
Abstract
Allogeneic haematopoietic stem cell transplantation (alloHSCT) offers a potentially curative therapy for patients suffering from diseases of the haematopoietic system but requires a high level of expertise and is both resource intensive and expensive. A frequent and life-threatening complication is graft-versus-host disease (GvHD). Acute GvHD (aGvHD) generally causes skin, gastrointestinal and liver symptoms, but chronic GvHD (cGvHD) has a different pathophysiology and may affect nearly every organ or tissue of the body. In Europe, GvHD prophylaxis is generally a calcineurin inhibitor in combination with methotrexate, with high-dose systemic steroids used for advanced GvHD treatment. Between 39% and 59% of alloHSCT patients will develop aGvHD and around 36–37% will develop cGvHD. Steroid response decreases with increasing disease severity, which in turn leads to an increase in non-relapse mortality. GvHD imposes a financial burden on healthcare systems, significantly increasing post-alloHSCT costs. Increased GvHD disease severity magnifies this. Balancing immunosuppression to control the GvHD whilst maintaining a degree of immunocompetence against infection is critical. European GvHD guidelines acknowledge the lack of evidence to support a standard second-line therapy, and improved long-term outcomes and quality-of-life (QoL) remain an unmet need. Evidence generation for potential treatments is challenging. Issues to overcome include choice of comparator (extensive off-label usage); blinding; selection of relevant patient-reported outcome measures (PROMs); and rarity of the condition, which may infeasibly increase timescales to achieve clinical and statistical relevance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Allogeneic haematopoietic stem cell transplantations are complex and expensive but increasingly used as a potentially curative therapy. |
Following alloHSCT, GvHD is the most life-threatening complication, and treating GvHD places a significant burden on patients and healthcare resources, and increases costs. High-dose systemic steroids are used first-line for advanced GvHD but many patients will become refractory to steroid treatment. A wide range of second-line and subsequent lines of therapies are recommended for GvHD treatment, but clinical evidence remains stubbornly disappointing. |
Evidence-based treatment options are needed, particularly for second-line treatment. |
1 Introduction
For certain life-threatening haematological diseases and non-malignant diseases, an allogeneic haematopoietic stem cell transplantation (alloHSCT) offers patients the opportunity of a potentially curative therapy [1, 2]. Several advances in alloHSCT procedures including better tolerated and safer transplantation therapies have allowed more patients to reach complete disease response, with a lower patient burden. Subsequently, more patients have become eligible for alloHSCT, which has expanded access and also led to a reduction in transplant-related morbidity and mortality, as well as improved outcomes [3]. Increasingly, older patients with higher risk factors and those with unrelated and/or mismatched donors may now benefit from alloHSCT [4].
According to the European Society for Blood and Marrow Transplantation (EBMT) activity survey report, there were 19,798 alloHSCTs performed in Europe in 2019, with these figures coming from 700 reporting centres in 51 countries [5].
AlloHSCT procedures are extremely complex and typically can be divided into several consecutive steps:
-
Selection of patients and collection of stem cells: Assessment of the eligibility for transplantation including basic investigations for fitness of both donor and recipient. Tissue typing and collection of stem cells from the donor and processing of the cells in the laboratory [6].
-
Conditioning therapy: Conditioning prepares the recipient’s marrow for transplantation, for example, myeloablation or immunosuppression [7].
-
Transplantation and engraftment: Donor cells are infused and the patients are then kept in hospital until they have recovered sufficient neutrophil numbers to reduce the risk of infection. Engraftment occurs when the donor’s cells are successfully integrated with the host’s [8].
-
Post-graft immunosuppression and post-transplant follow-up: Complications such as infection and potentially life-threatening graft-versus-host disease (GvHD) are monitored and treated [6].
Disease- and patient-related factors such as initial diagnosis, disease status (e.g. remission, refractory or relapsed disease), patient age, functional status, comorbid conditions and general condition are all key factors that influence the choice of alloHSCT approach and timing, as well as the conditioning regimen [9, 10]. Physicians need to find a delicate balance between patient characteristics, the risk of relapse/progression of the underlying malignant disease, and the late effects of the procedure.
Given the complexity of the procedure, alloHSCT requires a high level of expertise before, during and after transplantation administration, and alloHSCTs are therefore generally only administered within specialised centres operating a multidisciplinary team of physicians, laboratory scientists, pharmacists and nurses [11].
AlloHSCT is a resource-intensive and expensive procedure [12], but there are scant data on the cost. A German retrospective claims database study found the mean direct costs of alloHSCT were €246,266 and the indirect costs were €52,939 per adult patient with acute lymphoblastic leukaemia [13]. A similar US study found that for myeloablative alloHSCT the median total healthcare cost at day 100 was $289,283 [12].
For patients, alloHSCT is an intensive potentially curative treatment that is associated with significant treatment failure, psychological morbidity and physical morbidities such as organ complications or death [14]. Failure of the alloHSCT, for whatever reason (e.g. malignant disease relapse or graft failure) leaves the patient with a poor prognosis and few options for potentially successful salvage treatment [15].
2 Graft-versus-Host Disease (GvHD) is a Life-Threatening and Expensive Complication of Allogeneic Haematopoietic Stem Cell Transplantation (alloHSCT)
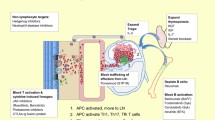
GvHD is a multisystem disorder that is broadly divided into acute GvHD (aGvHD) and chronic GvHD (cGvHD) based on signs and symptoms, rather than the time of onset (i.e. less or more than 100 days after HSCT) [16]. The newer category of chronic overlap cGvHD may also be seen, along with further categories of persistent, recurrent and late-onset aGvHD [17].
Acute GvHD is driven mainly by mature donor T cells [18] and is typified by rapid onset as a result of a cytokine storm that manifests in three organs [19]:
-
Skin with a maculopapular skin rash;
-
Liver with bile duct damage leading to cholestasis and jaundice;
-
Gastrointestinal (GI) tract with nausea, upper GI symptoms such as vomiting and anorexia and lower GI symptoms such as watery or bloody diarrhoea and abdominal pain.
Chronic GvHD is more complex and involves both T and B cells and can affect nearly every organ or tissue in the body because of its different pathophysiology from aGvHD [20].
An International task force has recommended that the MAGIC (Mount Sinai Acute GvHD International Consortium) criteria are utilised for staging aGvHD [21] and the NIH (National Institutes of Health) 2014 [22] criteria for cGvHD [16]. Regardless of the grading system used, there has been a shift from time to onset to the severity for staging [23].
Despite the administration of GvHD prophylaxis, between 39% and 59% of alloHSCT patients will develop aGvHD [24, 25], and around 36% will develop cGvHD [25, 26]. The frequency of GvHD reported in the individual studies depends mainly on the degree of match between donor and recipient, graft type, T-cell depletion/repletion strategies, conditioning regimen, GvHD prophylaxis, and the age of the patient/donor [27].
In some studies, GvHD triples mortality rates [28, 29] and approximately 80% of patients with refractory aGvHD die [1]. Whilst GvHD may not always be fatal, the morbidity it causes places a heavy burden on the patients and the healthcare teams through their survivorship. Post-alloHSCT, GvHD is currently the most challenging issue for physicians to manage [30]. Table 1 illustrates a summary of the percentage of alloHSCT recipients developing GvHD and refractory GvHD and the associated mortality/survival. It is heartening to note that progressively over time, meaningful progress has been made in both reducing the incidence of GvHD and increasing the survival of patients following alloHSCT [31].
In HSCT survivors, GvHD is a major determinant of long-term quality-of-life (QoL) [37], but it remains to be better documented. Lee et al. [37] in 2006 reported that patients who develop aGvHD show a profound and lasting decline in their QoL compared to those who do not develop GvHD. Patients with acute or chronic GvHD may feel uncertain about the nature of GvHD and its progression, as well as struggling to deal with their symptoms and to accept new restrictions to their daily life caused by GvHD and related treatment [38].
GvHD also has a significant impact on healthcare resource utilisation, with patients experiencing more and longer hospitalisations and incurring greater healthcare costs [39,40,41,42]. A study in the USA found significantly greater 1-year post-alloHSCT costs for patients with aGvHD compared to those without aGvHD ($466,720 vs. $263,568; P < 0.001), with inpatient care being the primary cost driver [43]. A Swedish real-world study found that the cumulative direct medical costs over 3 years post-alloHSCT for patients with moderate-to-severe cGvHD were approximately fourfold higher than for patients without cGvHD [41].
As GvHD severity increases, post-alloHSCT costs are significantly increased, for example, grade III versus grade IV aGvHD, P = 0.03 [41, 44]. This may be because increasing severity indicates steroid-resistant GvHD. Such steroid-resistant GvHD had been shown to double the costs of patients compared to those without GvHD [45]. The mean total initial hospitalisation costs of patients with aGvHD who were either steroid-resistant or high risk were $205,880 compared to $97,417 (P < 0.001) for those with no aGvHD as reported in a US retrospective claims database analysis [46]. For patients with steroid-resistant cGvHD, mean total costs were $532,673 versus no cGvHD costs of $252,909 (P < 0.001) in the 2 years following alloHSCT in another US retrospective claims database analysis [45].
3 Current and Potential Treatments
Currently in Europe, the immunosuppressive prophylaxis regimen for GvHD is provided after myeloablative conditioning and relies on a calcineurin inhibitor (CNI) in combination with methotrexate [30]. Typically, patients undergoing reduced-intensity conditioning will be given cyclosporine A alone or in combination with mycophenolate mofetil (MMF) as prophylaxis, although these have not been tested with a large, randomized, prospective clinical trial [30]. In recent times, antithymocyte globulin (ATG) or post-transplant cyclophosphamide is more frequently used to decrease the occurrence of acute and chronic GVHD [30, 47, 48].
For patients who develop Grade I aGvHD (involving skin only), first-line treatment is with topical steroids alone, but for Grades II–IV, first-line treatment is with high-dose systemic steroids (e.g. methyl-prednisolone) [1]. However, the likelihood of steroid response decreases with increasing aGvHD severity [1], with around a third to half of patients becoming steroid-resistant and having a poor prognosis [30]. Lack of response, or steroid resistance, is typically seen within a few weeks of treatment initiation and will increase the patients’ risk of non-relapse mortality compared to those responding to treatment [49].
Following alloHSCT, cGvHD first-line treatment includes steroids either alone or in combination with a CNI [4]. For mild cGvHD (with one to two involved organs), topical steroids, topical CNI or phototherapy may be considered, however, for moderate or severe cGvHD (≥ three involved organs), systemic steroid (prednisone) treatment should be considered [4].
For physicians treating GvHD, the key aim is to balance the need for immunosuppression in order to control the GvHD, whilst maintaining a degree of immunocompetence against infection [1, 4]. This means that viral and fungal infections are frequent complications of prolonged steroid therapy, and anti-infective prophylaxis including revaccinations should be considered for these patients [1].
With second-line and subsequent lines of therapies for GvHD, a wide range of pharmaceutical agents are currently recommended [50]. However, there is no accepted standard-of-care treatment for patients with steroid-refractory aGvHD, with treatment choice generally down to physician experience [51] (Figs. 1 and 2). Currently (in the USA only) ruxolitinib is indicated for steroid-refractory aGvHD and ruxolitinib may therefore be used second line, although efficacy evidence in this population is currently limited to an open-label phase III clinical trial [52]. However, with new and ongoing trials [53, 54], the treatment landscape in Europe is expected to change, with new options becoming available and current second-line therapies becoming third and subsequent lines of therapies.

Source: Modified from Dignan, Clark, et al. [55] and Penack et al. [57]. aRuxolitinib has US FDA approval for patients with aGvHD based on phase II trials only (although a phase III trial (NCT02913261; REACH2) has subsequently been completed) [52, 54]. CSA cyclosporine, ECP extracorporeal photopheresis, FDA Food and Drug Administration, GI gastrointestinal), IL-2 interleukin-2, IV intravenous, mTOR mechanistic target of rapamycin, MMF mycophenolate mofetil, TNF tumour necrosis factor
Overview of treatment options for acute graft-versus-host disease (aGvHD).

Source: Modified from Dignan, Amrolia, et al. [56] and Saidu et al. [58]. aA phase III trial for ruxolitinib in patients with cGvHD (NCT03112603; REACH3) is expected to complete in 2022 [59], although results have been published [60]. bIrbrutinib is US FDA approved for patients with cGVHD after failure of one or more systemic therapies; however, this was based on phase I/II trials. cBelumosudil is US FDA approved for patients with cGVHD after failure of at least two prior systemic therapies [61]. ECP extracorporeal photopheresis, mTOR mechanistic target of rapamycin, MMF mycophenolate mofetil
Overview of treatment options for chronic graft-versus-host disease (cGvHD).
For steroid-refractory patients, physicians may add in another therapy in combination with steroids, but the goal would be to taper the dose of steroids down as soon as possible, to avoid the detrimental side effects on patients associated with prolonged use of corticosteroids. Subsequent lines of therapy might be combined, for example etanercept plus basiliximab [51]. The short-term duration of the effect with these subsequent lines of therapy can be extremely disappointing, with around an 80% mortality rate seen, especially if the lower gastrointestinal tract is involved in patients with aGvHD [1].
Other non-pharmacological therapies such as extracorporeal photopheresis (ECP) are recommended as second-line treatment in some guidelines, for example the British Committee for Standards in Haematology guidelines for both aGvHD [55] and cGvHD [56].
The most recent update of the EBMT recommendations on GvHD prophylaxis and management were published in 2020 [57]. These guidelines acknowledged recent research and recommend initiation of systemic treatment only for grade II or higher aGvHD and lower steroid doses for grade II aGvHD with isolated skin or upper gastrointestinal tract manifestations [57].
However, the long-standing debate over a suitable choice of second-line therapy for GvHD was not resolved in the current guidelines due to a lack of evidence [57]. Therefore, the consensus group were unable to recommend a standard second-line treatment for aGvHD despite the wide range of second-line treatment options including alemtuzumab, α1-antitripsin, basiliximab, cellular therapies such as mesenchymal stromal cells and regulatory T cells, daclizumab, ECP, faecal microbiota transplantation, JAK inhibitors, mycophenolate mofetil, methotrexate, pentostatin, rabbit anti-thymocycte globulin, sirolimus and vedolizumab [57]. The best recommendation the group could offer for second-line treatment of GvHD was that physicians should follow their institutional guidelines and patients should be treated within clinical trials when possible [57].
In this field, a few drugs show promise and have been approved in the USA by the Food and Drug Administration (FDA):
-
In 2017, ibrutinib was approved for steroid-resistant cGvHD based on a phase I/II trial (NCT02195869) [62], and a phase III trial (NCT02959944; iNTEGRATE) was completed in 2020 [63, 64].
-
In 2019, ruxolitinib was approved for steroid-refractory aGvHD based on phase II trials only (although a phase III trial (NCT02913261; REACH2) has subsequently been completed) [52, 54].
-
In 2021, belumosudil, a kinase inhibitor, was approved for the treatment of adult and paediatric patients with cGvHD after failure of at least two prior lines of systemic treatment based on a phase II trial (NCT03640481; ROCKstar) [61].
However, it is important to note that FDA approval, especially when based on phase I or II data, demonstrates the unmet clinical need but not necessarily the actual clinical benefit when utilised outside of a carefully controlled clinical trial. For example, one can consider the recent furore over aducanumab for Alzheimer’s disease, which was granted accelerated FDA approval based on a surrogate endpoint with some disputing if it will actually lead to clinical benefit [65].
Ideally, GvHD should be prevented but there are still no European Medicines Agency (EMA)-approved agents for the prevention or treatment of aGvHD. According to the EBMT Handbook, what may be required is earlier risk-adapted or pre-emptive treatment strategies [1], as treatment of aGvHD may currently be initiated too late, when tissue destruction by the allogenic immune response is already advanced [19]. Therefore, biomarkers might be useful in identifying high-risk patients at an earlier stage [19] so that current treatments can be better utilised [66]. The so-called MAGIC biomarkers (ST2 and REG3α) [67] are the most advanced and best validated to predict steroid-refractory aGvHD long-term outcomes [66].
Researchers are utilizing advances in the understanding of the pathological mechanisms of aGvHD to investigate new treatment strategies for aGvHD. For instance, lymphocyte trafficking to affected organs plays an important role leading to aGvHD, and vedolizumab inhibits the migration of both naïve and activated lymphocytes and is therefore being investigated further [68, 69]. Table 2 provides a summary of the products currently being investigated for both aGvHD and cGvHD.
Whilst many agents have been evaluated either alone or in combination with steroids for first- or second-line use in GvHD, there is a scarcity of randomized, double-blind, controlled trials and for most of the agents, long-term efficacy still evades researchers [1]. This means that numerous products are being used off-label [75] and it is hoped that some of the new treatment strategies currently being evaluated might be able to definitively prove their efficacy and be recommended by the EBMT rather than just offered as options.
4 Conclusion
Despite significant progress in the understanding and management of alloHSCT complications, the high incidence rate of GvHD and its severe consequences remain a major setback in the face of both the patient’s personal and the healthcare system’s investment in the transplant. To ensure optimal clinical benefit from alloHSCT, the long-term goal is to treat according to individual disease biology, and it is imperative to prevent or control GvHD whilst preserving the beneficial effects of the graft (i.e., the graft-versus-cancer effects against residual disease) particularly for high-risk transplant populations [76].
Newer treatment strategies to improve long-term post-transplant outcomes and the QoL of the HSCT recipients remain an unmet need in this therapeutic field [18]. It would be appropriate to develop targeted treatment of the root cause of GvHD, rather than focussing on the consequence, such as systemic inflammation (e.g. cytokine storm) and the resulting immunosuppression. This strategy would allow selective inhibition of alloreactive T cells whilst preserving the anti-tumour adaptive immune responses, that is, maintaining the impact on cancer while limiting the damage to tissues. In the meantime, patients and clinicians would settle for better and more evidence-based therapeutic options that improve clinical outcomes and QoL for patients who develop GvHD.
Guidance on cGVHD trials was issued by the National Institutes of Health (NIH) Consensus Conference that was convened in 2020 [77]. However, generating a suitable evidence base will be challenging, and trade off may be required to achieve the most appropriate trial design. For example, an adaptive trial design may be considered to increase the number of participants allocated to the most effective treatment arm, although such trial designs may be more appropriate for early drug development [77]. Randomised, double-blind, controlled trials are the gold standard, but have proven to be challenging in rare conditions [78]. Choice of comparator is difficult when there is no EMA-approved product for steroid-refractory GvHD and products are used off-label, with different policies in different institutions and European countries. Comparison to the physician’s best available therapy (BAT) may be a solution [77], but this may compromise the feasibility of the blinding or require a burdensome complex trial protocol, which may not be possible. Finally, when non-pharmacological comparators such as ECP are considered, blinding becomes unrealistic.
Patient-relevant endpoints fit for purpose should also be identified to assess the impact of such therapies. Very little is known so far on the adequacy of patient-relevant endpoints for GvHD trials, and many health technology assessment (HTA) agencies do not accept PROMs when trials are not blinded. In addition, sensitivity to disease states may be an issue with many multi-domain instruments [78], which need to be assessed over a sufficient timescale.
We are therefore left mostly with objective clinical endpoints rather than those subject to a clinician’s judgement, which significantly limits the endpoint possibilities. Survival remains an important criterion, but it may require long-term follow-up, which is not easily managed because of the rarity of the condition. Extending the trial duration to achieve a sufficient sample size to generate clinically and statistically relevant data may not be feasible. Composite endpoints of morbidity and mortality such as GvHD-free/relapse-free survival (GRFS) may be used as a surrogate marker but the subjective nature of the diagnosis of GvHD makes these a poor surrogate, especially in open-label studies.
From a HTA standpoint, longer-term overall survival (12 months or 24 months) is seen as the gold standard for demonstrating efficacy, and if it is not possible to use overall survival as a primary endpoint, it should be considered as a secondary endpoint. HTA agencies are also keen to examine cost-effectiveness, often in terms of quality-adjusted life years (QALYs) or life years gained. However without the appropriate studies, such calculations might be meaningless.
Until high-quality evidence for a standard, second-line GvHD treatment is available, the choice of therapeutic options is based on individual physician experience. Improving and expanding the evidence base should be a priority for those investigating new therapeutic agents to challenge the current standards of care.
References
Holler E, Greinix H, Zeiser R. Acute graft-versus-host disease. EBMT Handb Hematop Stem Cell Transplant Cell Ther. 2019;2019:323–30.
Carreras E, Rambaldi A. Evaluation and counseling of candidates. EBMT Handb Hematop Stem Cell Transplant Cell Ther. 2019;2019:77–86.
Styczyński J, Tridello G, Koster L, Iacobelli S, van Biezen A, van der Werf S, et al. Death after hematopoietic stem cell transplantation: changes over calendar year time, infections and associated factors. Bone Marrow Transplant. 2020;55:126–36.
Wolff D, Lawitschka A. Chronic graft-versus-host disease. EBMT Handb Hematop Stem Cell Transplant Cell Ther. 2019;2019:331–45.
Passweg JR, Baldomero H, Chabannon C, Basak GW, de la Cámara R, Corbacioglu S, et al. Hematopoietic cell transplantation and cellular therapy survey of the EBMT: monitoring of activities and trends over 30 years. Bone Marrow Transplant. 2021;56:1651–64.
Rasheed W, Niederwieser DW, Aljurf M. The HSCT unit. EBMT Handb Hematop Stem Cell Transplant Cell Ther. 2019;2019:27–34.
Nagler A, Shimoni A. Conditioning. EBMT Handb Hematop Stem Cell Transplant Cell Ther. 2019;2019:99–107.
McCune JS, Bemer MJ, Long-Boyle J. Pharmacokinetics, Pharmacodynamics, and Pharmacogenomics of Immunosuppressants in Allogeneic Hematopoietic Cell Transplantation: Part II. Clin Pharmacokinet. 2016;55(5):551-93.
Deeg HJ, Sandmaier BM. Who is fit for allogeneic transplantation? Blood. 2010;116:4762–70.
De Witte T, Bowen D, Robin M, Malcovati L, Niederwieser D, Yakoub-Agha I, et al. Allogeneic hematopoietic stem cell transplantation for MDS and CMML: recommendations from an international expert panel. Blood. 2017;129:1753–62.
Langebrake C, Admiraal R, van Maarseveen E, Bonnin A, Bauters T, Bauters T, et al. Consensus recommendations for the role and competencies of the EBMT clinical pharmacist and clinical pharmacologist involved in hematopoietic stem cell transplantation. Bone Marrow Transplant. 2020;55:62–9.
Broder MS, Quock TP, Chang E, Reddy SR, Agarwal-Hashmi R, Arai S, et al. The cost of hematopoietic stem-cell transplantation in the United States. Am Heal Drug Benefits. 2017;10:366–73.
Mayerhoff L, Lehne M, Hickstein L, Salimullah T, Prieur S, Thomas SK, et al. Cost associated with hematopoietic stem cell transplantation: a retrospective claims data analysis in Germany. J Comp Eff Res. 2019;8:121–31.
Pidala J, Anasetti C, Jim H. Health-related quality of life following haematopoietic cell transplantation: patient education, evaluation and intervention. Br J Haematol. 2010;148:373–85
Barrett AJ, Battiwalla M. Relapse after allogeneic stem cell transplantation. Expert Rev Hematol. 2010;2010:429–41.
Schoemans HM, Lee SJ, Ferrara JL, Wolff D, Levine JE, Schultz KR, et al. EBMT−NIH−CIBMTR Task Force position statement on standardized terminology & guidance for graft-versus-host disease assessment. Bone Marrow Transplant. 2018;53:1401–15.
Lee SJ. Classification systems for chronic graft-versus-host disease. Blood. 2017;129:30–7.
Hill L, Alousi A, Kebriaei P, Mehta R, Rezvani K, Shpall E. New and emerging therapies for acute and chronic graft versus host disease. Ther Adv Hematol. 2018;9:21–46.
Ghimire S, Weber D, Mavin E, Wang XN, Dickinson AM, Holler E. Pathophysiology of GvHD and other HSCT-related major complications. Front Immunol. 2017;2017:1.
Cooke KR, Luznik L, Sarantopoulos S, Hakim FT, Jagasia M, Fowler DH, et al. The biology of chronic graft-versus-host disease: a task force report from the national institutes of health consensus development project on criteria for clinical trials in chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2017;2017:211–34.
Harris AC, Young R, Devine S, Hogan WJ, Ayuk F, Bunworasate U, et al. International, multicenter standardization of acute graft-versus-host disease clinical data collection: a report from the mount sinai acute GVHD international consortium. Biol Blood Marrow Transplant. 2016;22:4–10.
Jagasia MH, Greinix HT, Arora M, Williams KM, Wolff D, Cowen EW, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in chronic graft-versus-host disease: I. The 2014 diagnosis and staging working group report. Biol Blood Marrow Transplant. 2015;21:389–401.
Chao N. Clinical manifestations, diagnosis, and grading of acute graft-versus-host disease [Internet]. UpToDate. 2021. https://www.uptodate.com/contents/clinical-manifestations-diagnosis-and-grading-of-acute-graft-versus-host-disease. Accessed 28 Jan 2021.
Jagasia M, Arora M, Flowers MED, Chao NJ, McCarthy PL, Cutler CS, et al. Risk factors for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119:296–307.
Axt L, Naumann A, Toennies J, Haen SP, Vogel W, Schneidawind D, et al. Retrospective single center analysis of outcome, risk factors and therapy in steroid refractory graft-versus-host disease after allogeneic hematopoietic cell transplantation. Bone Marrow Transplant. 2019;54:1805–14.
Arai S, Arora M, Wang T, Spellman SR, He W, Couriel DR, et al. Increasing incidence of chronic graft-versus-host disease inallogeneic transplantation: a report from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant. 2015;21:266–74.
Flowers MED, Inamoto Y, Carpenter PA, Lee SJ, Kiem HP, Petersdorf EW, et al. Comparative analysis of risk factors for acute graft-versus-host disease and for chronic graft-versus-host disease according to National Institutes of Health consensus criteria. Blood. 2011;117:3214–9.
Yu J, Parasuraman S, Shah A, Weisdorf D. Mortality, length of stay and costs associated with acute graft-versus-host disease during hospitalization for allogeneic hematopoietic stem cell transplantation. Curr Med Res Opin. 2019;35:983–8.
Grube M, Holler E, Weber D, Holler B, Herr W, Wolff D. Risk factors and outcome of chronic graft-versus-host disease after allogeneic stem cell transplantation—results from a single-center observational study. Biol Blood Marrow Transplant. 2016;22:1781–91.
Michonneau D, Socié G. GVHD prophylaxis (immunosuppression). EBMT Handb Hematop Stem Cell Transplant Cell Ther. 2019;2019:177–82.
Greinix HT, Eikema D-J, Koster L, Penack O, Yakoub-Agha I, Montoto S, et al. Improved outcome of patients with graft-versus-host disease after allogeneic hematopoietic cell transplantation for hematologic malignancies over time: an EBMT mega-file study. Haematologica. 2021. https://doi.org/10.3324/haematol.2020.265769
Khoury HJ, Wang T, Hemmer MT, Couriel D, Alousi A, Cutler C, et al. Improved survival after acute graft-versus-host disease diagnosis in the modern era. Haematologica. 2017;102:958–66.
Bashey A, Zhang X, Morris LE, Holland HK, Solh M, Solomon SR. Improved survival of patients diagnosed with severe (Grade 3–4) acute GVHD or severe NIH Grade Chronic GVHD in the current era compared to historic controls. Blood. 2019;134:2006–2006.
El-Jawahri A, Li S, Antin JH, Spitzer TR, Armand PA, Koreth J, et al. Improved treatment-related mortality and overall survival of patients with grade IV acute GVHD in the modern years. Biol Blood Marrow Transplant. 2016;22:910–8.
Phelan R, Arora M, Chen M. CIBMTR summary slides—HCT trends and survival data [Internet]. 2020. https://www.cibmtr.org/referencecenter/slidesreports/summaryslides/Pages/index.aspx. Accessed 16 Sep 2021.
Granot N, Storb R. History of hematopoietic cell transplantation: challenges and progress. Haematologica. 2020;2020:2716–29.
Lee SJ, Kim HT, Ho VT, Cutler C, Alyea EP, Soiffer RJ, et al. Quality of life associated with acute and chronic graft-versus-host disease. Bone Marrow Transplant. 2006;38:305–10.
de Vere HI, Kilgour JM, Danby R, Peniket A, Matin RN. “Is this the GVHD?” A qualitative exploration of quality of life issues in individuals with graft-versus-host disease following allogeneic stem cell transplant and their experiences of a specialist multidisciplinary bone marrow transplant service. Health Qual Life Outcomes. 2021;2021:19.
Labopin M, Ruggeri A, Gorin NC, Gluckman E, Blaise D, Mannone L, et al. Cost-effectiveness and clinical outcomes of double versus single cord blood transplantation in adults with acute leukemia in France. Haematologica. 2014;99:535–40.
Ricci A, Jin Z, Broglie L, Bhatia M, George D, Garvin JH, et al. Healthcare utilization and financial impact of acute-graft-versus host disease among children undergoing allogeneic hematopoietic cell transplantation. Bone Marrow Transplant. 2020;55:384–92.
Schain F, Batyrbekova N, Liwing J, Baculea S, Webb T, Remberger M, Mattsson J. Real-world study of direct medical and indirect costs and time spent in healthcare in patients with chronic graft versus host disease. Eur J Health Econ. 2021;22(1):169-180.
Farhadfar N, Leather HL, Wang S, Burton N, Gatell VI, Itzler R, Salloum RG, Wingard JR. Severity of Acute Graft-versus-Host Disease and Associated Healthcare Resource Utilization, Cost, and Outcomes. Transplant Cell Ther. 2021:S2666-6367(21)01226-4. https://doi.org/10.1016/j.jtct.2021.09.004
Yu J, Lal L, Anderson A, DuCharme M, Parasuraman S, Weisdorf D. Healthcare resource utilization and costs associated with acute graft-versus-host disease following allogeneic hematopoietic cell transplantation. Support Care Cancer. 2020;28:5491–9.
Svahn B-M, Ringdén O, Remberger M. Treatment costs and survival in patients with grades III-IV acute graft-versus-host disease after allogenic hematopoietic stem cell transplantation during three decades. Transplantation. 2006;81:1600–3.
Yu J, Lal LS, Anderson A, DuCharme M, Parasuraman S, Weisdorf D. Healthcare resource utilization and costs among patients with steroid-resistant chronic graft-versus-host disease in the United States: a retrospective claims database analysis. Curr Med Res Opin. 2021;37:755–9.
Yu J, Judy JT, Parasuraman S, Sinha M, Weisdorf D. Inpatient healthcare resource utilization, costs, and mortality in adult patients with acute graft-versus-host disease, including steroid-refractory or high-risk disease, following allogeneic hematopoietic cell transplantation: HCRU, costs, and mortality. Biol Blood Marrow Transplant. 2020;26:600–5.
Chao NJ, Zeiser R. Prevention of acute graft-versus-host disease [Internet]. UpToDate. 2021. https://www.uptodate.com/contents/prevention-of-acute-graft-versus-host-disease?search=Prevention. Accessed 16 Sep 2021.
Luznik L, O’Donnell PV, Symons HJ, Chen AR, Leffell MS, Zahurak M, et al. HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. 2008;14:641–50.
Westin JR, Saliba RM, De Lima M, Alousi A, Hosing C, Qazilbash MH, et al. Steroid-refractory acute GVHD: predictors and outcomes. Adv Hematol. 2011;2011:601953. https://doi.org/10.1155/2011/601953
Velickovic VM, McIlwaine E, Zhang R, Spelman T. Adverse events in second- and third-line treatments for acute and chronic graft- versus -host disease: systematic review. Ther Adv Hematol. 2020;11:204062072097703.
Malard F, Huang XJ, Sim JPY. Treatment and unmet needs in steroid-refractory acute graft-versus-host disease. Leukemia. 2020;34:1229–40.
Food and Drug Administration. FDA approves ruxolitinib for acute graft-versus-host disease [Internet]. 2019. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ruxolitinib-acute-graft-versus-host-disease. Accessed 28 Jan 2021.
Jagasia M, Zeiser R, Arbushites M, Delaite P, Gadbaw B, Von BN. Ruxolitinib for the treatment of patients with steroid-refractory GVHD: an introduction to the REACH trials. Immunotherapy. 2018;10:391–402.
Zeiser R, von Bubnoff N, Butler J, Mohty M, Niederwieser D, Or R, et al. Ruxolitinib for glucocorticoid-refractory acute graft-versus-host disease. N Engl J Med. 2020;382:1800–10.
Dignan FL, Clark A, Amrolia P, Cornish J, Jackson G, Mahendra P, et al. Diagnosis and management of acute graft-versus-host disease. Br J Haematol. 2012;158:30–45.
Dignan FL, Amrolia P, Clark A, Cornish J, Jackson G, Mahendra P, et al. Diagnosis and management of chronic graft-versus-host disease. Br J Haematol. 2012;158:46–61 (John Wiley & Sons, Ltd).
Penack O, Marchetti M, Ruutu T, Aljurf M, Bacigalupo A, Bonifazi F, et al. Prophylaxis and management of graft versus host disease after stem-cell transplantation for haematological malignancies: updated consensus recommendations of the European Society for Blood and Marrow Transplantation. Lancet Haematol. 2020;7:e157–67.
Saidu NEB, Bonini C, Dickinson A, Grce M, Inngjerdingen M, Koehl U, et al. New Approaches for the treatment of chronic graft-versus-host disease: current status and future directions. Front Immunol. 2020;11:578314. doi: 10.3389/fimmu.2020.578314.
ClinicalTrials.gov. A Study of Ruxolitinib vs Best Available Therapy (BAT) in Patients With Steroid-refractory Chronic Graft vs. Host Disease (GvHD) After Bone Marrow Transplantation (REACH3). 2021. https://clinicaltrials.gov/ct2/show/NCT03112603. Accessed 7 Sep 2021.
Zeiser R, Polverelli N, Ram R, Hashmi SK, Chakraverty R, Middeke JM, et al. Ruxolitinib for glucocorticoid-refractory chronic graft-versus-host disease. N Engl J Med. 2021;385:228–38.
Food and Drug Administration. FDA approves belumosudil for chronic graft-versus-host disease [Internet]. 2021. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-belumosudil-chronic-graft-versus-host-disease. Accessed 30 Jul 2021.
FDA. FDA expands ibrutinib indications to chronic GVHD [Internet]. 2017. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-expands-ibrutinib-indications-chronic-gvhd. Accessed 28 Jan 2021.
ClinicalTrials.gov. Ibrutinib in combination with corticosteroids vs placebo in combination with corticosteroids in participants with new onset chronic graft versus host disease (cGVHD). 2021. https://clinicaltrials.gov/ct2/show/study/NCT02959944. Accessed 7 Sep 2021.
Miklos D, Zaid MA, Cooney JP, Albring J, Flowers M, Skarbnik AP, et al. Ibrutinib vs placebo in combination with corticosteroids in patients with new-onset chronic graft-versus-host disease (CGVHD): results from the randomized, double-blind phase 3 integrate study [internet]. Eur. Hematol. Assoc. Open Access Libr. 2021. https://library.ehaweb.org/eha/2021/eha2021-virtual-congress/324643/david.miklos.ibrutinib.vs.placebo.in.combination.with.corticosteroids.in.html. Accessed 16 Sep 2021.
Walsh S, Merrick R, Milne R, Brayne C. Aducanumab for Alzheimer's disease? BMJ. 2021 Jul 5;374:n1682. doi: 10.1136/bmj.n1682.
Srinagesh HK, Özbek U, Kapoor U, Ayuk F, Aziz M, Ben-David K, et al. The MAGIC algorithm probability is a validated response biomarker of treatment of acute graft-versus-host disease. Blood Adv. 2019;3:4034–42.
Major-Monfried H, Renteria AS, Pawarode A, Reddy P, Ayuk F, Holler E, et al. MAGIC biomarkers predict long-term outcomes for steroid-resistant acute GVHD. Blood. 2018;131:2846–55.
Gooptu M, Koreth J. Translational and clinical advances in acute graft-versus-host disease. Haematologica. 2020;2020:2550–60.
Fløisand Y, Lazarevic VL, Maertens J, Mattsson J, Shah NN, Zachée P, et al. Safety and effectiveness of vedolizumab in patients with steroid-refractory gastrointestinal acute graft-versus-host disease: a retrospective record review. Biol Blood Marrow Transplant. 2019;25:720–7.
Frisone P. Novel therapies for the treatment of GvHD [Internet]. 2020. https://gvhdhub.com/medical-information/editorial-theme-or-novel-therapies-for-the-treatment-of-gvhd. Accessed 18 Aug 2021.
Watkins B, Qayed M, McCracken C, Bratrude B, Betz K, Suessmuth Y, et al. Phase II trial of costimulation blockade with abatacept for prevention of acute GVHD. J Clin Oncol. 2021;39:1865–77.
Socié G, Vigouroux S, Yakoub-Agha I, Bay JO, Fürst S, Bilger K, et al. A phase 3 randomized trial comparing inolimomab vs usual care in steroid-resistant acute GVHD. Blood. 2017;129:643–9.
ClinicalTrials.gov. GRAVITAS-309: Itacitinib and Corticosteroids as Initial Treatment for Chronic Graft-Versus-Host Disease [Internet]. 2021. https://clinicaltrials.gov/ct2/show/NCT03584516. Accessed 8 Sep 2021.
Zeiser R, Polverelli N, Ram R, Hashmi SK, Chakraverty R, Flowers MED, et al. Ruxolitinib (RUX) vs best available therapy (BAT) in patients with steroid-refractory/steroid-dependent chronic graft-vs-host disease (cGVHD): primary findings from the phase 3, randomized REACH3 study. Blood. 2020;136:22–4.
Kuwatsuka Y, Atsuta Y, Hirakawa A, Uchida N, Inamoto Y, Najima Y, et al. Use of unapproved or off-label drugs in Japan for the treatment of graft-versus-host disease and post-transplant viral infection. Int J Hematol. 2020;112:841–50.
Villa NY, Rahman MM, McFadden G, Cogle CR. Therapeutics for Graft-versus-Host Disease: From Conventional Therapies to Novel Virotherapeutic Strategies. Viruses. 2016 Mar 22;8(3):85.
DeFilipp Z, Couriel DR, Lazaryan A, Bhatt VR, Buxbaum NP, Alousi AM, et al. National institutes of health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: III The 2020 treatment of chronic GVHD Report. Transplant Cell Ther. 2021;27:729–37.
Martin PJ, Lee SJ, Przepiorka D, Horowitz MM, Koreth J, Vogelsang GB, et al. National institutes of health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: VI. The 2014 clinical trial design working group report. Biol Blood Marrow Transplant. 2015;21:1343–59.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this article was supported by Medac GmbH, Germany.
Conflict of interest
Kristin Grabe-Heyne and Christof Henne are both employees of Medac GmbH, a company working in the field of allogeneic HSCT. Deborah Hooker and Jim Furniss are independent market access consultants who were supported by Medac GmbH for the preparation of this article and other work. They provide consulting to many different pharmaceutical companies. Peter Bader was not funded by Medac for this article. He declares grants received from Medac, Novii and Riemser and consulting fees or honorarium from Amgen, Jazz, Medac, Novartis, and Riemser, and payment for lectures including service on speakers bureaus from Novartis. PB is a patent holder of chemical grade mesenchymal stem cells and receives royalties from Medac. Mondher Toumi was not funded by Medac for this article. He has no conflicts of interest to declare.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Authors' contributions
DH performed the literature search, wrote the draft manuscript, edited the draft manuscript and approved the final version. KG-H, CH, PB, MT and JF critically examined the published data, edited the draft manuscript and approved the final version.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analysed.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Hooker, D.S., Grabe-Heyne, K., Henne, C. et al. Improved Therapeutic Approaches are Needed to Manage Graft-versus-Host Disease. Clin Drug Investig 41, 929–939 (2021). https://doi.org/10.1007/s40261-021-01087-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-021-01087-6