
Abstract
Background and Objective
The safety, tolerability, and pharmacokinetics of the once-daily human glucagon-like peptide-1 (GLP-1) analog liraglutide have been evaluated in pediatric patients aged greater than 10 years with type 2 diabetes (T2D). In this study, a population pharmacokinetic analysis was compared to the pediatric pharmacokinetic data with those from two clinical pharmacology trials in adults with T2D.
Methods
A one-compartment pharmacokinetic model previously found to adequately describe the pharmacokinetics of liraglutide in adults with T2D was applied to the evaluation of 13 pediatric subjects (10–17 years of age) with T2D. Steady-state estimates for apparent clearance (CL/F) for individual subjects and corresponding dose were used to derive the area under the plasma–concentration time curve from 0–24 h (AUC24) and investigate dose proportionality in the pediatric trial. A covariate analysis evaluated the effects of body weight, gender, and age category (pediatric/adult) on liraglutide exposure.
Results
Dose proportionality in the dose range of 0.3–1.8 mg was indicated by the model-derived AUC24 slope: 1.05 (95 % CI 0.96–1.15). Consistent with findings from adult trials, body weight and gender were relevant covariates for liraglutide exposure in the pediatric population. The CL/F estimates, and thus exposure, for the pediatric subjects with T2D were similar to those in the adult trials.
Conclusion
Based on this population pharmacokinetic analysis, the liraglutide dose regimen that was found to be clinically effective in adults is predicted to achieve the same range of exposure in the pediatric population (10–17 years of age) with a pre-trial body weight range of 57–214 kg.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
This model predicts that the liraglutide dose regimen used in adults will result in the same range of exposures in the pediatric population (10–17 years) with type 2 diabetes (T2D). |
This pharmacokinetic analysis of liraglutide in pediatric subjects with T2D could help broaden the range of treatment options for this population. |
1 Introduction
The human glucagon-like peptide-1 (GLP-1) analog liraglutide is currently approved for the treatment of adults with type 2 diabetes (T2D). Clinical trials in adult subjects with T2D demonstrated that liraglutide 1.2 mg and 1.8 mg (administered alone or in combination with other oral antidiabetic drugs and/or insulin) improved glycemic control (mean decrease in glycated hemoglobin [HbA1c] up to −1.5 %), reduced body weight (mean weight loss up to −3.4 kg for liraglutide 1.8 mg and −2.9 for liraglutide 1.2 mg) and had an acceptable safety and tolerability profile, with the most prevalent adverse events being gastrointestinal in nature (mainly mild, transient nausea) [1–7]. The pharmacokinetics of liraglutide in adults showed dose–exposure proportionality and properties consistent with once-daily dose administration. While advanced age (>65 years), race, and ethnicity did not affect drug exposure in adults, body weight and gender were identified as relevant covariates for liraglutide exposure [8]; however, these characteristics did not affect clinical response to the 1.2 mg and 1.8 mg doses.
The prevalence of T2D in youth is increasing [9]. Currently, in addition to the emphasis on lifestyle change, metformin and insulin are the only pharmacological treatments approved for the medical management of pediatric subjects with this disease [10]. However, as metformin therapy alone is often unable to maintain glycemic control in pediatric subjects [11], there is a growing need to broaden the range of treatment options for this population.
A recent clinical trial was the first to evaluate the safety, tolerability, and pharmacokinetics of liraglutide in pediatric subjects (10–17 years of age) with T2D [12]. While the pharmacokinetic profile of liraglutide in pediatric subjects was consistent with the profile in adults, there were several limitations for the analysis of pharmacokinetic data in the study [12]. The pediatric population with T2D is still relatively small compared with the adult population, which hindered recruitment of pediatric patients into this trial. Additionally, a minimized number and volume of blood samples (sparse sampling) and time limits on trial participation can be encountered [12]. Given these limitations, a traditional non-compartmental approach to data analysis cannot always be employed to evaluate pediatric exposure or liraglutide dose–exposure proportionality. However, these limitations can be overcome by using the population pharmacokinetics analysis approach that allows the comparison of drug pharmacokinetics from pediatric trials with those previously obtained in adults [13].
In this study, in order to compare the pharmacokinetics of liraglutide in children and adolescents (referred to herein as ‘pediatric’) and adult subjects, and to determine whether the adult dosing regimen is appropriate for future clinical trials in this pediatric population, we undertook a population pharmacokinetic analysis comparing the data from the above-mentioned pediatric trial with liraglutide with those from two previously published liraglutide trials in adult subjects with T2D [14, 15].
2 Methods
2.1 Data Sources Used to Populate the Pharmacokinetics Model
Population demographics and pharmacokinetic data from previously reported clinical trials (one in pediatric subjects [Trial 1, n = 13 subjects] and two in adult subjects [Trials 2 and 3, n = 12 and n = 32 subjects, respectively]) were used as data sources for this population pharmacokinetic analysis (see Online Resource Table 1 and, Figs. 1, 2) [12, 14, 15]. The adult trials were pre-selected and were chosen as they included adults with T2D and had pharmacokinetic sampling at steady-state (24 h). Trial 3 had a lower proportion of women compared with Trials 1 and 2 (28 % vs. 62 and 50 %, respectively). The median body weight of pediatric subjects (Trial 1) was greater than the weight of adult subjects in Trials 2 and 3 (106 kg vs. 83 and 96 kg, respectively); this was true mainly because of one outlier (baseline body weight of 214 kg), which commonly occurs with small sample sizes. The adult trials were selected as comparators prior to the unblinding of the pediatric trial due to the similarity of their study populations (all subjects with T2D) and the availability of full steady-state pharmacokinetic profiles with liraglutide 1.8 mg. All of these trials were conducted in accordance with the Declaration of Helsinki [16] and adhered to the Good Clinical Practice Guidelines [17], with subjects (or their legal age representatives) providing written informed consent. Pediatric subjects treated in Europe also provided written assent.
2.1.1 Pediatric Trial
The pediatric trial (Trial 1) was a clinical pharmacology, randomized, multicenter, double-blind, placebo-controlled trial of once-daily liraglutide in pediatric subjects (10–17 years of age) with T2D (trial registration number: NCT00943501). Details of trial design and assessments have been described previously [12] and are briefly summarized below. Eligible participants had HbA1c 6.5–11 % and fasting plasma glucose (FPG) 6.1–13.3 mmol/L (110–240 mg/dL). The first four participants had different glycemic inclusion criteria (HbA1c 7–10 % and FPG 7.2–12.2 mmol/L [130–220 mg/dL]), which were later broadened to those above in order to facilitate recruitment. Additional eligibility criteria were: body mass index (BMI) >85th percentile for their age and gender; subjects were treated with diet and exercise alone or in combination with a stable dose of metformin for at least 4 weeks before screening. The majority of subjects were post-pubertal based on Tanner stage; only two subjects in the liraglutide group were pre-pubertal (at Tanner stage I or II). Subjects with impaired renal function defined as serum-creatinine greater than the upper reference range for age in children were excluded.
Liraglutide dose was increased weekly (with 1 week at each dose level) starting at 0.3 mg/day to doses of 0.6, 0.9, 1.2, and 1.8 mg/day or corresponding placebo over 5 weeks, based on tolerability and FPG (average of three consecutive measurements >6.1 mmol/L [110 mg/dL]). If the dose was not increased, participants continued on their highest attained dose level for the rest of the treatment period. The safety and side effect profile of liraglutide in pediatric subjects with T2D was similar to that in adults; gastrointestinal adverse events were reported most commonly [12]. Samples for pharmacokinetic assessments were drawn on the last day of the week before dose escalation at liraglutide doses of 0.3 mg (week 1), 0.6 mg (week 2), 1.2 mg (week 4) and after the final drug dose for 1.8 mg (week 5). The first four participants had a 24-h blood sampling schedule during weeks 1, 2, and 4, which was amended to a 13-h schedule for the remaining participants, in order to reduce their time spent in the clinic. Blood samples were taken 15 min before drug administration and at 2, 5, 8, 10, and 13 h post-dosing. Blood samples were also taken at 24, 48, and 72 h after administration of the final drug dose in week 5 (1.8 mg or the highest attained dose) to determine the concentration of liraglutide during the 0–72 h post-dose period.
Of the 14 subjects randomized to liraglutide, one subject withdrew prior to data collection due to procedural issues and therefore did not contribute any pharmacokinetic data. Thus, data for 13 subjects treated with liraglutide were included in the population pharmacokinetic analysis. One subject remained at liraglutide 0.3 mg throughout the trial period, and three subjects did not escalate beyond liraglutide 0.6 mg; these subjects continued on these dose levels until the end of the trial. One subject self-administered liraglutide 1.5 mg by mistake for 6 days prior to the day of pharmacokinetic sampling, whereas drug doses for two subjects were escalated from 0.6 mg to 0.9 mg by mistake on the day of pharmacokinetic blood sampling. Although these doses (1.5 mg and 0.9 mg) deviated from the trial protocol, liraglutide concentration measurements from these samples were included in this population pharmacokinetic analysis, even if the subjects were not in steady-state. Information regarding the amount of drug and time of administration was available for all doses taken during the trial [12].
2.1.2 Adult Trials
Trial 2 (NCT00993304) was a pharmacokinetic study that examined the effect of steady-state liraglutide 1.8 mg on postprandial lipid levels [14]. It was a randomized, double-blind, placebo-controlled, two-period, crossover trial in adult subjects with T2D with HbA1c between 6.5 and 10 % and BMI between 18.5 and 40 kg/m2, and subjects with impaired renal function, defined as creatinine clearance <60 mL/min, were excluded. Blood samples were drawn at steady-state after subjects had received liraglutide 1.8 mg for 1 week. In addition, serial samples were taken over a 60-h period after the administration of the last dose. Pharmacokinetic sampling and profiling details for the liraglutide 1.8 mg dose are shown in Table 1.
Trial 3 (NCT00873223) was an open-label trial investigating the pharmacokinetics and pharmacodynamics of a single dose of insulin detemir and steady-state liraglutide 1.8 mg in adult subjects with T2D [15]. Subjects with HbA1c between 7 and 9.5 % were eligible for this trial, and subjects with impaired renal function, defined as serum creatinine ≥1.5 mg/dL for males and ≥1.3 mg/dL for females, were excluded. Only pharmacokinetic data obtained after administration of liraglutide 1.8 mg alone for 1 week, with serial blood samples drawn 0–24 h after the last dose of liraglutide 1.8 mg, were used in the present analysis (Table 1).
For all three trials, the levels of liraglutide in plasma samples were measured using a validated enzyme-linked immunosorbent assay (ELISA) method [18].
2.2 Pharmacokinetic Model
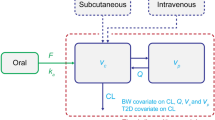
A one-compartment pharmacokinetic model with first-order absorption and elimination parameters (absorption rate constant [k a], apparent clearance [CL/F], and apparent volume of distribution [Vd/F]) was previously found to adequately describe liraglutide pharmacokinetic data in adult subjects with T2D [8]. In order to demonstrate that the model developed previously for adults with T2D would be applicable for the study of pediatric subjects, model qualification was performed by means of goodness-of-fit plots as part of the analysis, following the recommendations in the US FDA guidance for industry regarding population pharmacokinetic analyses [19]. The pharmacokinetic model was fitted to the pooled data from the three trials to obtain parameter estimates for this particular dataset. Between-subject variability parameters were included for CL/F and Vd/F (data not shown). Residual error was described using a proportional error model.
Estimates for steady-state drug CL/F for each individual pediatric subject and the corresponding dose level were used to derive area under the plasma–concentration time curve from 0–24 h (AUC24) [i.e. liraglutide exposure] using the following calculation: AUC24 = dose (in nmol)/(CL/F); AUC24 was used to evaluate the dose proportionality for liraglutide in the dose range 0.3–1.8 mg. Estimated CL/F and the derived AUC24 for pediatric and adult subjects were compared to support the selection of starting and maintenance doses in future pediatric clinical trials. Model-derived liraglutide concentration–time profiles for those pediatric (Trial 1) and adult (Trials 2 and 3) subjects who received the 1.8 mg dose were compared.
The effect of pre-selected covariates (body weight, gender, and age category [i.e. pediatric/adult]) on liraglutide exposure was investigated using a pooled dataset from the three trials (Trials 1, 2, and 3). By pooling all data, the pharmacokinetic model allowed for testing the effect of all covariates simultaneously. The parameterization of the covariates (power model) is as shown in Online Resource Table 1. Body weight and gender were chosen because they were previously identified as important covariates for liraglutide exposure in adults, although neither parameter impacted clinical response to the liraglutide 1.2 and 1.8 mg doses [8]. Due to a narrow age range in pediatric subjects (10–17 years), and an age gap between the oldest pediatric subjects (17 years) and the youngest adult subject (33 years), the inclusion of age as a continuous covariate was not feasible (data not shown). A covariate modeling approach emphasizing parameter estimation rather than stepwise hypothesis testing was implemented [20, 21], where the impact of each covariate was assessed in a full model estimation approach by examining the change in mean (90 % confidence interval [CI]) AUC24 relative to a reference subject which represented a composite of selected reference covariates (a 90 kg adult female). Body weight of 90 kg was chosen as it represents the approximate average body weight for patients with T2D in the trial populations analyzed here. For discrete parameters, one of the parameters was selected for gender (i.e. female) and age group (i.e. adult). Covariate effects were considered relevant for the exposure if the 90 % CIs were outside the range of bioequivalence limits (0.8–1.25) [20].
2.3 Software
NONMEM 7.1.2 software (ICON Development Solutions, Ellicott City, MD, USA) was used for all population pharmacokinetic analyses, and S-plus, 8.0 (TIBCO, Palo Alto, CA, USA) and R (Revolution R Enterprise 6.0) was used for data processing and graphical analyses.
3 Results
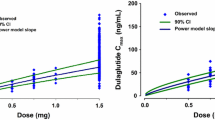
A goodness-of-fit plot was used to compare the observed and model-derived liraglutide concentration–time profiles in the pediatric trial at each dose level (0.3, 0.6, 1.2, and 1.8 mg; Fig. 1). The large degree of overlap between the observed and model-derived concentrations indicated that the current model could adequately describe liraglutide pharmacokinetics in pediatric subjects with T2D. Additional goodness-of-fit plots are shown in Online Resource Figs. 3 and 4.

Observed and model-derived liraglutide concentration profiles in the pediatric population by dose level. Figures with bars geometric mean (95% confidence interval) of the observed liraglutide concentration. Lines geometric means of the individual (post hoc) model-derived concentration–time profiles
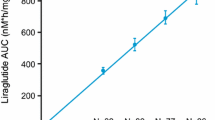
In order to confirm pediatric dose proportionality, estimates of CL/F for each individual and the corresponding liraglutide dose level were used to derive AUC24. In the pediatric population, model-estimated liraglutide exposure (AUC24) increased proportionally with increasing liraglutide dose (0.3–1.8 mg; Fig. 2). The geometric mean (95 % CI) exponent was 1.05 (0.96–1.15); the inclusion of 1.0 in the 95 % CI indicated dose proportionality, consistent with previous findings in adults [8], also strongly supported dose proportionality.

Dose proportionality test based on model-estimated AUC24, which, in turn, was derived from estimated CL/F. Solid line represents geometric mean AUC24, as estimated by the linear mixed-effects model of log (AUC24) versus log (dose). Model-derived AUC24 slope: 1.05 [0.96–1.15]95 % CI. AUC area under the plasma–concentration time curve, AUC 24 AUC from zero to 24 h, CL/F apparent clearance, CI confidence interval
Body weight and gender have been previously identified as important covariates for liraglutide exposure in adults [8]. In order to determine whether these factors also affected liraglutide exposure in pediatric subjects, the impact of these covariates, as well as age category (pediatric vs. adult), was investigated by examining the mean AUC24 (90 % CI) relative to a reference subject (a 90 kg adult female). Similar to previous observations in adults, both body weight and gender had a pharmacokinetically relevant impact on mean liraglutide exposure. The relevance of the identified covariates (body weight and gender) for the pediatric population was confirmed in a sensitivity analysis which did not include adult data (data not shown). Age group (pediatric vs. adult) was not considered a pharmacokinetically relevant factor since it was of minor magnitude (10 % decrease in AUC24 compared with an adult subject of the same weight and gender), although the 90 % CI (0.78–1.03) was slightly outside the 80–125 % range (Fig. 3). AUC24 decreased with increasing body weight; geometric mean AUC24 was 64 % higher at the lowest observed body weight (53 kg) and 56 % lower at the highest observed body weight (216 kg) compared with the exposure at a reference body weight of 90 kg (Fig. 3). Furthermore, AUC24 was lower for males; a 31 % lower drug exposure compared with the reference female subject of the same body weight was observed. The inclusion of covariates reduced between-subject variability in CL/F by 50 % compared with the base model without covariates.

Geometric mean ratios and 90 % CI of effect of demographic covariates (gender, body weight, and age group) on AUC24 relative to a reference subject (90 kg adult female). The solid line represents the ratio of 1 (i.e. no pharmacokinetic relevance). Broken lines delineate the acceptance interval for bioequivalence (0.8–1.25); these limits were used to determine drug exposure equivalence in this analysis. AUC area under the plasma–concentration time curve, AUC 24 AUC from zero to 24 h, CI confidence interval
At the liraglutide 1.8 mg dose, the CL/F estimate for pediatric subjects was similar to that in the two adult trials (median [range, L/h] 1.55 [0.67–3.83], 0.91 [0.59–2.35], and 1.48 [1.04–3.72] for Trials 1, 2, and 3, respectively), leading to an overlapping range of estimated AUC24 values following the 1.8 mg dose in pediatric and adult subjects (Fig. 4a). The range of estimated AUC24 in pediatric subjects used in this analysis as well as in the dose proportionality evaluation (Fig. 2) was comparable to the range in adult subjects and is in line with previous studies [21, 22]. The differences in CL/F between the two adult trials were partly due to the differences in body weight and gender distribution (see Table 1). As demonstrated in the covariate analysis that identified body weight as a relevant factor for explaining between-subject variability in liraglutide exposure (Fig. 3), AUC24 decreased with increasing body weight (Fig. 4a). Upon adjusting for possible confounding by body weight and gender differences, liraglutide concentration–time profiles for pediatric and adult subjects treated with the 1.8 mg dose appeared very similar (Fig. 4b).

a AUC24 versus body weight for liraglutide 1.8 mg once daily, according to age category (pediatric subjects from Trial 1 vs. adult subjects from Trials 2 and 3). The completely superimposed lines show the model-predicted AUC24 versus body weight for pediatric and adult subjects, respectively. The estimated AUC24 has been normalized to a 1.8 mg dose for four pediatric subjects, reaching a maximum dose of 0.3 (n = 1) or 0.6 mg (n = 3). b Model-derived typical steady-state concentration–time profiles for pediatric and adult subjects receiving 1.8 mg liraglutide. The figure shows results for pediatric and adult populations with body weight and gender adjusted (both populations with body weight of 90 kg and 50 % female gender composition) in order to minimize confounding. AUC area under the plasma–concentration time curve, AUC 24 AUC from zero to 24 h
4 Discussion
Herein, we report the application of a population pharmacokinetic model to pharmacokinetic data from pediatric subjects administered the subcutaneous GLP-1 agonist, liraglutide, over a 5-week period. As in adults, this model was able to describe well the observed concentrations of liraglutide in pediatric subjects, indicating its suitability for analysis of pediatric liraglutide pharmacokinetics. Moreover, dose proportionality across a range of doses was observed in the pediatric population, as in adults [21]. The liraglutide dose regimen used in adults is predicted to achieve the same range of exposures in the tested pediatric population. Thus, the results of this analysis support the use of the same starting, escalation, and maintenance doses of liraglutide currently recommended for adults in future clinical trials in pediatric patients who have similar age and body weight as the children investigated in this trial. As a result, the adult dosing regimen is currently being used in the ongoing liraglutide phase III trial (Ellipse), which is investigating the efficacy and safety of liraglutide (added to metformin) over 52 weeks in pediatric subjects with T2D.
Body weight and gender were previously identified in adult trials as pharmacokinetically relevant covariates for liraglutide exposure, although they did not impact clinical response in patients with T2D, including exploratory pharmacodynamic data from this study which showed short-term improvement in glycemic control [8]. The findings of the present analysis demonstrate that there is an indication of the same covariates being relevant in the pediatric population as in the adult population, with lower liraglutide exposure observed with increasing body weight and with male gender. The means and ranges of body weight and BMI for the population in this trial exceed those of reference standards for the same age group. However, the population is representative of the population of adolescents intended for treatment with liraglutide for T2D. Additionally, while the reason for the difference in exposure between male and female subjects is unknown, it is consistent across populations and trials [22, 23]. The model-estimated reduction in liraglutide exposure for male subjects (31 %) in this analysis was comparable to previous estimates in adults [8]. There were no signs of differences in the exposure of liraglutide between the pediatric group and adults in the present analysis.
While this population pharmacokinetic model can adequately describe the pharmacokinetics of liraglutide in pediatric subjects, it may have some limitations. The use of a descriptive model limits the applicability of the findings for liraglutide pharmacokinetics to pediatric subjects with T2D of similar age (range 10–17 years) and weight (57–214 kg). The population of children and adolescents with T2D, in addition to being difficult to recruit and retain, have unique physical characteristics which separate them from the typical adult population with T2D and therefore mandate these confirmatory pharmacokinetic studies. In addition, body composition varies with age until adulthood. Thus, given these distinct characteristics, long-term studies of GLP-1 efficacy in this population are required. It is possible that drug exposure for younger and leaner subjects may be different. Of note, since the prevalence of T2D is very low in youth aged <10 years (0.01 cases per 1,000 individuals) and higher in older youths (0.42 cases per 1,000 individuals), it was a regulatory requirement to investigate the pharmacokinetics of liraglutide in children or adolescents aged >10 years [24]. These studies begin to address the need for data that support the proposal that one might ‘extrapolate’ effects of GLP-1 agonists into the pediatric population. However, great variability in body composition, as seen in this T2D population, may mitigate against this possibility, requiring pharmacodynamic studies to support these conclusions. Therefore, the Ellipse phase III study is currently underway.
There were also some limitations of the covariate analysis. The low number of pediatric subjects results in relatively few subjects in each subpopulation, leading to larger confidence intervals and less certain conclusions compared with larger datasets. Additionally, age was analyzed as a categorical covariate, as it was in the previous analysis of adult data [8], due to the age distribution in the joint population. This trial protocol allowed for subjects not adhering to the planned liraglutide dosing escalation regimen; while the model accounts for the different dose levels, there were fewer samples for the analysis of the maximum liraglutide 1.8 mg dose than initially planned. Furthermore, although the designs of the adult and pediatric trials were different, the model focuses on steady-state exposure (24 h), and thus the difference in design at the end of the interval (>24 h) is of less importance and should not influence the results of the analysis.
Both the FDA and the European Medicines Agency (EMA) recognize the importance of model-based drug development to improve decision making for effectiveness and safety [19, 25]. Accordingly, we used a population pharmacokinetic approach comparing the available pediatric data with data from similar adult trial subjects (i.e. subjects with T2D) to select the doses for the presently ongoing 52-week phase III efficacy and safety trial in pediatric subjects with T2D (NCT01541215). In this larger, longer trial, population pharmacokinetics will be used to determine whether the similar liraglutide exposure in pediatric and adult subjects results in a similar clinical response.
References
Zinman B, Gerich J, Buse JB, Lewin A, Schwartz S, Raskin P, et al. Efficacy and safety of the human glucagon-like peptide-1 analog liraglutide in combination with metformin and thiazolidinedione in patients with type 2 diabetes (LEAD-4 Met+TZD). Diabetes Care. 2009;32:1224–30.
Russell-Jones D, Vaag A, Schmitz O, Sethi BK, Lalic N, Antic S, et al. Liraglutide vs insulin glargine and placebo in combination with metformin and sulfonylurea therapy in type 2 diabetes mellitus (LEAD-5 met+SU): a randomised controlled trial. Diabetologia. 2009;52:2046–55.
Nauck M, Frid A, Hermansen K, Shah NS, Tankova T, Mitha IH, et al. Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (liraglutide effect and action in diabetes)-2 study. Diabetes Care. 2009;32:84–90.
Marre M, Shaw J, Brandle M, Bebakar WM, Kamaruddin NA, Strand J, et al. Liraglutide, a once-daily human GLP-1 analogue, added to a sulphonylurea over 26 weeks produces greater improvements in glycaemic and weight control compared with adding rosiglitazone or placebo in subjects with type 2 diabetes (LEAD-1 SU). Diabet Med. 2009;26:268–78.
Garber A, Henry R, Ratner R, Garcia-Hernandez PA, Rodriguez-Pattzi H, Olvera-Alvarez I, et al. Liraglutide versus glimepiride monotherapy for type 2 diabetes (LEAD-3 Mono): a randomised, 52-week, phase III, double-blind, parallel-treatment trial. Lancet. 2009;373:473–81.
Buse JB, Rosenstock J, Sesti G, Schmidt WE, Montanya E, Brett JH, et al. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: a 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6). Lancet. 2009;374:39–47.
Pratley RE, Nauck M, Bailey T, Montanya E, Cuddihy R, Filetti S, et al. Liraglutide versus sitagliptin for patients with type 2 diabetes who did not have adequate glycaemic control with metformin: a 26-week, randomised, parallel-group, open-label trial. Lancet. 2010;375:1447–56.
Ingwersen SH, Khurana M, Madabushi R, Watson E, Jonker DM, Le Thi TD, et al. Dosing rationale for liraglutide in type 2 diabetes mellitus: a pharmacometric assessment. J Clin Pharmacol. 2012;52:1815–23.
Pinhas-Hamiel O, Zeitler P. The global spread of type 2 diabetes mellitus in children and adolescents. J Pediatr. 2005;146:693–700.
Vaidyanathan J, Choe S, Sahajwalla CG. Type 2 diabetes in pediatrics and adults: thoughts from a clinical pharmacology perspective. J Pharm Sci. 2012;101:1659–71.
Zeitler P, Hirst K, Pyle L, Linder B, Copeland K, Arslanian S, et al. A clinical trial to maintain glycemic control in youth with type 2 diabetes. N Engl J Med. 2012;366:2247–56.
Klein DJ, Battelino T, Chatterjee DJ, Hale PM, Chang CT, Arslanian SA. Liraglutide trial in pediatric type 2 diabetes: safety, tolerability and pharmacokinetics/pharmacodynamics. Diabetes Technol Ther. 2014;16(10):679–8.
De Cock RF, Piana C, Krekels EH, Danhof M, Allegaert K, Knibbe CA. The role of population PK-PD modelling in paediatric clinical research. Eur J Clin Pharmacol. 2011;67(Suppl 1):5–16.
Hermansen K, Baekdal TA, During M, Pietraszek A, Mortensen LS, Jorgensen H, et al. Liraglutide suppresses postprandial triglyceride and apolipoprotein B48 elevations after a fat-rich meal in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, crossover trial. Diabetes Obes Metab. 2013;15(11):1040–8.
Morrow L, Hompesch M, Guthrie H, Chang D, Chatterjee DJ. Co-administration of liraglutide with insulin detemir demonstrates additive pharmacodynamic effects with no pharmacokinetic interaction. Diabetes Obes Metab. 2011;13:75–80.
World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. Last amended by the 59th WMA General Assembly, Seoul. 2008. http://www.wma.net/en/30publications/10policies/b3/.
International Conference on Harmonisation. ICH Harmonised Tripartite Guideline: Guideline For Good Clinical Practice E6 (R1), current Step 4 version, dated 10 June 1996. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf.
Agerso H, Jensen LB, Elbrond B, Rolan P, Zdravkovic M. The pharmacokinetics, pharmacodynamics, safety and tolerability of NN2211, a new long-acting GLP-1 derivative, in healthy men. Diabetologia. 2002;45:195–202.
US Food and Drug Administration. Guidance for industry: general considerations for pediatric pharmacokinetic studies for drugs and biological products. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072114.pdf. Accessed 30 May 2013.
Hu C, Zhang J, Zhou H. Confirmatory analysis for phase III population pharmacokinetics. Pharm Stat. 2011;10:14–26.
Knebel W, Tammara B, Udata C, Comer G, Gastonguay MR, Meng X. Population pharmacokinetic modeling of pantoprazole in pediatric patients from birth to 16 years. J Clin Pharmacol. 2011;51:333–45.
Gottschalk M, Danne T, Vlajnic A, Cara JF. Glimepiride versus metformin as monotherapy in pediatric patients with type 2 diabetes: a randomized, single-blind comparative study. Diabetes Care. 2007;30:790–4.
Al-Shareef MA, Sanneh AF, Aljoudi AS. Clinical effect of metformin in children and adolescents with type 2 diabetes mellitus: a systematic review and meta-analysis. J Family Community Med. 2012;19:68–73.
Liese AD, D’Agostino RB Jr, Hamman RF, Kilgo PD, Lawrence JM, Liu LL, et al. The burden of diabetes mellitus among US youth: prevalence estimates from the SEARCH for Diabetes in Youth Study. Pediatrics. 2006;118:1510–8.
European Medicines Agency. Guideline on the role of pharmacokinetics in the development of medicinal products in the paediatric population. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003066.pdf. Accessed 16 Aug 2013.
Acknowledgments
This study was supported by Novo Nordisk, who provided medical writing assistance (Leanne Johnson-Huang and Irina Nayvelt, Novo Nordisk Inc, Plainsboro, NJ, USA), and funded technical editing (Watermeadow Medical, Witney, UK). The data are from NCT00943501, which was run by Novo Nordisk.
Kristin C. C. Petri and Lisbeth V. Jacobsen are employees of, and hold stocks in, Novo Nordisk; David Klein was an investigator in the Novo-Nordisk sponsored trial, and is now a Medical Director with Medpace.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Petri, K.C.C., Jacobsen, L.V. & Klein, D.J. Comparable Liraglutide Pharmacokinetics in Pediatric and Adult Populations with Type 2 Diabetes: A Population Pharmacokinetic Analysis. Clin Pharmacokinet 54, 663–670 (2015). https://doi.org/10.1007/s40262-014-0229-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-014-0229-z