
Abstract
Background and Objectives
Viloxazine extended-release (viloxazine ER, SPN-812) is a novel non-stimulant with activity at serotonin receptors and the norepinephrine transporter, which is under investigation as a potential treatment for attention-deficit/hyperactivity disorder. Given the potential for viloxazine ER to be coadministered with other pharmacotherapies, this trial investigated the pharmacokinetics and safety of combination viloxazine ER + methylphenidate versus viloxazine ER or methylphenidate alone.
Methods
In this single-center, crossover, open-label trial, healthy adult participants received oral administration of 700 mg viloxazine ER alone, 36 mg methylphenidate alone, and combination viloxazine ER (700 mg) + methylphenidate (36 mg), with blood samples collected over 4 days post-administration. The active drug in viloxazine ER (viloxazine) and methylphenidate was measured using chromatographic tandem mass spectrometry. Safety assessments included adverse events (AEs), vital signs, echocardiograms, and clinical laboratory evaluations.
Results
Of 36 healthy adults who were enrolled, 34 completed the trial. The geometric least squares mean ratios are reported as [combination/single drug (90% confidence intervals)]. For viloxazine ER, maximum measured plasma concentration (Cmax) = 100.98% (96.21–105.99), area under the concentration–time curve from time zero to the last measurable time (AUCt) = 98.62% (96.21–101.08), and area under the concentration–time curve from time zero to infinity (AUC∞) = 98.96% (96.55–101.44). For methylphenidate, Cmax = 103.55% (97.42–110.07), AUCt = 106.67% (101.01–112.64), and AUC∞ = 106.61% (100.99–112.54). All reported AEs were mild in severity.
Conclusions
Coadministration of viloxazine ER and methylphenidate did not impact the pharmacokinetics of viloxazine or methylphenidate relative to administration of either drug alone. The combination appeared to be safe and well tolerated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Viloxazine extended-release (ER) is under investigation as a non-stimulant treatment for attention-deficit/hyperactivity disorder and could be coadministered with other medications. |
Viloxazine ER and methylphenidate were coadministered and the potential for pharmacokinetic interactions was evaluated. |
Coadministration of viloxazine ER with methylphenidate did not impact the pharmacokinetics of either drug alone, and the combination appeared to be safe and well-tolerated. |
1 Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a neurobehavioral disorder characterized by a developmentally inappropriate and persistent pattern of inattention, hyperactivity, and impulsivity that occurs across multiple settings and leads to various degrees of functional impairment [1,2,3]. An estimated 6.1 million US children and adolescents aged 2–17 years (9.4%) have ever received an ADHD diagnosis [4], and, while the prevalence is lower in adults (estimated to be between 2.5 and 4.4% [5,6,7]), ADHD often persists as a chronic, lifelong disorder that may require flexible treatment approaches across the lifespan [8].
Current US FDA-approved ADHD pharmacotherapies are classified as either stimulants (e.g. methylphenidate, lisdexamfetamine, amphetamine) or non-stimulants (e.g. atomoxetine, guanfacine, clonidine) [9, 10]. Stimulant pharmacotherapy is generally used as first-line therapy, as stimulants are associated with greater efficacy and near-immediate onset of effect relative to non-stimulants in improving ADHD outcomes [11,12,13]. Nevertheless, stimulants are associated with several key limitations that may be addressed by prescribing non-stimulants or a combination of stimulant and non-stimulant therapy to treat ADHD. For instance, stimulant therapy must be carefully monitored in patients with many ADHD comorbidities (estimated to occur in up to 52% of youths and 87% of adults with ADHD [14]), such as substance use disorders [15, 16], bipolar disorder [17], and sleep disorders [18]. In patients for whom stimulants are an option, the benefits of stimulant therapy can be limited by their generally short (e.g. 4–12 h) duration of action [19]; the risk of common adverse effects such as weight loss, decreased appetite, and insomnia [19, 20]; the potential for symptom rebound [21]; and the potential for abuse and diversion [22]. In light of these limitations, non-stimulant therapies or stimulant and non-stimulant concomitant therapy may address ADHD symptoms and provide better symptom control when presented with signs, symptoms, or insufficient efficacy that preclude a stimulant-only treatment approach [11, 12].
Viloxazine has demonstrated activity at serotonin (5-HT) receptors and the norepinephrine transporter (NET) [23]. Via in vivo microdialysis in a rodent model, therapeutic concentrations of viloxazine were shown to potentiate five- to sixfold increases in 5-HT, norepinephrine, and dopamine levels in the prefrontal cortex (PFC) [23], a region strongly implicated in ADHD pathophysiology [24]. In vitro, viloxazine also exhibits antagonistic activity at 5-HT2B receptors and agonistic activity at 5-HT2C receptors [23], although the downstream effect of this activity remains to be fully elucidated. Viloxazine increases norepinephrine and dopamine in the PFC via NET inhibition, with no apparent activity at the dopamine transporter [23]. In the nucleus accumbens, viloxazine only minimally increases extracellular dopamine (approximately 60% above saline controls) [23], suggesting a low potential for abuse liability. An extended-release form of viloxazine, viloxazine extended-release (viloxazine ER, SPN-812), is currently under development as a non-stimulant treatment for ADHD [25,26,27,28,29].
The purpose of the current study was to determine whether coadministration of viloxazine ER and methylphenidate impacts the pharmacokinetics of either drug alone. Using clinically relevant doses, the pharmacokinetics of a single dose of viloxazine ER coadministered with a single dose of methylphenidate were assessed and compared with the pharmacokinetics of a single dose of viloxazine ER or methylphenidate alone in healthy adults. Safety and tolerability were also assessed.
2 Methods
This trial was conducted in accordance with the Helsinki Declaration and the International Council for Harmonization (ICH) Note for Guidance on Good Clinical Practice, and all participants provided written informed consent. The trial conduct was reviewed and approved by IntegReview IRB (Austin, TX, USA), and the trial was conducted by Worldwide Clinical Trials Early Phase Services (Morrisville, NC, USA).
2.1 Participants
Healthy adult male and female participants (n = 36) aged 18–55 years were recruited for the trial. Inclusion criteria required participants to be non-smokers, with a body mass index (BMI) of 18–30 kg/m2 (inclusive), and females of childbearing potential had to be abstinent or using acceptable birth control. Criteria for exclusion included a history or presence of significant diseases, a history of seizures, clinically significant safety laboratory or electrocardiogram (ECG) abnormalities, infection with HIV or hepatitis B or C, alcohol or drug abuse, a need for prescription medication (other than topical or hormonal agents), use of drugs known to notably induce or inhibit hepatic drug metabolism, pregnancy or lactation, allergy to viloxazine ER or methylphenidate-containing products, or any other qualifier that had the potential to interfere with participation in the trial as determined by the investigator.
2.2 Study Design
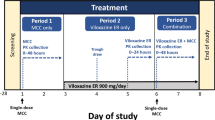
This was a single-center, randomized, crossover, open-label trial in healthy adult participants that evaluated a single dose of viloxazine ER alone, methylphenidate alone, and combination viloxazine ER + methylphenidate. Eligibility was confirmed within 28 days of screening, and patients began one continuous 13-day clinic residency for treatment. Patients received each of the three treatments in a counterbalanced sequence (Fig. 1a), where blood for pharmacokinetic analysis was collected for 4 days following each treatment (Fig. 1b). End-of-study procedures were completed after the last day of period 3 (study day 13) or prior to study discontinuation in the case of early withdrawal and discharge.

Treatment sequences and study design. a Participants received each of the three treatments in one of six possible sequences (counterbalanced). b After screening, participants were randomized and began one continuous 13-day clinic residency for treatment, where blood for pharmacokinetic analysis was collected for 4 days following each treatment. A viloxazine ER (700 mg), B methylphenidate (36 mg), C combination viloxazine ER (700 mg) + methylphenidate (36 mg), ER extended-release, E entry and laboratory tests to confirm eligibility, EOS end-of-study procedures, PK blood draws for pharmacokinetic analyses (0–96 h), SD single dose
2.3 Treatments
Participants received three treatments: single-dose viloxazine ER alone (700 mg; manufactured by Supernus Pharmaceuticals, Inc., Rockville, MD, USA), single-dose methylphenidate alone (36 mg; distributed by Janssen Pharmaceuticals, Inc., Belgium, under the brand name Concerta®), and the combination of single-dose viloxazine ER (700 mg) + single-dose methylphenidate (36 mg). The highest daily doses of viloxazine ER evaluated for the treatment of ADHD in children (6–12 years of age) [25,26,27] and adolescents (12–17 years of age) [28, 29] are 400 mg and 600 mg, respectively, corresponding to approximately 700 mg following multiple-dose administration in healthy adults based on overall exposure extrapolation. Treatment was administered orally following an overnight fast of a minimum of 10 h, and fasting continued for at least 4 h after dosing. With the exception of 240 mL of water at the time of dosing, fluid was restricted from 1 h before to 1 h after dosing. Participants received all three treatments in one of six possible sequences (Fig. 1a). Participants received a single dose of the assigned treatment on the morning of day 1 of each period, followed by a washout period of 4 days between doses (Fig. 1b).
2.4 Sample Collection
A total of 63 blood draws (approximately 4 mL blood per timepoint) were collected over each of three dosing periods, starting on day 1 at time 0 (pre-dose), and 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 16, 20, 24, 30, 36, 48, 60, 72, 84, and 96 h post-dose. Each blood sample for pharmacokinetic analysis was collected in a K2-EDTA tube and gently inverted. Blood samples were then centrifuged at approximately 3000 rpm for 10 min at approximately 4 °C and the resulting plasma was aliquoted into two polypropylene screw-cap tubes. Within 60 min of collection, samples were frozen at − 20 °C or lower pending shipment for analysis.
2.5 Bioanalytical Assay
Viloxazine was extracted from 50 µL of human plasma by supported liquid extraction (SLE) in a 96-well plate format, using viloxazine-d5 as an internal standard. Extracts were analyzed by ultra-performance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS) using positive electrospray ionization and multiple reaction monitoring (precursor/product ion, m/z: 238/100 for viloxazine; 243/56 for viloxazine-d5). The main chromatographic parameters were Waters Acquity BEH C8 UPLC 1.7 μm, 2.1 × 50 mm column, 10 mM ammonium bicarbonate (pH 9.0) as the aqueous mobile phase, acetonitrile as the high organic mobile phase, and gradient elution. Waters Quattro Premier XE mass spectrometer (Waters Corporation, Milford, MA, USA) was used for the detection. The validated range was 0.0100–10.0 µg/mL, with a lower limit of quantitation (LLOQ) of 0.0100 µg/mL. The inter- and intra-run precision (relative standard deviation [SD]) were ≤ 9% and ≤ 10%, respectively, while the intra-run accuracy was 97–107%.
5-hydroxyviloxazine glucuronide (5-HVLX-gluc, the primary metabolite of viloxazine) was extracted from 100 µL of human plasma by mixed-mode cation-exchange solid-phase extraction (SPE) in a 96-well plate format, using 5-HVLX-G-d5 as an internal standard. Extracts were analyzed by UPLC-MS/MS using positive electrospray ionization and multiple reaction monitoring (precursor/product ion, m/z: 430/254 for 5-HVLX-G, 435/259 for 5-HVLX-G-d5). The main chromatographic parameters were Restek Pinnacle DB Biphenyl 1.9 μm, 2.1 × 50 mm column, 0.1% formic acid in water as the aqueous mobile phase, methanol as the high organic mobile phase, and isocratic elution. Waters Quattro Premier XE or Waters Xevo TQ-S Micro mass spectrometer was used for the detection. The validated range was 0.00500–10.0 µg/mL, with a LLOQ of 0.00500 µg/mL. The inter- and intra-run precision were both ≤ 14%, while the intra-run accuracy was 94–115%.
Methylphenidate was extracted from 150 µL of human plasma by SLE in a 96-well plate format, using methylphenidate-d9 as an internal standard. Extracts were analyzed by UPLC-MS/MS using positive electrospray ionization and multiple reaction monitoring (precursor/product ion, m/z: 234/84 for methylphenidate; 243/93 for methylphenidate-d9). The main chromatographic parameters were Waters Acquity UPLC BEH C8 1.7 μm, 2.1 × 50 mm column, 2 mM ammonium formate (pH 4.0) in water as the aqueous mobile phase, 2 mM ammonium formate (pH 4.0) in 5:95 water:acetonitrile as the organic mobile phase, and gradient elution. Waters Quattro Premier XE mass spectrometer (Waters Corporation, Milford, MA, USA) was used for the detection. The validated range was 0.0500–5.00 ng/mL, with an LLOQ of 0.0500 ng/mL. The inter- and intra-run precision (relative SD) were both ≤ 3%, while the intra-run accuracy was 89–98%.
2.6 Safety Monitoring and Assessments
Baseline (pre-dose) measurements included medical history, physical examination, ECG recording, clinical laboratory tests (i.e. serum chemistry, complete blood count, urinalysis), and the Columbia-Suicide Severity Rating Scale (C-SSRS) suicidality assessment. ECGs were performed 7 h after each dose on day 1 of each treatment cycle (the expected time of the maximum measured plasma concentration [Tmax] of each treatment), and vital signs were assessed 6 h post-dose. Adverse events (AEs) were classified into standardized terminology from the verbatim description (investigator term) according to the Medical Dictionary for Regulatory Activities (MedDRA) version 21.0, and summarized by System Organ Class and MedDRA preferred term. An AE was defined as “any untoward medical occurrence associated with the use of a drug in humans, whether or not considered drug related,” consistent with United States Code of Federal Regulations Title 21, Part 312 (21CFR312). AEs were monitored over the course of treatment. Prior to discharge on day 13 or upon study withdrawal, participants underwent a final ECG, physical examination, recording of vital signs, C-SSRS suicidality assessment, a blood draw and urine sample collection, and a final assessment of AEs.
2.7 Pharmacokinetic and Statistical Analyses
The pharmacokinetic analyses compared viloxazine ER alone and methylphenidate alone with the combination of viloxazine ER + methylphenidate. The primary outcome was the relative bioavailability of viloxazine and methylphenidate, i.e. comparing drug plasma exposure when administered alone relative to that of the combination. The pharmacokinetic parameters were derived using non-compartmental analyses: area under the concentration–time curve from time zero to the last measurable time (AUCt), area under the concentration–time curve from time zero to infinity (AUC∞), and maximum measured plasma concentration (Cmax). Secondary outcomes were the pharmacokinetics of viloxazine, methylphenidate, and 5-HVLX-gluc, as measured by the following parameters: AUCt, AUC∞, Cmax, Tmax, apparent volume of distribution (Vd/F), apparent total clearance of drug (CL/F), terminal elimination rate constant (λz), and terminal phase elimination half-life (t½). Methylphenidate and viloxazine ER are each formulated to deliver methylphenidate and viloxazine, respectively, in the freebase quantities specified by their dosages, i.e. a 36 mg methylphenidate tablet delivers 36 mg freebase, and 700 mg viloxazine ER (administered as 1 × 100 mg and 3 × 200 mg viloxazine ER capsules) delivers 700 mg freebase. Therefore, 36 mg methylphenidate and 700 mg viloxazine were used to calculate their CL/F and Vd/F. Secondary outcomes also included safety assessments as measured by reported AEs, ECGs, clinical laboratory tests, vital signs, physical examinations, and C-SSRS suicidality assessment.
The safety population (n = 36) included all participants who received at least one dose of study medication; the pharmacokinetic population (n = 35) included all participants who had an adequate pharmacokinetic profile in any treatment period with no major protocol deviations that could impact pharmacokinetic data (e.g. emesis); and the bioanalysis population (n = 34) included all participants who had completed all three treatments with adequate pharmacokinetic profiles, with no evidence of a major protocol deviation that may have impacted pharmacokinetic data.
The pharmacokinetic parameters and AEs are presented using descriptive statistics. Concentration–time data for plasma concentrations of viloxazine, 5-HVLX-gluc, and methylphenidate that were below the limit of quantification were entered as zero in the tabulation of these data; there were no values above the limits of quantification. Concentration–time data were summarized by subject, treatment, and analyte using descriptive statistics (n, arithmetic mean, SD, median, minimum, maximum, and coefficient of variation) at each scheduled collection time with actual sampling time. The relative bioavailability of viloxazine and methylphenidate following treatment with viloxazine ER or methylphenidate alone versus the combination treatment was assessed by analysis of variance (ANOVA) on the log-transformed pharmacokinetic parameters Cmax, AUCt, and AUC∞. The model included treatment period and sequence as fixed effects, with subjects nested within sequence as a random effect. The least squares mean (LSM) for each treatment group and the difference in the LSMs were calculated, and then back-exponentiated to the original scale to obtain the 90% confidence intervals (CIs) for the geometric LSM ratios. The absence of an impact of combination treatment on the bioavailability of viloxazine or methylphenidate was declared if 90% CIs were contained within predefined no-difference limits of 80–125% for Cmax, AUCt, and AUC∞ ratios. All pharmacokinetic analyses were conducted using Phoenix™ WinNonlin® version 6.3 or higher (Certara, L.P., Princeton, NJ, USA), and all statistical analyses and clinical data presentations were performed using SAS® 9.3 or higher (SAS Institute, Inc., Cary, NC, USA).
3 Results
3.1 Patient Demographics
Healthy adult participants (n = 36; 20 males and 16 females) were enrolled in the study and were included in the safety population. Of these, 34 participants met the pharmacokinetic criteria for evaluation and were included in the bioanalysis population. Of the participants in the safety population, 50.0% were White, 47.2% were Black, and 2.8% were Asian. Participants averaged 35.2 ± 10.12 years of age (mean ± SD; range 19–55), with an average BMI of 24.23 ± 2.70 kg/m2 (mean ± SD).
3.2 Descriptive Pharmacokinetics of Viloxazine, 5-Hydroxyviloxazine Glucuronide (5-HVLX-Gluc), and Methylphenidate
After treatment with viloxazine ER alone, the Cmax of viloxazine was 4.73 ± 0.86 µg/mL (mean ± SD) (Table 1) and occurred 5 h after dosing (Tmax median; range 3–10 h), while after treatment with the combination, viloxazine Cmax was 4.84 ± 0.89 µg/mL (mean ± SD) and occurred 5 h after dosing (Tmax median; range 4–10 h). After treatment with viloxazine ER alone, 5-HVLX-gluc Cmax was 3.43 ± 0.92 µg/mL (mean ± SD) [Table 2] and occurred 6 h after treatment (Tmax median; range 4–12 h), while 5-HVLX-gluc Cmax after combination treatment was 3.48 ± 0.99 µg/mL (mean ± SD), which occurred 5 h after treatment (Tmax median; range 4–10 h). Plasma concentration–time curves of viloxazine (Fig. 2) and 5-HVLX-gluc (Fig. 3) after a single dose of 700 mg viloxazine ER alone were similar to those after combination viloxazine ER + methylphenidate (36 mg), suggesting no drug interaction. This observation was confirmed by statistical analysis (Table 3).

Viloxazine plasma concentration–time profiles after viloxazine ER alone (blue open circles) and combination viloxazine ER + methylphenidate (black diamonds) on a linear and b semi-logarithmic scales. For clarity, symbols and lines for the combination (black diamonds) have been nudged to the right 0.2 points without changing the overall shape of the line (mean ± standard deviation). ER extended-release

5-HVLX-gluc plasma concentration–time profiles after viloxazine ER alone (blue open circles) and combination viloxazine ER + methylphenidate (black diamonds) on a linear and b semi-logarithmic scales. For clarity, symbols and lines for the combination (black diamonds) have been nudged to the right 0.2 points without changing the overall shape of the line (mean ± standard deviation). 5-HVLX-gluc 5-hydroxyviloxazine glucuronide, ER extended-release
After treatment with methylphenidate alone, methylphenidate Cmax was 10.8 ± 5.00 ng/mL (mean ± SD) [Table 1], which occurred 6 h after treatment (Tmax median; range 1–10 h), while after combination treatment, methylphenidate Cmax was 11.1 ± 4.44 ng/mL (Cmax mean ± SD) and occurred 6 h after treatment (Tmax median; range 1–12 h). Plasma concentration–time curves of methylphenidate (Fig. 4) after a single dose of 36 mg methylphenidate alone were similar to those after combination viloxazine ER (700 mg) + methylphenidate, suggesting no drug interaction, which was confirmed by statistical analysis (Table 3).

Methylphenidate plasma concentration–time profiles after methylphenidate alone (red open circles) and combination viloxazine ER + methylphenidate (black diamonds) on a linear and b semi-logarithmic scales. For clarity, symbols and lines for the combination (black diamonds) have been nudged to the right 0.2 points without changing the overall shape of the line (mean ± standard deviation). ER extended-release
3.3 Relative Bioavailability of Viloxazine and Methylphenidate
All 90% CIs were within the predetermined no-difference criteria of 80–125% (Table 3). For viloxazine, the geometric LSM ratios (90% CI) were Cmax 100.98% (96.21–105.99), AUCt 98.62% (96.21–101.08), and AUC∞ 98.96% (96.55–101.44). For methylphenidate, the geometric LSM ratios (90% CI) were Cmax 103.55% (97.42–110.07), AUCt 106.67% (101.01–112.64), and AUC∞ 106.61% (100.99–112.54). Therefore, combination viloxazine ER + methylphenidate did not significantly alter the bioavailability of viloxazine or methylphenidate relative to a single dose of viloxazine ER or methylphenidate alone, respectively.
3.4 Safety
AEs considered related to study treatment were reported by 58.3% (n = 21) of participants: 28.6% (n = 10) of participants following viloxazine ER alone, 8.8% (n = 3) of participants following methylphenidate alone, and 44.4% (n = 16) of participants receiving the combination. All reported AEs were mild in severity, and no participant reported any moderate or serious AEs, nor did any participant discontinue the study due to AEs or death. The most commonly reported AEs (≥ 5%) related to study treatment are listed in Table 4.
There were no clinically significant abnormal results for clinical laboratory test results, vital signs, or physical examinations, and no increase in suicidality as measured by the C-SSRS. Three participants (8.3%) had a normal ECG at baseline but abnormal ECG at the end of the study; all abnormal ECG results were determined by the investigator to be not clinically significant. There were no differences in ECG results for participants treated with combination viloxazine ER + methylphenidate versus viloxazine ER or methylphenidate alone.
4 Discussion
We report no impact of the viloxazine ER and methylphenidate combination treatment on the pharmacokinetics of viloxazine, 5-HVLX-gluc, or methylphenidate, relative to each drug’s administration alone, as reflected in our primary endpoints Cmax, AUCt, and AUC∞. All three treatments appeared to be safe and well tolerated, with a low incidence of AEs and the absence of any reported serious AEs. There were no substantial differences between numbers of participants reporting AEs after treatment with viloxazine ER versus methylphenidate alone, nor when comparing viloxazine ER versus the combination. Although methylphenidate carries warnings of serious cardiovascular events [30], multiple clinical trials [25,26,27,28,29] and a recent study on cardiac repolarization [31] suggest viloxazine ER is not associated with increased cardiovascular risk, even at supratherapeutic doses. In the present study, the combination did not appear to increase these risks, as measured by blood pressure, pulse rate, or ECG parameters. Overall, the combination did not increase the severity of any AE, as all reported AEs were mild, and no patient discontinued the study due to an AE.
Pharmacokinetic drug–drug interactions (DDIs) may cause significant changes in safety and/or efficacy; therefore, they are strongly considered in the decision to co-prescribe ADHD medications. Viloxazine ER is primarily metabolized to 5-hydroxyviloxazine by the cytochrome P450 (CYP) system isoenzyme CYP2D6 as a first step before its glucuronidation, with minor involvement of several other CYP isoenzymes. Subsequent glucuronidation to 5-HVLX-gluc is mediated by uridine 5′-diphospho-glucuronosyltransferase (UGT) 1A9 and UGT2B15 [32]. Viloxazine ER also functions as a reversible inhibitor of CYP1A2 (half maximal inhibitory concentration [IC50] 0.269 µM) [32]. Conversely, methylphenidate is metabolized primarily by carboxylesterase 1 via de-esterification into ritalinic acid, which has little, if any, pharmacologic activity [30, 33]. While clinical data suggest that methylphenidate does not recruit the CYP system for its metabolism [34], non-clinical studies have shown that methylphenidate impacts several CYP isoforms [35, 36]. Methylphenidate has been shown to inhibit CYP2D1 in canine striatal cells and human liver microsomes in vitro (Ki value approximately 15 µM), as well as HepG2 cells transfected with human CYP2D6 (IC50 value approximately 25 µM) [36]. In a rodent model, methylphenidate administered as a single high dose (modeling illicit methylphenidate abuse in humans) decreased total hepatic CYP, as well as the catalytic activity and/or polypeptide levels of the three isoenzymes measured (CYP1A, 2E1, and 3A) [35]. When methylphenidate was administered at lower doses over 2 weeks (modeling therapeutic dosing) only total hepatic CYP, CYP1A catalytic activity, and CYP1A polypeptide levels were reduced, with CYP3A and 2E1 remaining relatively unaffected.
These data notwithstanding, in clinical use methylphenidate is not known to have any appreciable ability to induce, inhibit, or be a substrate for the CYP system [30]. As such, methylphenidate is unlikely to result in any clinically meaningful interactions with other drugs based on interactions within this metabolic pathway. Consistent with this expectation, the current study did not detect any significant effect of concurrent administration on the bioavailability of viloxazine or methylphenidate. Although more subjects (16 vs. 3) reported mild treatment-related AEs during combination treatment relative to treatment with methylphenidate alone, respectively, this effect is unlikely to be due to a pharmacokinetic DDI, as viloxazine and methylphenidate plasma exposures did not increase with a single-dose concurrent administration.
While stimulants are generally considered first-line therapy for ADHD, clinicians often transition from mono- to combination pharmacotherapy for ADHD with certain clinical cases. Most commonly, this occurs in the approximately 30% of patients who are partial or non-responders [37], or who experience dose-limiting adverse effects or comorbid diagnoses, especially in patients with hyperactivity [13]. Polypharmacy in this setting frequently combines different drug types or classes, such as medium- or long-acting stimulants coadministered with short-acting stimulants or non-stimulants, in order to most effectively address symptoms, adverse effects, or comorbidities [11,12,13].
The non-stimulant viloxazine ER is a novel compound with activity at 5-HT receptors and the NET [23], which appeared to be well tolerated in several recent randomized, double-blind, placebo-controlled phase II [25] and III [26,27,28,29] clinical trials. Preliminary results from the phase II and III trials reported significant, clinically meaningful, and rapid reductions in ADHD symptoms in children aged 6–11 years (NCT02633527 [25], NCT03247530 [26], NCT03247543 [27]) and adolescents aged 12–17 years (NCT03247517 [28]), relative to placebo, as assessed by the ADHD Rating Scale-IV [25] and -5 [26,27,28]. Across all five trials, viloxazine ER was well tolerated, with the most frequently reported AEs being somnolence, decreased appetite, and headache. Discontinuation rates due to treatment-emergent AEs (TEAEs) were low (< 7% across all trials), with fewer than 2% of subjects reporting a serious TEAE, and no reported deaths.
The present analysis is subject to limitations. For instance, the environment in which this study was conducted may not fully capture how these drugs are administered in practice; in the real world, ADHD pharmacotherapies are typically titrated slowly and administered chronically over weeks, months, or years of treatment. This long-term treatment may produce pharmacokinetic outcomes not captured in this study with large, acute, single administration of one drug or administration of two drugs in combination. Additionally, we report only pharmacokinetic results from healthy adults, as no children were included in this study, nor did any participants have a known ADHD diagnosis. Finally, diet and activity were tightly controlled in this inpatient study, while such factors may vary considerably across patient populations.
5 Conclusions
We evaluated the pharmacokinetics and safety of the non-stimulant viloxazine ER (700 mg) and the stimulant methylphenidate (36 mg), each administered alone and in combination. We report no DDIs as measured by the relative bioavailability of viloxazine or methylphenidate, pharmacokinetic profiles, or safety assessments when administering the combination, compared with treatment with either drug alone. Although the clinical benefit of viloxazine ER combined with stimulant therapy remains to be investigated, our data suggest the combination is unlikely to result in any clinically significant DDIs.
References
Singh A, Yeh CJ, Verma N, Das AK. Overview of attention deficit hyperactivity disorder in young children. Health Psychol Res. 2015;3(2):23–35. https://doi.org/10.4081/hpr.2015.2115.
Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
Adesman AR. The diagnosis and management of attention-deficit/hyperactivity disorder in pediatric patients. Prim Care Companion J Clin Psychiatry. 2001;3(2):66–77.
Danielson ML, Bitsko RH, Ghandour RM, Holbrook JR, Kogan MD, Blumberg SJ. Prevalence of parent-reported ADHD Diagnosis and Associated Treatment Among U.S. Children and Adolescents, 2016. J Clin Child Adolesc Psychol. 2018;47(2):199–212. https://doi.org/10.1080/15374416.2017.1417860.
Simon V, Czobor P, Balint S, Meszaros A, Bitter I. Prevalence and correlates of adult attention-deficit hyperactivity disorder: meta-analysis. Br J Psychiatry. 2009;194(3):204–11. https://doi.org/10.1192/bjp.bp.107.048827.
Kessler RC, Adler LA, Barkley R, Biederman J, Conners CK, Demler O, et al. The prevalence and correlates of adult ADHD in the United States: results from the national comorbidity survey replication. Am J Psychiatry. 2006;163:716–23.
Polanczyk GV, de Lima MS, Horta BL, Biederman J, Rohde LA. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry. 2007;164:942–8.
Turgay A, Goodman DW, Asherson P, Lasser RA, Babcock TF, Pucci ML, et al. Lifespan persistence of ADHD: the life transition model and its application. J Clin Psychiatry. 2012;73(2):192–201. https://doi.org/10.4088/JCP.10m06628.
Briars L, Todd T. A review of pharmacological management of attention-deficit-hyperactivity disorder. J Pediatr Pharmacol Ther. 2016;21(3):192–206.
Catala-Lopez F, Hutton B, Nunez-Beltran A, Page MJ, Ridao M, Macias Saint-Gerons D, et al. The pharmacological and non-pharmacological treatment of attention deficit hyperactivity disorder in children and adolescents: a systematic review with network meta-analyses of randomised trials. PLoS One. 2017;12(7):e0180355. https://doi.org/10.1371/journal.pone.0180355.
Adler LA, Reingold LS, Morrill MS, Wilens TE. Combination pharmacotherapy for adult ADHD. Curr Psychiatry Rep. 2006;8:409–15.
Wilens T, Spencer T, Biederman J, Wozniak J, Connor D. Combined pharmacotherapy: an emerging trend in pediatric psychopharmacology. J Am Acad Child Adolesc Psychiatry. 1995;34(1):110–2.
Molife C, Bernauer MJ, Farr AM, Haynes VS, Kelsey D. Combination therapy patterns and predictors of ADHD in commercially insured and Medicaid populations. Postgrad Med. 2012;124(5):7–22. https://doi.org/10.3810/pgm.2012.09.2586.
Clemow DB, Bushe C, Mancini M, Ossipov MH, Upadhyaya H. A review of the efficacy of atomoxetine in the treatment of attention-deficit hyperactivity disorder in children and adult patients with common comorbidities. Neuropsychiatr Dis Treat. 2017;13:357–71. https://doi.org/10.2147/NDT.S115707.
Biederman J, Wilens T, Mick E, Milberger S, Spencer TJ, Faraone SV. Psychoactive substance use disorders in adults with attention deficit hyperactivity disorder (ADHD): effects of ADHD and psychiatric comorbidity. Am J Psychiatry. 1995;152:1652–8.
Wilens T. Impact of ADHD and its treatment on substance abuse in adults. J Clin Psychiatry. 2004;65:38–45.
Soutullo C, DelBello MP, Ochsner JE, McElroy SL, Taylor SA, Strakowski SM, et al. Severity of bipolarity in hospitalized manic adolescents with history of stimulant or antidepressant treatment. J Affect Disord. 2002;70:323–7.
Corkum P, Moldofsky H, Hogg-Johnson S, Humphries T, Tannock R. Sleep problems in children with attention-deficit/hyperactivity disorder: impact of subtype, comorbidity, and stimulant medication. J Am Acad Child Adolesc Psychiatry. 1999;38(10):1285–93.
Nafees B, Setyawan J, Lloyd A, Ali S, Hearn S, Sasane R, et al. Parent preferences regarding stimulant therapies for ADHD: a comparison across six European countries. Eur Child Adolesc Psychiatry. 2014;23(12):1189–200. https://doi.org/10.1007/s00787-013-0515-6.
Efron D, Jarman F, Barker M. Side effects of methylphenidate and dexamfetamine in children with attention defict hyperactivity disorder: a double-blind, crossover trial. Pediatrics. 1997;100(4):662–6.
Carlson GA, Kelly KL. Stimulant rebound: how common is it and what does it mean? J Child Adolesc Psychopharmacol. 2003;13(2):137–42.
Wilens TE, Adler LA, Adams J, Sgambati S, Rotrosen J, Sawtelle R, et al. Misuse and diversion of stimulants prescribed for ADHD: a systematic review of the literature. J Am Acad Child Adolesc Psychiatry. 2008;47(1):21–31. https://doi.org/10.1097/chi.0b013e31815a56f1.
Yu C, Garcia-Olivares J, Candler S, Schwabe S, Maletic V. New insights into the mechanism of action of viloxazine: serotonin and norepinephrine modulating properties. J Exp Pharmacol. 2020;12:285–300.
Levy F. Pharmacological and therapeutic directions in ADHD: specificity in the PFC. Behav Brain Funct. 2008;4:12. https://doi.org/10.1186/1744-9081-4-12.
Johnson JK, Liranso T, Saylor K, Tulloch G, Adewole T, Schwabe S, et al. A phase II double-blind, placebo-controlled, efficacy and safety study of SPN-812 (extended-release viloxazine) in children With ADHD. J Atten Disord. 2020;24(2):348–58.
Nasser A, Liranso T, Adewole T, Fry N, Hull JT, Chowdhry F, et al. A phase 3, randomized, placebo-controlled trial to assess the efficacy and safety of once-daily SPN-812 (viloxazine extended release) in the treatment of ADHD in school-age children. Clin Ther. 2020;42(8):1452–66.
Nasser A, Hull JT, Chowdhry F, Adewole T, Liranso T, Marcus R, et al. Extended-release viloxazine (SPN-812) 200 mg or 400 mg for the treatment of ADHD in children: Topline results of a phase 3, randomized, double-blind, placebo-controlled study (P303). 32nd Annual Psych Congress: 3–6 October 2019; San Diego, CA.
Nasser A, Hull JT, Chowdhry F, Adewole T, Liranso T, Marcus R, et al. Extended-release viloxazine (SPN-812) 200 mg or 400 mg for the treatment of ADHD in adolescents: topline results of a phase 3, randomized, double-blind, placebo-controlled study (P302). 32nd Annual Psych Congress: 3–6 October 2019; San Diego, CA.
Nasser A, Hull JT, Chowdhry F, Adewole T, Liranso T, Marcus R, et al. Phase 3, randomized, double-blind, placebo-controlled study evaluating efficacy and safety of extended-release viloxazine (SPN-812) for pediatric ADHD: Update on the second adolescent study (P304). 21st ASENT Annual Meeting: 25–28 March 2019; Rockville (MD).
Concerta (Methylphenidate HCl) extended release [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2017.
Nasser A, Faison SL, Liranso T, Adewole T, Busse GD, Fava M, et al. Evaluation of the effect of SPN-812 (viloxazine extended release) on QTc interval in healthy adults. J Clin Psychiatry. 2020;81(6):e1–6.
Yu C. Metabolism and in vitro drug-drug interaction assessment of viloxazine. Xenobiotica. 2020;50(11):1285–300.
Sun Z, Murry DJ, Sanghani SP, Davis WI, Kedishvili NY, Zou Q, et al. Methylphenidate is stereoselectively hydrolyzed by human carboxylesterase CES1A1. J Pharmacol Exp Ther. 2004;310(2):469–76. https://doi.org/10.1124/jpet.104.067116.
DeVane CL, Markowitz JS, Carson SW, Boulton DW, Gill HS, Nahas Z, et al. Single-dose pharmacokinetics of methylphenidate in CYP2D6 extensive and poor metabolizers. J Clin Psychopharmacol. 2000;20(3):347–9. https://doi.org/10.1097/00004714-200006000-00009.
Le Nedelec MJ, Rosengren RJ. Methylphenidate inhibits cytochrome P450 in the Swiss Webster mouse. Hum Exp Toxicol. 2002;21:273–80.
Tyndale RF, Sunahara R, Inaba T, Kalow W, Gonzalez FJ, Niznik HB. Neuronal cytochrome P450IID1 (debrisoquine/sparteine-type): potent inhibition of activity by (-)-cocaine and nucleotide sequence identity to human hepatic P450 gene CYP2D6. Mol Pharmacol. 1991;40(1):63–8.
Spencer T, Biederman J, Wilens T, Harding M, O’Donnell D, Griffin S. Pharmacotherapy of attention-deficit hyperactivity disorder across the life cycle. J Am Acad Child Adolesc Psychiatry. 1996;35(4):409–32. https://doi.org/10.1097/00004583-199604000-00008.
Acknowledgements
Editorial support was provided by IMPRINT Science, New York, NY, USA, and funded by Supernus Pharmaceuticals, Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was fully sponsored by Supernus Pharmaceuticals, Inc.
Conflicts of interest
Nicholas Fry, Toyin Adewole, Oyinkansola Odebo, Stefan Schwabe, Zhao Wang, Azmi Nasser are employees of Supernus Pharmaceuticals, Inc. Shamia L. Faison was an employee of Supernus Pharmaceuticals, Inc. at the time of this work, but is now an employee of Certara Strategic Consulting. Vladimir Maletic is an employee of the University of South Carolina School of Medicine, and is a consultant for ACADIA Pharmaceuticals Inc.; Alfasigma USA, Inc.; Alkermes, Inc.; Allergan; Eisai-Purdue; Intra-Cellular Therapies; Janssen; H. Lundbeck A/S; Otsuka America Pharmaceutical, Inc.; Sage Pharmaceuticals; Sunovion Pharmaceuticals Inc.; Supernus Pharmaceuticals, Inc.; and Takeda Pharmaceutical Company Ltd. He also serves on the speakers bureau of ACADIA Pharmaceuticals Inc.; Alkermes, Inc.; Allergan; Ironshore; Intra-Cellular Therapies; Janssen; H. Lundbeck A/S; Otsuka America Pharmaceutical, Inc.; Sunovion Pharmaceuticals Inc.; and Takeda Pharmaceutical Company Ltd; and his spouse serves on the speakers bureau of Otsuka America Pharmaceutical, Inc.
Availability of data and material
The data are not available in a repository, but reasonable requests can be directed to Azmi Nasser at anasser@supernus.com.
Ethics approval
This trial was conducted in accordance with the Helsinki Declaration and the International Council for Harmonization (ICH) Note for Guidance on Good Clinical Practice. The trial conduct was reviewed and approved by IntegReview IRB, Austin, TX, USA.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent for publication
Patients signed informed consent regarding the publication of their data.
Code availability
Not applicable.
Author contributions
Conceptualization: Shamia L. Faison, Azmi Nasser, Stefan Schwabe. Methodology: Shamia L. Faison, Azmi Nasser, Nicholas Fry, Toyin Adewole, Oyinkansola Odebo. Data analysis: Zhao Wang, Azmi Nasser. Writing: Vladimir Maletic, Azmi Nasser.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Faison, S.L., Fry, N., Adewole, T. et al. Pharmacokinetics of Coadministered Viloxazine Extended-Release (SPN-812) and Methylphenidate in Healthy Adults. Clin Drug Investig 41, 149–159 (2021). https://doi.org/10.1007/s40261-020-00992-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-020-00992-6