
Abstract
Background and Objectives
BI 1358894, a novel small-molecule inhibitor of transient receptor potential canonical ion channels, is under development for treatment of major depressive disorder. Phase I trials assessing the safety and pharmacokinetics of BI 1358894 in Caucasian male healthy volunteers (HVs) have been performed. This Phase I, double-blind, placebo-controlled, parallel-group trial assessed the safety, tolerability and pharmacokinetics of BI 1358894 in Japanese male HVs.
Methods
Male HVs were randomized to receive oral BI 1358894 (n = 18) or placebo (n = 6) after a high-fat, high-calorie meal within three dose groups (50 mg, 100 mg, 200 mg), administered sequentially in dose-ascending order. The primary endpoint was number of HVs with drug-related adverse events (DRAEs). Secondary endpoints were the pharmacokinetic parameters of BI 1358894.
Results
Overall, 24 male HVs entered the trial [mean (standard deviation) age: 30.0 (7.6) years]. DRAEs occurred in 3/18 HVs (BI 1358894 100 mg group: one HV experienced dizziness and headache; BI 1358894 200 mg group: one HV experienced headache, another reported sleep disorder). BI 1358894 exposure increased dose dependently and proportionally, peaking 4–6 h after administration before declining in a multiphasic manner with a terminal elimination half-life of ~70 h in the 50 mg and 100 mg dose groups, and 203 h in the 200 mg dose group.
Conclusion
BI 1358894 was well tolerated with a favorable pharmacokinetic profile in Japanese male HVs, similar to findings from a previous study in Caucasian male HVs.
Trial Registration
ClinicalTrials.gov (NCT03875001; 08-Mar-2019).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Single doses of BI 1358894 were well tolerated with a favorable pharmacokinetic profile in Japanese male healthy volunteers similar to previous findings in Caucasian male healthy volunteers |
These findings support the inclusion of the Japanese population in future global studies of BI 1358894 |
1 Introduction
Major depressive disorder (MDD) is a psychiatric condition characterized by depressed mood, loss of interest or pleasure, insomnia or hypersomnia, reduced concentration and attention, reduced self-confidence, and suicidal ideation [1, 2]. Pharmacological treatment options for MDD are currently limited. While several first-line treatment options are available for patients with MDD, no specific existing treatment strategy, such as switching to a different drug class or introducing an adjunct therapy, has been identified for treating patients who do not respond to initial antidepressant treatment [3, 4]. Despite the availability of numerous antidepressants, the inadequate treatment responses, therapeutic lag, and safety and tolerability issues suggest there is an unmet treatment need for patients with MDD [5].
Affective instability is a hallmark of MDD that has been linked to hyperactivity of the amygdala [6, 7], an area of the brain associated with the processing of emotion and mood. Transient receptor potential canonical (TRPC) ion channels modulate neuronal excitability via integrating signals derived from G protein-coupled receptors, and are highly expressed in brain regions such as amygdala and frontal cortex [8,9,10]. Preclinically, it has been demonstrated that lack of TRPC ion channels or its pharmacological inhibition attenuated fear and anxiety without impairing other behaviors in mice [10, 11].
BI 1358894, a small-molecule inhibitor of TRPC ion channels, is currently under development for the treatment of MDD. BI 1358894 is metabolized by the enzymes uridine 5'-diphospho-glucuronosyltransferase (UGT) 1A3, UGT2B7, cytochrome P450 3A4, aldehyde oxidase, and, to a minor extent, by aldehyde dehydrogenase (unpublished data). BI 1358894 is not a substrate of the transporters, P-glycoprotein, breast cancer resistance protein, organic anion transporting polypeptide (OATP) 1B1, or OATP1B3. Previous studies have indicated no clinically relevant effect of BI 1358894 on midazolam, and thus CYP3A activity [12]. Similarly, a less than twofold increase in BI 1358894 bioavailability observed when co-administered with itraconazole (NCT03843151) indicates that concomitant CYP3A4 inhibitor use should not be restricted in future clinical studies of BI 1358894 [13]. Other drug-drug interaction studies revealed an increased bioavailability of rosuvastatin (Cmax increased by 74%) and dabigatran (Cmax increased by 19%) when co-administered with BI 1358894 (NCT04099732) with moderate tolerability [14].
A recent Phase I study of 73 patients with MDD using functional magnetic resonance imaging indicated that BI 1358894 attenuated activity in several cortico-limbic brain regions, including the bilateral amygdala region [15]. Cholecystokinin-tetrapeptide (CCK-4), a neuropeptide, stimulates amygdala neuronal activity and can induce anxiety/panic attacks; BI 1358894 was shown to reduce the psychological and physiological responses of CCK-4-induced anxiety and panic symptoms in healthy volunteers [16, 17].
Phase I clinical trials have shown BI 1358894 to be well tolerated at doses ≤ 200 mg, with a favorable pharmacokinetic profile in healthy Caucasian volunteers [12, 18]. Further, the administration of BI 1358894 after a high-fat, high-calorie breakfast resulted in ~1.6 and ~ 2.5-fold higher exposure at the 50 mg and 100 mg doses, respectively, than in the fasted state [12, 19]. For both fasted and fed parts of the study, the incidence of drug-related adverse events (DRAEs) between doses, and fed versus fasted groups, were similar [12]. However, to evaluate the potential for including Japan in a global Phase II study of BI 1358894, there is a need to assess the pharmacokinetic profile in this population to confirm that interethnic differences in drug absorption, metabolism, and excretion do not exist. Here, the results from a Phase I clinical study investigating the safety and pharmacokinetic profiles of BI 1358894 in healthy Japanese male volunteers are presented. The pharmacokinetic profile of BI 1358894 and its metabolite (Metabolite A), a pharmacologically inactive compound, was also analyzed.
2 Methods
2.1 Objectives and Study Design
The primary objective of this single-site, Phase I study (NCT03875001) was to assess the safety and pharmacokinetic profile of BI 1358894 using a single-rising dose (SRD) schedule in healthy Japanese male volunteers.
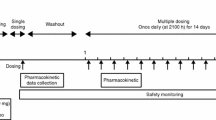
Eligible volunteers were enrolled into a double-blind, randomized, placebo-controlled parallel group trial with an SRD design. Following a 28-day screening period, volunteers were allocated to the BI 1358894 50 mg, 100 mg, or 200 mg group, then randomized 3:1 within each group to receive a single oral dose of BI 1358894 or placebo. The placebo group served as a control for evaluation of safety and tolerability as is standard for trials involving healthy volunteers. The BI 1358894 200 mg dose group was subdivided into two randomized sequential cohorts. Each dose was administered sequentially in dose-ascending order, with each subsequent dose group only being investigated following a dose-escalation review (Fig. 1A). A time interval of at least 6 days was maintained between administration of the last trial drug in the preceding dose group, and the first trial drug administration in the next dose group. In accordance with US Food and Drug Administration (FDA) guidelines, 30 min prior to receiving study treatment, volunteers received a high-fat, high-calorie meal of approximately 900 kcal consisting of ~150 kcal as protein, ~250 kcal as carbohydrate, and ~500–600 kcal as fat. This was to ensure that pharmacokinetics were assessed under conditions in which the impact of food on gastrointestinal physiology, resulting in an effect on the bioavailability of the drug, was at a sufficiently high level [20].

Study design depicting the single-rising dose allocation (a) and the study flow chart (b). aA time interval of at least 6 days was maintained between administration of the last trial drug in the preceding dose group and the first trial drug administration in the next dose group. bThe BI 1358894 200 mg dose group was divided into two sequential cohorts. A allocation, EOT end of trial, S screening, SD single dose
For the BI 1358894 50 mg and 100 mg dose groups, blood samples of approximately 3 mL for pharmacokinetic assessment of BI 1358894 were collected pre-dose and post dose at 10 min, 20 min, and 30 min; and at 1 h, 1.5 h, 2 h, 2.5 h, 3 h, 4 h, 5 h, 6 h, 7 h, 8 h, 10 h, 12 h, 16 h, 24 h, 34 h, and 48 h; then at 24-h intervals, until 192 h post dose (Fig. 1B). For the BI 1358894 200 mg group, samples were collected as per the 50 mg/100 mg groups up to and including Day 7, then one sample was taken on Day 9 at 192 h post-treatment, while additional single samples were taken on Day 11 (240 h post-treatment), Day 15 (336 h post-treatment), Day 22 (504 h post-treatment), and Day 29 (672 h post-treatment; Fig. 1B). Urine samples for pharmacokinetic assessment were collected within 3 h prior to treatment on Day 1, and at 0–4 h, 4–8 h, 8–12 h, 12–24 h, and at 24–48 h, 48–72 h, and 72–96 h on Days 2–5 for all dose groups (Fig. 1B). Both blood and urine samples were stored upright at – 20 °C at the trial site until transfer on dry ice to the analytical laboratory (Covance Laboratory Limited, UK).
2.2 Participants
Healthy male volunteers aged 20–45 years with a body mass index (BMI) of 18.5–25 kg/m2 were enrolled in this study. Due to unavailable developmental and reproductive toxicity data during study design, females were excluded from this study. Further, volunteers were excluded if they had any findings in the medical examination that deviated from normal, any laboratory value outside the reference range, or any evidence of concomitant disease judged to be clinically relevant by the investigator. Further exclusion criteria included the use of drugs that might influence the results of the trial within 30 days prior to administration of trial medication, smoking (more than ten cigarettes or three cigars or three pipes per day), a previous history of suicidal behavior or ideation, or drug or alcohol abuse.
2.3 Bioanalysis
Samples were analyzed using the validated Covance Analytical Method B894HPP (Covance Laboratories Ltd, UK). Blood was collected, processed to plasma by centrifugation, and stored at – 20 \(^\circ\)C until analyzed. The thawed plasma samples underwent centrifugation at 2500g for 10 min at 4–8 °C. An aliquot (50 μL) was taken and 25 μL of an internal standard solution (150 nmol/L BI 1358894 and 200 nmol/L Metabolite A) was added except for the blank control, which had 25 μL of acetonitrile:water (30:70, v/v) added. The samples were vortex mixed and subjected to protein precipitation extraction. Acetonitrile (175 μL) was added to each sample, the plate was sealed, vortex mixed, and centrifuged at 3000g for 5 min at room temperature. The supernatant (100 μL) was transferred into a 96-well plate using an automated liquid handling device, and 100 μL of water:formic acid (100:0.2, v/v) was added to each sample and vortex mixed. Chromatography was performed on a Phenomenex Kinetex C8 column under a gradient elution for a total run time of approximately 4.2 min. Plasma concentrations of BI 1358894 and its metabolite (Metabolite A) were determined simultaneously using a validated liquid chromatography tandem mass spectrometry (LC-MS/MS) assay. Calibration curves covered a range of 1–2000 nmol/L for BI 1358894, and 2–2000 nmol/L for Metabolite A, in 50 μL undiluted plasma samples.
Urine concentrations of BI 1358894 were determined using a validated LC-MS/MS assay. Calibration curves for BI 1358894 in urine covered a range of 1–1000 nmol/L in undiluted samples.
The overall (mean) accuracy and precision of the plasma and urine samples of all accepted runs were required to be ≤ 15.0% and ± 15%, respectively.
2.4 Endpoints and Assessments
The primary endpoint was the number of volunteers with DRAEs. Adverse events (AEs), serious adverse events (SAEs), and adverse events of special interest (AESIs; drug-induced liver injury) were monitored and reported using the Medical Dictionary for Regulatory Activities (MedDRA) version 22.0. DRAEs, AEs, SAEs, and AESIs are defined in Online Supplementary Material (OSM) Resource 3. Safety was further assessed by laboratory tests, continuous electrocardiogram (ECG) monitoring, regular evaluation of vital signs, and routine physical examinations. Possible psychedelic effects of BI 1358894 on volunteers were monitored using the Bowdle visual analogue scale (B-VAS), which was administered at screening, 1 h prior to treatment; at 2 h, 4 h, 8 h, 24 h, 34 h, and 48 h after treatment; and at the end of the trial. An additional B-VAS assessment was also performed at 96 h after treatment (for the 50 mg and 100 mg groups), and at 144 h after treatment (for the 200 mg group). Suicidality was assessed using the Columbia suicide severity rating scale (C-SSRS) at screening, 96 h after treatment (for the 50 mg and 100 mg groups), or 144 h after treatment (for the 200 mg group), and at the end of the trial.
The secondary pharmacokinetic endpoints were: the area under the concentration–time curve (AUC) of BI 1358894 in plasma from administration time (t = 0) to the last quantifiable data point (AUC0–tz); AUC of BI 1358894 in plasma from t = 0 extrapolated to infinity (AUC0-∞); and maximum measured concentration of BI 1358894 in plasma (Cmax).
Further pharmacokinetic endpoints of interest were apparent clearance of BI 1358894 in plasma after extravascular administration (CL/F), apparent volume of distribution during the terminal phase after extravascular administration (Vz/F), mean residence time of the analyte in the body extravascular administration (MRTex), terminal half-life of BI 1358894 in plasma (t1/2), time from last dose to the maximum measured concentration of BI 1358894 in plasma (tmax), and urinary parameters. For Metabolite A, AUC, Cmax, tmax, t1/2, and metabolic ratio were calculated.
2.5 Statistical Analysis
All primary safety and pharmacokinetic endpoints were assessed descriptively. Pharmacokinetics were assessed in samples from patients receiving BI 135889. Safety was assessed for patients receiving BI 1358894 and placebo. The pharmacokinetic parameters of AUC0–tz, AUC0-∞, and Cmax were analyzed for dose proportionality using a linear regression model on a logarithmic scale that yielded a slope (β). Based on the estimate for the slope, a 90% confidence interval (CI) was calculated with perfect dose proportionality corresponding to a slope of 1. Exposure ratio of test versus reference treatment (geometric mean [gMean]) was calculated.
The study planned to include 24 volunteers and was not based on a power calculation. The size of 8 volunteers per group used here (6 receiving BI 1358894 and 2 receiving placebo) is commonly used in SRD studies and is considered acceptable for the exploratory evaluation of single-dose safety and pharmacokinetic endpoints.
3 Results
3.1 Disposition of Volunteers
A total of 24 volunteers entered the trial and all completed the planned observation period (OSM Resource 1). The mean (standard deviation [SD]) age of volunteers was 30.0 (7.6) years, and the mean (SD) BMI was 21.6 (1.4) kg/m2 (OSM Resource 2). Mean (SD) age was slightly lower in the BI 1358894 50 mg dose group (26.3 [8.4] years) compared with the other treatment groups (29.8 [7.0] years – 32.3 [8.4] years). Baseline BMI was similar across all treatment groups.
3.2 Safety Profile
Four on-treatment AEs were observed in 3 volunteers receiving BI 1358894 (OSM Resource 3); 1 volunteer (1/6 [16.7%]) in the BI 1358894 100 mg group presented with dizziness and headache, and 2 volunteers displayed DRAEs in the BI 1358894 200 mg group, which were headache (1/6 [16.7%]) and sleep disorder (1/6 [16.7%]). All AEs were deemed DRAEs by the trial investigator; all were of mild intensity and all resolved by the end of the trial (OSM Resource 4). No SAEs, AESIs, AEs leading to discontinuation of trial drug, or AEs of severe intensity were observed (OSM Resource 3).
There were no clinically relevant findings observed during the evaluation of vital signs, ECGs, laboratory tests, and B-VAS; and no concerns related to suicidality were raised following C-SSRS assessment.
3.3 Pharmacokinetics
3.3.1 BI 1358894
Pharmacokinetic analysis included 18 volunteers who were treated with BI 1358894 and provided at least one pharmacokinetic endpoint. BI 1358894 exposure in terms of AUC0-∞, AUC0-tz, and Cmax increased dose dependently over the entire dose range of 50–200 mg, and dose proportionality was statistically exhibited for AUC0-∞ and Cmax, indicated by slopes of 1.1351 and 1.0050, respectively. The slope for AUC0-tz was 1.1757, indicating a more than dose proportional relationship (Table 1).
The interindividual variabilities of exposure were low to moderate across all three dose groups, and the geometric coefficient of variation (gCV) ranged from 10.9% to 26.1%. Total clearance was consistent among dose groups and ranged from 189 to 228 mL/min. Vz/F was high, with gMean values between 1290 and 3320 L (Table 2).
Following single oral administrations of BI 1358894 50–200 mg, BI 1358894 reached Cmax within 4.00–5.50 h (median tmax). The median tmax for the 50 mg, 100 mg, and 200 mg dose groups was 4.00, 5.50, and 5.00 h, respectively. After reaching Cmax, mean plasma concentrations exhibited a multiphasic decline with a parallel terminal phase, and t1/2 were 66.9 h and 74.0 h in the BI 1358894 50 mg and 100 mg dose groups, respectively. For the BI 1358894 200 mg dose group, another late phase with a slow decline in low plasma concentrations was observed that was associated with a t1/2 of 203 h. This was based on a longer pharmacokinetic sampling timeframe of up to 672 h for the BI 1358894 200 mg dose group, compared with up to 192 h for the BI 1358894 50 mg and 100 mg dose groups (Figs. 2A and B; Table 2).

Mean plasma concentration–time profiles of BI 1358894 after single oral administration of 50 mg, 100 mg, and 200 mg doses on a linear scale (a) and semi-logarithmic scale (b). The insert shows the 0- to 24-h time interval on a linear scale. Note: Error bars indicate standard deviation
Urinary excretion of BI 1358894 was low. Cumulative amounts of BI 1358894 in the urine were below 2 nmol/L and the cumulative fraction excreted in urine was below 0.001% of dose in 200 mg dose group.
3.3.2 Metabolite A
Metabolite A reached Cmax within 17–24 h (median tmax) before plasma concentrations declined in a multiphasic manner, with a parallel terminal phase for the three dose groups (Fig. 3A, B). Plasma concentrations of Metabolite A were higher than those of BI 1358894, with the ratio of metabolite to parent substance almost independent of doses 50 mg, 100 mg, and 200 mg. Exposure to Metabolite A in terms of AUC0-∞, AUC0-tz, and Cmax increased as BI 1358894 dose increased. The interindividual variabilities of exposure were low to moderate, and the gCV ranged from 17.7% to 31.8% (Table 2).

Mean plasma concentration–time profiles of Metabolite A after single oral administration of 50 mg, 100 mg, and 200 mg doses on a linear scale (a) and semi-logarithmic scale (b). Note: Error bars indicate standard deviation
4 Discussion
The Phase I clinical study described here evaluated the safety and pharmacokinetic profile of BI 1358894 in Japanese healthy male volunteers. Single doses of BI 1358894 at ≤ 200 mg were well tolerated and showed no dose dependency in terms of DRAE frequency under fed conditions. These safety findings coincide with those from other Phase I studies of BI 1358894 in Caucasian healthy male volunteers [12, 18, 19, 21] and support the inclusion of the Japanese population in a future global Phase II study of BI 1358894 in patients with MDD.
Exposure to BI 1358894 significantly increased dose dependently and dose proportionally for AUC0-∞ and Cmax. Pharmacokinetics was characterized by moderate absorption and slow elimination. BI 1358894 was administered in the fed state as a positive food effect had been demonstrated in a Phase I study whereby consumption of a high-fat, high-calorie breakfast prior to BI 1358894 administration increased BI 1358894 exposure in a dose proportional manner across all dose groups in Caucasian male volunteers [12, 19]. This was in contrast to the fasted state when BI 1358894 exposure was less than dose proportional over the entire dose range [12] potentially due to low intestinal permeability [20].
In the present study, following oral administration, the plasma concentration of BI 1358894 reached a peak at approximately 4–6 h post dose and then declined in a multiphasic manner with terminal elimination half-life of approximately 70 h in the 50 mg and 100 mg dose groups. In the BI 1358894 200 mg group, another late phase with a slow decline in low plasma concentrations associated with a terminal elimination half-life of approximately 203 h was observed, but this was based on a longer pharmacokinetic sampling timeframe of up to 672 h for the BI 1358894 200 mg dose group, compared with up to 192 h for the BI 1358894 50 mg and 100 mg dose groups. Extended sampling of the 200 mg dose group to 672 h was carried out to allow for comparison with the Caucasian study [12] under identical conditions to assess any potential ethnic differences at the 200 mg dose.
Overall, the pharmacokinetic results from this study indicate an increase in systemic exposure of BI 1358894 in Japanese healthy male volunteers when administered as a single dose under fed conditions. Though Metabolite A showed higher exposures than the parent compound in all dose groups, it should be noted that it is pharmacologically inactive and does not likely contribute to the clinical activity of BI 1358894. Similar to BI 1358894, accumulation was anticipated for Metabolite A. The pharmacokinetics of Metabolite A after multiple-dose administration has been characterized in clinical studies including drug-drug interaction studies (unpublished data) and will also be measured in an ongoing Phase II trial (NCT04521478) [12]. Genetic and environmental factors can cause ethnic differences in pharmacokinetics to a varying extent. These ethnic differences may affect dosage, dosage regimen, safety, and efficacy, and can cause a reluctance to rely on clinical data from other ethnic groups for drug approval [22]. Encouragingly, similar exposures were observed in both the present study (AUCtz range for BI 1358894 50–200 mg: 6320–32,200) and in the Caucasian study (range 2520–32,800) [12]. Dose dependence and proportionality, which was comparable with the Caucasian study, was also observed across the BI 1358894 dose range of 50–200 mg under fed conditions (slopes in current study: AUC0-∞, 1.1351; AUC0-tz, 1.1757; Cmax, 0.9396; slopes in Caucasian study: AUC0-∞, 1.0745; AUC0-tz, 1.1237; Cmax, 0.9396) [12]. Additionally, given that multiple pathways are involved in the metabolism of BI 1358894, and that BI 1358894 is not a substrate of four transporters (P-gp, BCRP, OATP1B1, and OATP1B3) (unpublished data), the likelihood of ethnic differences in pharmacokinetics is considered to be low. There were also no ethnic differences observed with regards to safety in healthy volunteers.
The consistent safety and pharmacokinetic results observed across Japanese and Caucasian ethnicities supports the inclusion of Japanese patients without dose adjustment within larger global clinical studies of BI 1358894.
There is an unmet therapeutic need for patients with MDD. The findings of a previous review of 30 years of research show that despite reliance on antidepressants, only 54% of patients respond. Furthermore, the response rates are only 12% higher than placebo, highlighting the need for a different therapeutic approach [5]. Novel antidepressants with greater effectiveness over symptoms, more rapid therapeutic effects, and fewer adverse effects are needed to allow optimal functional recovery in more patients [5]. It is hoped that BI 1358894 through inhibition of TRPC4/5 ion channels will help to address the unmet clinical need for patients with MDD. In a Phase I study, BI 1358894 reduced cholecystokinin-tetrapeptide-induced panic symptoms, indicating target engagement and functional mechanistic effects [17]. In another Phase I study of patients with MDD, BI 1358894 attenuated activity in the bilateral amygdala region, an area of the brain associated with the processing of emotion and mood implicated in MDD [15,16,17]. These studies indicate that TRPC4/5 ion channel modulation of the amygdala by BI 1358894 may represent a novel mechanism of action for the treatment of patients with MDD and as such should be investigated further.
Several Phase I clinical studies have investigated the safety and efficacy of BI 1358894 in healthy volunteers (NCT03843151, NCT04099732, and NCT03754959). The results from this study add to this knowledge base and support the progression of BI 1358894 into Phase II clinical trials in patients with MDD.
Some limitations of the study should be considered. As this was a Phase I trial in healthy male volunteers, the generalizability of the data to female patients with MDD or patients from different ethnicities is limited. Further, the healthy volunteers received a single treatment for a short trial duration (33 days) and the sample size of each treatment group was small. As such, these safety and pharmacokinetic findings will require confirmation in larger trials in the future. A Phase II trial of BI 1358894 in male and female patients with MDD of various ethnicities is ongoing (NCT04521478) and has a treatment duration of 3 months. This trial will assess the efficacy, long-term safety, and the factors affecting the variability of pharmacokinetics of BI 1358894, as well as patient-reported outcomes. Further, the dose-response relationship for BI 1358894 and its optimal dose will be assessed in the Phase II trial. The present study design did not incorporate fasted dose groups, therefore the pharmacokinetics and safety of BI 1358894 in Japanese male volunteers cannot be compared with Caucasian male volunteers in the fasted state. However, as the previous Phase I study demonstrated a more favorable pharmacokinetic profile in the fed state and not the fasted state, these groups were not deemed necessary for inclusion in the present study. Finally, it should be noted that study assessments in the present study were based only on single doses, which is not reflective of the dosing schedules used in standard clinical care practice.
5 Conclusion
In summary, single doses of BI 1358894 were well tolerated at doses up to 200 mg. The pharmacokinetic data demonstrated dose dependence and proportionality in terms of exposure to BI 1358894 in the fed state. Overall, these findings support the inclusion of the Japanese population in a future global Phase II study of BI 1358894 in patients with MDD.
References
Gautam S, Jain A, Gautam M, Vahia VN, Grover S. Clinical Practice guidelines for the management of depression. Indian J Psychiatry. 2017;59(Suppl 1):S34-s50.
American Psychiatric Association. American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders. Arlington, VA; 2013.
Davies P, Ijaz S, Williams CJ, Kessler D, Lewis G, Wiles N. Pharmacological interventions for treatment-resistant depression in adults. Cochrane Database Syst Rev. 2019 Dec 17;12:Cd010557.
Dupuy JM, Ostacher MJ, Huffman J, Perlis RH, Nierenberg AA. A critical review of pharmacotherapy for major depressive disorder. Int J Neuropsychopharmacol. 2011;14(10):1417–31.
Rakesh G, Pae CU, Masand PS. Beyond serotonin: newer antidepressants in the future. Expert Rev Neurother. 2017;17(8):777–90.
Koenigsberg HW, Denny BT, Fan J, Liu X, Guerreri S, Mayson SJ, et al. The neural correlates of anomalous habituation to negative emotional pictures in borderline and avoidant personality disorder patients. Am J Psychiatry. 2014;171(1):82–90.
Mandell D, Siegle GJ, Shutt L, Feldmiller J, Thase ME. Neural substrates of trait ruminations in depression. J Abnorm Psychol. 2014;123(1):35–48.
Fowler MA, Sidiropoulou K, Ozkan ED, Phillips CW, Cooper DC. Corticolimbic expression of TRPC4 and TRPC5 channels in the rodent brain. PLOS ONE. 2007;2(6):e573.
Riccio A, Li Y, Moon J, Kim KS, Smith KS, Rudolph U, et al. Essential role for TRPC5 in amygdala function and fear-related behavior. Cell. 2009;137(4):761–72.
Riccio A, Li Y, Tsvetkov E, Gapon S, Yao GL, Smith KS, et al. Decreased anxiety-like behavior and Galphaq/11-dependent responses in the amygdala of mice lacking TRPC4 channels. J Neurosci. 2014;34(10):3653–67.
Just S, Chenard BL, Ceci A, Strassmaier T, Chong JA, Blair NT, et al. Treatment with HC-070, a potent inhibitor of TRPC4 and TRPC5, leads to anxiolytic and antidepressant effects in mice. PLOS ONE. 2018;13(1):e0191225.
Fuertig R, Goettel M, Herich L, Hoefler J, Wiebe ST, Sharma V. Effects of single and multiple ascending doses of BI 1358894 in healthy male volunteers on safety, tolerability and pharmacokinetics: two phase I partially randomised studies. CNS Drugs. 2023;37(12):1081–97.
Fuertig R, Goettel M, Van der Plas M, Schlieker L, Sharma V. P.449 Relative bioavailability of a single oral dose BI 1358894 administered alone or in combination with multiple oral doses of itraconazole. Eur Neuropsychopharmacol. 2020;40:S255-S6.
ACNP 59th Annual Meeting: Poster Session II (T94). Neuropsychopharmacol. 2020;45(1):170–277.
Grimm S, Keicher C, Paret C, Niedtfeld I, Beckmann C, Mennes M, et al. The effects of transient receptor potential cation channel inhibition by BI 1358894 on cortico-limbic brain reactivity to negative emotional stimuli in major depressive disorder. Eur Neuropsychopharmacol. 2022;65:44–51.
Goettel M, Mack S, Fuertig R, Sharma V, Just S, Boer JD. P.448 BI 1358894 attenuates cholecystokinin tetrapeptide (CCK-4) induced anxiety and panic symptoms in healthy male volunteers. Eur Neuropsychopharmacol. 2020;40:S255.
Goettel M, Fuertig R, Mack SR, Just S, Sharma V, Wunder A, et al. Effect of BI 1358894 on cholecystokinin-tetrapeptide (CCK-4)-induced anxiety, panic symptoms, and stress biomarkers: a phase I randomized trial in healthy males. CNS Drugs. 2023;37(12):1099–109.
Goettel M, Höfler J, Fuertig R, Sharma V, Göttel M. First-in-human study of oral BI 1358894 in healthy male volunteers: a phase I study to investigate safety and tolerability. Biol Psychiatry. 2020;87(9):S289–90.
Fuertig R, Goettel M, Höfler J, Sharma V. P.450 A phase I single-rising-dose study of oral BI 1358894 in healthy male volunteers: safety, pharmacokinetics and food effect. Eur Neuropsychopharmacol. 2020;40:S256-S7.
US Department of Health and Human Services and Food and Drug Administration Center for Drug Evaluation and Research. Guidance for industry: food-effect bioavailability and fed bioequivalence studies. Office of Training and Communications, Division of Drug Information, document HFD-240. Washington DC: DHHS and FDA, CDER; 2002.
Goettel M, Fuertig R, Wiebe S, Herich L, Sharma V. P.447 Multiple rising doses of oral BI 1358894 in healthy male volunteers: a phase I study investigating safety, tolerability and pharmacokinetics. Eur Neuropsychopharmacol. 2020;40:S254-S5.
Kim K, Johnson JA, Derendorf H. Differences in drug pharmacokinetics between East Asians and Caucasians and the role of genetic polymorphisms. J Clin Pharmacol. 2004;44(10):1083–105.
Acknowledgements
The authors would like to thank Kerstin Bader for leading the bioanalysis within the study. The authors would also like to thank Dr Ryuzo Hanada for his contribution to the manuscript development. Editorial support (in the form of writing assistance, assembling figures, collating author comments, grammatical editing and referencing) was provided by Katie Baker, PhD, and Blessing Anonye, PhD, of Fishawack Communications Ltd, UK, (part of Avalere Health) and was funded by Boehringer Ingelheim International GmbH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Boehringer Ingelheim (BI study number 1402-0008; NCT03875001). The funders of the study had a role in the study design, data analysis, data interpretation, and writing of the report.
Conflict of Interest
AH is an employee of Nippon Boehringer Ingelheim Co., Ltd. JY was an employee of Nippon Boehringer Ingelheim Co., Ltd at the time of the study conduct. VS was an employee of Boehringer Ingelheim International GmbH at the time of the study conduct. Authors did not receive any direct compensation relating to the development of the manuscript.
Data availability
The datasets generated and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
Code availability
Not applicable.
Ethics approval
The study was carried out in accordance with the principles of the Declaration of Helsinki, the International Conference for Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Good Clinical Practice (GCP) guidelines, applicable regulatory requirements, and Boehringer Ingelheim standard operating procedures. The study protocol, the subject information form and the informed consent form were reviewed and approved by the institutional review board of the Souseikai Hakata Clinic, Fukuoka, Japan on 22 March 2019.
Consent to participate
All participants provided informed written consent in accordance with ICH GCP and local procedures.
Consent for publication
The informed consent form included permission to publish.
Author contributions
JY, AH, and VS contributed to the study concept and design, and were responsible for data analyses and interpretation. All authors contributed towards the preparation of the manuscript, approved the final submitted version, and agreed to be listed as authors.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Yoon, J., Sharma, V. & Harada, A. Safety, Tolerability, and Pharmacokinetics of Oral BI 1358894 in Healthy Japanese Male Volunteers. Clin Drug Investig 44, 319–328 (2024). https://doi.org/10.1007/s40261-024-01357-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-024-01357-z