, et al. Absorption, distribution, metabolism, and excretion of macitentan, a dual endothelin receptor antagonist, in humans. Xenobiotica. 2012;42:901–10.
Sidharta PN, Lindegger N, Ulc I, Dingemanse J. Pharmacokinetics of the novel dual endothelin receptor antagonist macitentan in subjects with hepatic or renal impairment. J Clin Pharmacol. doi:10.1002/jcph.193. Epub 3 Oct 2013.
Sidharta PN, van Giersbergen PL, Halabi A, Dingemanse J. Macitentan: entry-into-humans study with a new endothelin receptor antagonist. Eur J Clin Pharmacol. 2011;67:977–84.
OPSUMIT: EPAR—product information. Last updated Oct 2, 2014. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002697/human_med_001717.jsp&mid=WC0b01ac058001d124. Accessed Oct 2014.
FDA_guidance_2002. FDA Guidance for Industry: Food-effect bioavailability and fed bioequivalence studies. December 2002. http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM126833.pdf. Accessed 26 Feb 2015.
EMA. CPMP/EWP/560/95/Rev. 1 Corr. Guideline on the investigation of drug interactions. 21 June 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf. Accessed 26 Feb 2015.
Prescribing information: Opsumit® (macitentan) tablets, for oral use. South San Francisco: Actelion Pharmaceuticals US, Inc.; 2013.
Summary of product characteristics: Opsumit® (macitentan) 10 mg tablets, for oral use. London: Actelion Registration, Ltd; 2013.
Sidharta PN, van Giersbergen PL, Dingemanse J. Safety, tolerability, pharmacokinetics, and pharmacodynamics of macitentan, an endothelin receptor antagonist, in an ascending multiple-dose study in healthy subjects. J Clin Pharmacol. 2013;53:1131–8.
de Kanter, R, Sidharta P, Delahaye S, Gnerre C, Segrestaa J, Buchmann S, et al. Physiologically-based pharmacokinetic (PBPK) modeling of macitentan: prediction of drug–drug interactions (submitted).
Yasuda SU, Zhang L, Huang SM. The role of ethnicity in variability in response to drugs: focus on clinical pharmacology studies. Clin Pharmacol Ther. 2008;84:417–23.
Bruderer S, Marjason J, Sidharta PN, Dingemanse J. Pharmacokinetics of macitentan in Caucasian and Japanese subjects: the influence of ethnicity and sex. Pharmacology. 2013;91:331–8.
Ahn LY, Kim SE, Yi S, Dingemanse J, Lim KS, Jang IJ, et al. Pharmacokinetic–pharmacodynamic relationships of macitentan, a new endothelin receptor antagonist, after multiple dosing in healthy Korean subjects. Am J Cardiovasc Drugs. 2014;14:377–85.
Zisowsky J, Sidharta PN, Krause A, Dingemanse J. Pharmacokinetic/pharmacodynamic analyses in SERAPHIN, a randomized, controlled study of macitentan in patients with pulmonary arterial hypertension [abstract]. Clin Pharmacol Drug Dev. 2013;2:1–47.
Actelion Results Database. Study AC-055-201. http://trials.actelion.com/asp/Trial_Registry/RStudyInfo.asp?ST=AC-055-201. Accessed Oct 2014.
FDA. Guidance for industry: E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs, October 2005. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm073153.pdf. Accessed 26 Feb 2015.
EMA. CHMP/ICH/2/04 ICH note for guidance on the clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs (ICH E14). 1 Nov 2005. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002879.pdf. Accessed 26 Feb 2015.
Lindegger N, Sidharta PN, Reseski K, Dingemanse J. Macitentan, a dual endothelin receptor antagonist for the treatment of pulmonary arterial hypertension, does not affect cardiac repolarization in healthy subjects. Pulm Pharmacol Ther. 2014;29:41–8.
Fattinger K, Funk C, Pantze M, Weber C, Reichen J, Stieger B, et al. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther. 2001;69:223–31.
Treiber A, Aanismaa P, de Kanter R, Delahaye S, Treher M, Hess P, et al. Macitentan does not interfere with hepatic bile salt transport. J Pharmacol Exp Ther. 2014;350:130–43.
Weiss J, Theile D, Ruppell MA, Speck T, Spalwisz A, Haefeli WE. Interaction profile of macitentan, a new non-selective endothelin-1 receptor antagonist, in vitro. Eur J Pharmacol. 2013;701:168–75.
Treiber A, Schneiter R, Hausler S, Stieger B. Bosentan is a substrate of human OATP1B1 and OATP1B3: inhibition of hepatic uptake as the common mechanism of its interactions with cyclosporin A, rifampicin, and sildenafil. Drug Metab Dispos. 2007;35:1400–7.
Bruderer S, Aanismaa P, Homery MC, Hausler S, Landskroner K, Sidharta PN, et al. Effect of cyclosporine and rifampin on the pharmacokinetics of macitentan, a tissue-targeting dual endothelin receptor antagonist. AAPS J. 2012;14:68–78.
Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425–36.
Barst RJ, Gibbs JS, Ghofrani HA, Hoeper MM, McLaughlin VV, Rubin LJ, et al. Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S78–84.
Kaminsky LS, Zhang ZY. Human P450 metabolism of warfarin. Pharmacol Ther. 1997;73:67–74.
Sidharta PN, Dietrich H, Dingemanse J. Investigation of the effect of macitentan on the pharmacokinetics and pharmacodynamics of warfarin in healthy male subjects. Clin Drug Investig. 2014;34:545–52.
Costa IM, Soares PJ, Afonso M, Ratado P, Lanaot JM, Falcao AC. Therapeutic monitoring of warfarin: the appropriate response marker. J Pharm Pharmacol. 2000;52:1405–10.
Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–57.
Chaumais MC, Perrin S, Sitbon O, Simonneau G, Humbert M, Montani D. Pharmacokinetic evaluation of sildenafil as a pulmonary hypertension treatment. Expert Opin Drug Metab Toxicol. 2013;9:1193–205.
Sidharta PN, van Giersbergen PL, Wolzt M, Dingemanse J. Investigation of mutual pharmacokinetic interactions between macitentan, a novel endothelin receptor antagonist, and sildenafil in healthy subjects. Br J Clin Pharmacol. 2014;78(5):1035–42.
FDA. Guidance for industry: Drug interaction studies—study design, data analysis, implications for dosing and labeling recommendations. Draft Guidance, February, 2012. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292362.pdf. Accessed 26 Feb 2015.
Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Effects of the antifungal agents on oxidative drug metabolism: clinical relevance. Clin Pharmacokinet. 2000;38:111–80.
Atsmon J, Dingemanse J, Shaikevich D, Volokhov I, Sidharta PN. Investigation of the effects of ketoconazole on the pharmacokinetics of macitentan, a novel dual endothelin receptor antagonist, in healthy subjects. Clin Pharmacokinet. 2013;52:685–92.
Binet I, Wallnofer A, Weber C, Jones R, Thiel G. Renal hemodynamics and pharmacokinetics of bosentan with and without cyclosporine A. Kidney Int. 2000;57:224–31.
van Giersbergen PL, Bodin F, Dingemanse J. Cyclosporin increases the exposure to tezosentan, an intravenous dual endothelin receptor antagonist. Eur J Clin Pharmacol. 2002;58:243–5.
Kim RB. Organic anion-transporting polypeptide (OATP) transporter family and drug disposition. Eur J Clin Invest. 2003;33(Suppl 2):1–5.
Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–36.
Vavricka SR, Van Montfoort J, Ha HR, Meier PJ, Fattinger K. Interactions of rifamycin SV and rifampicin with organic anion uptake systems of human liver. Hepatology. 2002;36:164–72.
George J, Murray M, Byth K, Farrell GC. Differential alterations of cytochrome P450 proteins in livers from patients with severe chronic liver disease. Hepatology. 1995;21:120–8.
Verbeeck RK. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur J Clin Pharmacol. 2008;64:1147–61.
Prescott LF, Forrest JA, Adjepon-Yamoah KK, Finlayson ND. Drug metabolism in liver disease. J Clin Pathol Suppl (R Coll Pathol). 1975;9:62–5.
Yang LQ, Li SJ, Cao YF, Man XB, Yu WF, Wang HY, et al. Different alterations of cytochrome P450 3A4 isoform and its gene expression in livers of patients with chronic liver diseases. World J Gastroenterol. 2003;9:359–63.
Actelion Pharmaceuticals Ltd, Data on file, 2005.
Actelion Pharmaceuticals Ltd, Data on file, 2012.
Acknowledgments
Funding to support this study and preparation of this manuscript was provided by Actelion Pharmaceuticals Ltd. All of the authors are full-time employees of Actelion Pharmaceuticals Ltd. PNS, AT, and JD own stock in Actelion Pharmaceuticals Ltd.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Sidharta, P.N., Treiber, A. & Dingemanse, J. Clinical Pharmacokinetics and Pharmacodynamics of the Endothelin Receptor Antagonist Macitentan. Clin Pharmacokinet 54, 457–471 (2015). https://doi.org/10.1007/s40262-015-0255-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-015-0255-5