
Abstract
Background and Objective
Highly purified human menotrophin and urofollitrophin preparations obtained from human urine via a novel patented purification method have been tested over a timeframe of 14 years in the studies presented in this article. The objective of the studies was to investigate the pharmacokinetics and the pharmacodynamics of follicle-stimulating hormone (FSH) after single subcutaneous and intramuscular doses and multiple subcutaneous doses of the tested preparations in healthy fertile pituitary-suppressed women.
Designs
We performed five open, randomised, crossover, single-dose bioequivalence and/or bioavailability studies and one open, multiple-dose, pharmacokinetics and pharmacodynamics study.
Study Subjects and Treatments
The six studies included 121 healthy fertile women taking their usual combined oral contraceptives for 3 months before the study: Study 1: 300 international units (IU) of highly purified menotrophin as single subcutaneous and intramuscular doses. Study 2: 300 IU of highly purified menotrophin (test formulation vs. comparator) as single subcutaneous doses. Study 3: 300 IU of highly purified urofollitrophin (hp-FSH) (test formulation vs. comparator) as single subcutaneous doses. Study 4: 300 IU (2 × 150 IU vs. 4 × 75 IU) of hp-FSH as single subcutaneous doses. Study 5: 225 and 445 IU of hp-FSH as single subcutaneous doses. Study 6: daily 225 IU of hp-FSH as subcutaneous doses for 5 consecutive days.
Main Outcome Measures
The main outcome measures were the FSH pharmacokinetic parameters, estradiol concentrations, and the number and size of the follicles.
Results
FSH after single subcutaneous and intramuscular injections of menotrophin or urofollitrophin attained a systemic peak (maximum) concentration (C max) that was on average consistent throughout the first four studies and ranged from 4.98 to 7.50 IU/L. The area under the plasma concentration–time curve (AUC) from administration to the last observed concentration time t (AUCt) ranged from 409.71 to 486.16 IU/L·h and the elimination half-life (t ½) ranged from 39.02 to 53.63 h. After multiple doses of urofollitrophin (225 IU) for 5 days, FSH attained a mean C max of 14.93 ± 2.92 IU/L and had an AUC during the time interval τ between two consecutive doses at steady state (AUCτ) of 322.59 ± 57.92 IU/L·h, which was similar to the mean AUCt after a single subcutaneous dose of 225 IU of urofollitrophin in study 5 (306.82 ± 68.37 IU/L·h).
Conclusions
In our studies, the intramuscular and subcutaneous routes of menotrophin were equivalent; both menotrophin and urofollitrophin were bioequivalent to their marketed reference; FSH kinetic parameters following injection of urofollitrophin were dose proportional and independent from the administered concentration; and multiple doses of FSH increased estradiol levels and enhanced growth of follicles with a good dose–response correlation. Local tolerability was excellent throughout the six studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Two new highly purified human menotrophin and highly purified urofollitrophin (hp-FSH) preparations endowed with a higher purification grade administered as single and multiple doses to healthy women, pituitary suppressed by oral hormonal contraceptives, showed excellent local tolerability. |
The pharmacokinetics of follicle-stimulating hormone (FSH) showed bioequivalence of both new preparations to their marketed references. |
The pharmacokinetics of FSH following injection of hp-FSH was dose proportional and independent from the administered concentration. |
Multiple doses of hp-FSH stimulated estradiol elevation and enhanced growth of follicles. |
1 Background
Gonadotrophic hormones, including follicle-stimulating hormone (FSH), luteinising hormone (LH) and human chorionic gonadotrophin (hCG), have been used to promote fertility for over 30 years. Urofollitrophin is a biologically pure urinary FSH (uFSH) preparation. Human menotrophin (hMG) is a naturally occurring combination of gonadotrophins that contains both FSH and LH activity in equal amounts. In hMG, the follicle-stimulating activity is of pituitary origin while the luteinising activity is predominantly of pituitary origin, but it may also contain luteinising activity of placental origin. hMG is obtained from the urine of postmenopausal women but, where necessary, hCG obtained from the urine of pregnant women may be added.
The secretion of gonadotrophic hormones in normal fertile women occurs in a pulsatile manner in response to hypothalamic pulses. The levels of FSH and LH vary with the menstrual cycle and show characteristic preovulatory peaks called surges. Serum FSH levels at which follicle growth is initiated range individually between 5.7 and 12.0 international units (IU)/L [1–3]. Median serum FSH levels measured in a sample of 42 healthy volunteers with a normal menstrual cycle during the follicular phase were 4.0 (range 0.9–9.2) IU/L on cycle day 1, 6.6 (range 4.3–12.5) IU/L at the peak (maximum) concentration (C max) occurring on cycle day 5 (range 1–9) and 3.3 (range 0.8–5.7) IU/L at the minimum occurring on cycle day 13 (range 8–16) [3, 4]. After menopause, the gonadotrophin secretion is greatly increased in the absence of inhibitory signals. Therefore, postmenopausal women have high concentrations of FSH and LH. Median FSH was 51.9 (range 11.3–116) IU/L in a sample of 33 postmenopausal women [5] and FSH had a geometric mean of 45.7 IU/L in a sample of 157 postmenopausal women [6].
Urine from postmenopausal women has represented a source of natural human hMG and FSH for more than 60 years. The first use of gonadotrophins to induce ovulation in hypogonadic women dates back to the work of Gemzell et al. [7], and the first successful pregnancy resulting from treatment with urinary gonadotrophins was reported by Lunenfeld et al. [8, 9]. However, hMG was purified and isolated from crude extracts of large urine pools by Breadbury et al. [10]. The first preparation of urinary hMG was registered for clinical use in Italy in 1950. The older preparations of urinary hMG and FSH contained small peptidic impurities, which may affect follicle recruitment and development [11–13]. The urine contaminants, present in the older preparations of urinary hMG and FSH, prevented the use of the subcutaneous route due to potential local reactions [14, 15], though several clinical studies indicated that the risk of allergic reactions was minimal [16, 17]. More recently, the new highly purified FSH (hp-FSH) and highly purified hMG (hp-hMG) preparations have reached a higher purification grade equivalent to that of FSH obtained by recombinant DNA technology (rFSH) [18]. Their purification grade allows a safe and well-tolerated subcutaneous administration. With regard to efficacy, recent meta-analyses have confirmed that hp-FSH, hp-hMG and rFSH are equally effective in controlled ovarian stimulation within assisted reproductive technology (ART) programmes [19–23]. Therefore, the costs of the available formulations have been recently taken into account as discriminating features [24].
The preparations tested in the studies presented in this article contain hp-hMG and hp-FSH obtained via a novel patented purification method. IBSA (Institut Biochimique S.A., Pambio-Noranco, Switzerland) hp-FSH and hp-hMG preparations are commercially available worldwide, in 65 and 45 countries, respectively.
Briefly, the purification process starts from urine from postmenopausal women. After some initial ultrafiltration and solvent precipitation steps, a highly purified material is obtained after a series of chromatography (ion exchange, affinity and hydrophobic interaction) and filtration steps. A high level of viral safety of the purified material is also achieved by the application to the process of several validated virus clearance procedures. As previously noted, when necessary, hCG from urine of pregnant women is added to achieve the 1:1 FSH to LH activity ratio.
Previous literature studies concluded that FSH had a similar pharmacokinetic profile both after administration of older urinary preparations or of new highly purified preparations [25], suggesting that the novel urine purification processes to obtain highly purified preparations do not affect the pharmacokinetic profile of FSH. The main advantage of the new highly purified preparations is the almost total absence of contaminants, which allows subcutaneous administration, thus improving the patients’ comfort, ease of use of the product and the batch-to-batch consistency [26, 27].
Hp-hMG and hp-FSH are indicated for (1) the induction of ovulation in amenorrhoeic or anovulatory women who have not responded to treatment with clomiphene citrate; and (2) controlled ovarian hyperstimulation within an ART programme—induction of multiple follicular development in women undergoing assisted reproduction techniques such as in vitro fertilisation. Both gonadotrophins are also used in cases of male infertility due to hypogonadotropic hypogonadism.
2 Methods
2.1 Study Designs
Hp-hMG and hp-FSH were investigated in six phase I trials (Table 1). Single-dose studies were all designed as open-label, randomised, crossover studies with a washout interval of at least 14 days between the two consecutive doses in studies 1–3 and of at least 28 days in studies 4–5.
The first two trials investigated hMG. In detail, study 1 aimed at comparing the pharmacokinetic profile of FSH after subcutaneous and intramuscular injection of hp-hMG. Study 2 compared the pharmacokinetic profile of FSH after subcutaneous injection of hp-hMG and of a marketed reference.
The last four trials investigating hp-FSH were concurrent with the formulation development. The study aims were to compare the pharmacokinetic profile of FSH after subcutaneous injection of the tested formulation and of a marketed reference (study 3); to compare the pharmacokinetic profile of FSH after subcutaneous injection of the same dose of the tested formulation administered in two different concentrations (study 4); to investigate the dose linearity of FSH after single subcutaneous dose at two increasing strengths (study 5); and to investigate the pharmacokinetic profile and the pharmacodynamic effects of FSH after multiple subcutaneous doses (study 6).
2.1.1 Study Population and Criteria for Inclusion
All six studies were performed at the Phase I Unit of CROSS Research S.A. (Arzo, Switzerland).
It was planned that 121 healthy fertile women would be included in the six trials according to the following main inclusion criteria: (1) age of 18–40 years; (2) body mass index between 18 and 30 kg/m2; (3) good health based on medical history, physical examination, a 12-lead electrocardiogram (ECG) and routine haematology and blood chemistry tests; (4) use of a combined oral contraceptive (estrogen–progestin combined preparation) for at least 3 months prior to the study start; and (5) willingness to provide written informed consent.
The main exclusion criteria were (1) pregnancy; (2) intake of any medication; (3) history of drug, alcohol, caffeine or tobacco abuse; (4) history of an abnormal menstrual cycle; (5) history of any endocrine, ovarian or genitourinary abnormality or disease; and (6) a positive Pap test result.
In order to reduce the variability caused by the endogenous release of FSH and LH, all women participating in the studies used their usual oral combined contraceptive during the whole study period. After 1 week of withdrawal, the intake of the contraceptive started from about 20 days before the first dose of the tested drug until the end of the study to ensure pituitary suppression.
The sample size for the first three studies (n = 18) was calculated using the FSH area under the plasma concentration–time curve (AUC) of serum FSH versus time reported in the literature [28–30]. For the calculation of the sample size of the first three studies, the Westlake method for bioequivalence studies was applied [31, 32]. With a coefficient of variation (CV) of 0.2073, the α-error fixed at 0.05 and a 20 % equivalence limit difference, a crossover study enrolling 16 volunteers (32 observations) would have provided a 95 % power. The number of 18 subjects was considered to have sufficient power to give information on the actual pharmacokinetic response.
The sample size for study 4, n = 28, was calculated using the Schuirmann two one-sided t-test [33, 34] and the same data used for the previous studies [30]. Percentage CV (CV %) = 22 was set according to the hypothesis that a higher inter-subject variability was to be expected than that reported in the literature. With a hypothetical CV % = 22, α = 0.05 and an 80 % power, n was 24. Taking into account a rate of four subjects failing to meet the endogenous FSH suppression criterion, 28 healthy women were enrolled in order to have 22 subjects to be considered in the pharmacokinetic analysis.
The sample size of study 5 was calculated using Schuirmann two one-sided test equivalence analysis of mean ratios. Results of study 4 were used and the CV was set to 0.268 and the within-subject correlation to 0.0. The bioequivalence acceptance range of 80.00–125.00 % was used. A total of 26 pairs were required to achieve 80 % power with an α level of 5 %.
The sample size of study 6 was not calculated using any statistical procedure. A sample of 13 subjects was deemed sufficient to achieve the main study objective.
2.1.2 Investigational Treatments and Dose Regimens
In the first two studies, single doses of hp-hMG were administered in two subsequent periods. Single doses of 300 IU were administered using 2 × 150 IU vials and 4 × 75 IU vials in studies 1 and 2, respectively. In study 1, each subject received one intramuscular and one subcutaneous injection of hp-hMG (Merional®, IBSA). In study 2, each subject received two single subcutaneous injections [Merional®, IBSA, test formulation, and Menopur®, Ferring Pharmaceuticals (Saint-Prex, Switzerland), reference formulation].
In studies 3, 4 and 5, two single subcutaneous injections of hp-FSH were administered in two crossover periods. In study 3, single doses of 300 IU were administered using 4 × 75 IU vials (Fostimon®, IBSA, test formulation, and Bravelle™, Ferring Pharmaceuticals, reference formulation). In study 4, single doses of 300 IU were administered using 2 × 150 IU vials (Fostimon®, IBSA) in one period and 4 × 75 IU vials (Fostimon®, IBSA) in the other period. In study 5, single doses of 225 IU (3 × 75 IU vials) (Fostimon®, IBSA) and of 445 IU (3 × 150 IU vials) (Fostimon®, IBSA) were given in the two crossover periods.
In study 6, each subject received multiple subcutaneous doses of 225 IU of hp-FSH once a day for 5 days. Each dose was prepared by combining one 75 IU vial with one 150 IU vial (Fostimon®, IBSA). The multiple-dose treatment started on day 21 of intake of an oral combined contraceptive.
2.1.3 Ethical Procedures
The documentation of the six studies was reviewed and approved by the independent ethics committee of Canton Ticino before each study initiation (from 2000 to 2012). The Swiss Federal Health Authorities (Swissmedic) approved and authorised each study. All of the studies were conducted in compliance with the Swiss ordinance on clinical trials of therapeutic agents and in accordance with the Declaration of Helsinki and the general principles of International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Harmonised Tripartite Guidelines for Good Clinical Practice (GCP). Subjects of any study did not undergo any study procedure before signing the written informed consent form.
2.1.4 Pharmacokinetic and Pharmacodynamic Variables and Data Analysis
In the calculation of pharmacokinetic parameters, baseline FSH values were subtracted from each post-dose concentration value. Similarly, estradiol concentrations measured in study 6 were baseline subtracted.
In the first four studies, the parameters were also corrected for the actual administered dose, taking into account both the actual strength of the used finished products and the residual product measured in the used vials after injections. In study 5, the AUC from administration to the last observed concentration time t (AUC t ) and C max were compared between treatments, after normalisation for the dose.
The following pharmacokinetic parameters were measured and/or calculated for FSH, when feasible, using Kinetica version 4.0 or higher (Thermo Scientific, Philadelphia, PA, USA) or WinNonLin® 6.3 (Pharsight, Cary, NC, USA): C max, t max (time to achieve C max), t ½ (elimination half-life, calculated, as ln2/λ z, where λ z is the terminal elimination rate constant, calculated by log-linear regression using at least 3 points), AUCt calculated with the linear trapezoidal method, AUC∞ (AUC from time zero to infinity, calculated, if feasible, as AUC t + C t /λ z, where C t is the last measurable drug concentration) and AUCτ (AUC during the time interval τ between two consecutive doses at steady state).
In studies 1–5, the C max and AUC values of FSH were compared using analysis of variance (ANOVA) for a crossover design on log-transformed data. In study 5, the analysis was performed after dose normalisation. Schuirmann two one-sided t-tests at the level of significance of 5 % were also performed. The acceptance criterion for bioequivalence, and for linearity in study 5, was that the 90 % confidence intervals (CIs) of the ratios of the parameter geometric means were within the 80–125 % range.
The follicular growth stimulation was evaluated in study 6 as the pharmacodynamic effect of FSH. The number and the size of follicles were determined by ultrasonography. The development of follicles in both ovaries was evaluated at screening and on days 26 and 35–37, i.e. on the fifth treatment day and 10–12 days after the end of the treatment.
2.1.5 Sample Collection, Handling and Analytics
Venous blood samples were collected for the determination of FSH in serum (studies 1, 3, 4, 5 and 6) or plasma (study 2) and of estradiol in serum (study 6) at the sampling timepoints reported in Table 1. The suppression of endogenous FSH was verified measuring the endogenous FSH from 3 days to 1 day before the first dose in the first three studies, at the screening and then on the day before the first dose in studies 4 and 5, and on day 20 (i.e. 2 days before the start of treatment) in study 6.

Mean serum follicle-stimulating hormone concentration–time curves after single subcutaneous and intramuscular injections of highly purified human menotrophin (top study 1, n = 17; bottom study 2, n = 18). Error bars indicate +standard deviation. FSH follicle-stimulating hormone, hp-hMG highly purified human menotrophin, i.m. intramuscular, IU international units, s.c. subcutaneous

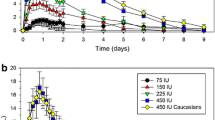
Mean follicle-stimulating hormone serum concentration–time profiles after single subcutaneous doses of highly purified follicle-stimulating hormone (top study 3, n = 18; centre study 4, n = 22; bottom study 5, n = 25). Error bars indicate +standard deviation. FSH follicle-stimulating hormone, hp-FSH highly purified follicle-stimulating hormone, IU international units, s.c. subcutaneous

Mean serum follicle-stimulating hormone (IU/L) and estradiol (pg/mL) original concentration–time profiles (study 6, n = 12). Error bars indicate +standard deviation. E2 estradiol, FSH follicle-stimulating hormone, IU international units
In the first three studies, one pre-dose sample was collected in each study period, while three pre-dose samples were collected in the last three studies. In the last three studies, baseline FSH was calculated as arithmetic mean of the three pre-dose values. In all six studies, blood samples were collected up to 192 h post-dose after each single dose and after the last dose in study 6. Serum and plasma samples were stored frozen at or below −20 °C until analysed.
FSH was determined at the following qualified bioanalytical laboratories:
-
Studies 1 and 2: Institut Dr Viollier, Basel, Switzerland, and Bio-Inova, Plaisir, France, respectively, both using a chemiluminometric sandwich immunoassay, Advia Centaur®, Bayer, with a lower quantification limit (LQL) of 0.3 IU/L and a precision CV ranging from 2.2 to 3.9 %.
-
Study 3: CentraLabS Clinical Research Ltd., Alconbury, UK using a validated modified radioimmunoassay (Coat-a-Count® IRMA, Euro DPC Ltd, UK) with a LQL of 0.4 IU/L. The calibration range covered 0.19–15 IU/L. Accuracy: bias ranged from −4.3 to 20.8 % for the quality control (QC) samples. Precision: CV ranged from 1.9 to 40.0 % for the QC samples.
-
Study 4: Nuvisan GmbH, Neu-Ulm, Germany using a validated two-site immunoradiometric assay with a LQL of 0.300 IU/L. The calibration range covered 0.000–28.0 IU/L. Accuracy: bias ranged from −3.92 to 3.57 %. Precision: CV ranged from 2.23 to 8.06 % for the QC samples.
-
Study 5: Analytical Biochemical Laboratory B.V., Assen, Netherlands. The applied validated method was a solid-phase two-site fluoroimmunometric assay in which two monoclonal antibodies were directed against two separate antigenic determinants on the FSH molecule. The LQL was 0.250 IU/L. The calibration range covered 0.250–500 IU/L. Accuracy: bias ranged from −4.4 to −2.4 % for the QC samples. The inter-day CV and mean bias values for the calibration standards ranged from 0.5 to 1.6 % and −1.0 to 1.1 %, respectively.
-
Study 6: Analytical Biochemical Laboratory B.V. The LQL and calibration range were as for study 5. Accuracy: bias ranged from −9.6 to −5.4 % for the QC samples. The inter-run CV and mean bias values for the calibration standards ranged from 0.4 to 1.9 % and −1.2 to 0.5 %, respectively. Estradiol was measured by the validated method Wallac AutoDelfia® Fluoroimmunoassay at Analytical Biochemical Laboratory B.V. with an LQL of 13.6 pg/mL. The calibration range covered 13.6–4059 pg/mL. Accuracy: bias ranged from −7.4 to −5.6 % for the QC samples. The inter-run CV values for the calibration standards was 0.0 % and mean bias ranged from −0.3 to 0.0 %.
2.1.6 Safety Variables
Safety measures included the recording of adverse events, the measurement of vital signs, ECG recording, full physical examinations and routine haematology, blood chemistry and urinalysis laboratory tests. In all studies, local reactions and pain at the injection site were evaluated by the volunteers. Adverse events were defined as any untoward medical occurrences in study subjects receiving the study treatment and which did not necessarily have to have a causal relationship with the study treatment. For each reported adverse event, the relationship with study treatment, when assessable, was classified as certain, probable, possible or unlikely/none and intensity was classified as mild, moderate or severe.
3 Results
3.1 Disposition of Subjects
Altogether, 121 healthy fertile women were enrolled and received at least one dose of the studied formulations. In study 1, one of 18 subjects withdrew prematurely from the study due to personal reasons. In study 5, one of 26 subjects prematurely withdrew consent to take part in the study for personal reasons. In study 6, one of 13 subjects wished to leave the study prematurely for personal reasons after receiving two doses of the investigational treatment. All other enrolled and randomised subjects completed as per protocol each study they were taking part in. No discontinued subject was included in any pharmacokinetic analysis. All completers were considered in the pharmacokinetic analysis of each study with the exception of study 4, in which 28 subjects were enrolled. Twenty-two of the subjects were included in the pharmacokinetic analysis, while the remaining six subjects were excluded from the pharmacokinetic analysis due to baseline FSH values ≥2 IU/L and were considered in the safety analysis only.
3.2 Follicle-Stimulating Hormone (FSH) Pharmacokinetic Profile After a Single Dose of Highly Purified Human Menotrophin
FSH mean serum levels after single subcutaneous and intramuscular injection of hp-hMG (study 1) and after single subcutaneous injection of the hp-hMG test formulation (study 2) are reported in Fig. 1. Systemic FSH concentrations, measured in study 1, are very similar after single intramuscular and subcutaneous injection and the pharmacokinetic analysis suggested the equivalence of the two administration routes for the tested formulation (90 % CI 105–122 for AUC t and 90 % CI 103–127 for C max).
Study 2 demonstrated the bioequivalence of the hp-hMG formulation versus the marketed reference (90 % CI 80.24–100.2 for AUC t and 90 % CI 81.5–99.01 for C max). Table 2 summarises the FSH pharmacokinetic parameters measured and calculated after baseline subtraction and correction for the actually administered dose after a single dose of hp-hMG in studies 1 and 2.
3.3 FSH Pharmacokinetic Profile After a Single Dose of Highly Purified FSH (hp-FSH)
Study 3 demonstrated the bioequivalence of the hp-FSH formulation versus the marketed reference (90 % CI 91.4–107.8 for AUC t and 90 % CI 92.6–111.9 for C max; see Table 3 for pharmacokinetic parameters and Fig. 2 for FSH mean serum levels after a single subcutaneous injection of the test formulation).
Study 4 proved the equivalence of the tested hp-FSH formulation manufactured at two different strengths (150 vs. 75 IU vials) administered at the same dose (300 IU) (90 % CI 82–94 for AUC t and 90 % CI 83–93 for C max; Table 3; Fig. 2).
Study 5 showed the pharmacokinetic linearity of FSH administered as one single dose of 225 IU (3 × 75 IU vials) and of 445 IU (3 × 150 IU vials) (see Fig. 2; Table 3).
3.4 FSH Pharmacokinetic Profile After Multiple Doses of hp-FSH
The mean serum levels of FSH and estradiol during treatment and after the fifth dose of hp-FSH are reported in Fig. 3.
Pharmacokinetic parameters of baseline-corrected serum FSH after the fifth (last) injection of hp-FSH are presented in Table 4. The mean AUCτ was similar to the mean AUC∞ of serum FSH after a single dose of 225 IU of hp-FSH measured in study 5 (compare data in Table 4 with that in Table 3), thus denoting the linearity and time independence of FSH clearance.
The number of follicles is depicted by size and by timepoint in Fig. 4.

Mean number of follicles by size category at baseline and after treatment with highly purified follicle-stimulating hormone (study 6, n = 12), counted at the screening visit on the fifth day of treatment and 10–12 days after the end of the treatment
3.5 Safety
The frequency of adverse events reported during the six studies of FSH is summarised in Table 5. Local reactions to the injection of hp-hMG and hp-FSH are summarised in Table 6.
4 Discussion
FSH pharmacokinetics and pharmacodynamics have been investigated in six clinical studies over a timeframe of 14 years while the two tested hp-hMG and hp-FSH formulations were being developed. The first five open-labelled, randomised, crossover single-dose studies differed slightly in their designs mainly due to regulatory requirements being updated over the years. In particular, a correction for the actually administered dose and for the residual product content in the used vials was applied to the calculation of FSH pharmacokinetic parameters in the first four studies. Later, this kind of correction was no longer required by the regulator and was not applied to studies 5 and 6. The sample size calculation for the first three studies was performed using literature data, whereas the data collected in first three studies advantaged the sample size calculation for studies 4 and 5. The sample size of study 6 was estimated without any statistical calculation. Healthy fertile women with a normal menstrual cycle were the population selected consistently in all six studies. All enrolled women were pituitary suppressed. The suppression of endogenous gonadotrophins aimed to reduce the bias in the pharmacokinetic analysis of FSH. Oral hormonal contraceptives were chosen for reasons of safety and compliance, whilst gonadotrophin-releasing hormone agonists were excluded due to their higher invasiveness towards healthy subjects. Moreover, the oral contraceptives inhibit endogenous FSH and LH to at least the same low concentrations observed in subjects with hypogonadotrophic hypogonadism [28–30, 35]. Although complete pituitary inhibition is not ensured with oral contraceptives [36], adequate suppression of the hypothalamic–pituitary–ovarian axis has been demonstrated in the literature [37–40]. Women with endogenous FSH <4 IU/L can be considered to be under appropriate gonadotrophin suppression [41]. Nevertheless, FSH <2 IU/L, reached by prolonged use of combined contraceptives, was considered to be an adequate degree of inhibition [37–40] in the first four studies. Afterwards, the acceptance level was increased to FSH <4 IU/L [41, 42]. Baseline values of endogenous FSH measured pre-dose were subtracted from each post-dose concentration value. In the last three studies, baseline FSH and estradiol values were calculated as arithmetic means of three pre-dose values. This approach was deemed appropriate on the basis of analogous literature studies such as those by le Cotonnec et al. [25, 42, 43] and considering that the risk of overcorrection of data is mitigated by the crossover design applied to studies 1–5 [41]. In studies 4–6, the evaluation of pre-dose levels of endogenous FSH and estradiol extended to three timepoints, which was also in compliance with the guidelines on bioequivalence and bioavailability updated at that time. A 24-h baseline serum FSH or estradiol profile was deemed unnecessary since relevant diurnal fluctuations can be excluded for both hormones on the basis of previous literature pharmacokinetic studies [42, 43]. In fact, FSH and LH are normally secreted in a pulsatile fashion that does not correlate with day or night, estrogen or progesterone levels, or waking or sleeping [44, 45]. More recent research has confirmed the absence of circadian rhythms of gonadotrophin secretion through constant routine protocols of sleep/wake, light, temperature, position and nutritional cues across a 24-h period in fertile and postmenopausal women [46, 47]. In the same study, estradiol also did not show any significant change in concentration between evening, night and morning [46]. The washout period was 14 days between two consecutive injections in studies 1–3, whilst it was prolonged to 28 days between administrations in studies 4–5. This change ensured that FSH concentrations returned below the quantification level in all subjects before administration of the second period, thus avoiding any carry-over effect.
After a single dose of 300 IU of hp-hMG and of hp-FSH, the FSH concentration versus time curves and pharmacokinetic parameters were substantially similar. Both the hp-hMG and the hp-FSH formulations were bioequivalent to their respective marketed references in terms of the C max and AUC of systemic FSH after a single subcutaneous injection. The equivalence between the subcutaneous and intramuscular routes of administration of hp-hMG was proven for FSH AUC t , the 90 % CIs of which met the usually applied acceptance range of 80.00–125.00 %. On the other hand, C max had a larger inter- and intra-individual variability and 90 % CI accounting for 103–127 %.This result proved the equivalence of the two administration routes in terms of the extent of absorption and is consistent with literature data for similar studies [30]. Furthermore, two different strength formulations (150 vs. 75 IU vials) also proved to be bioequivalent.
In the five single-dose studies after subcutaneous or intramuscular administration of hp-hMG and hp-FSH, the FSH pharmacokinetic profile in the dose range 225–445 IU showed (1) a slow absorption from the injection site (t max ≥20 h); (2) a sustained elimination from the central compartment (t ½ >40 h); (3) linear pharmacokinetics; and (4) bioavailability independent from the route and the mode of administration (subcutaneous vs. intramuscular route or use of different strength formulations). The pharmacokinetic results of FSH obtained in the studies reported here are consistent with those of the literature [48, 49]. In particular, FSH was dose proportional after single subcutaneous injections of hp-FSH in the dose range 225–445 IU according to the results of study 5, similar to results previously published in the literature [48, 49]. In detail, Mannaerts et al. [48] investigated the pharmacokinetics of FSH after multiple intramuscular doses of 75, 150 and 225 IU/day of rFSH for 7 days in gonadotrophin-deficient subjects and after multiple intramuscular doses of 75, 150 and 225 IU/day of rFSH and 150 IU/day of uFSH in pituitary-suppressed subjects. In the multiple rising dose study of rFSH in gonadotrophin-deficient subjects, serum FSH concentrations increased in a dose-dependent manner consistent with the results of study 5. Mannaerts et al. [50, 51] observed that FSH was at steady state within 5 days of treatment, which was consistent with the data in the literature. In the gonadotrophin-deficient subjects, steady state was achieved after 3–5 days [48]. In the multiple subcutaneous dose study (study 6), serum FSH increased over the physiological baseline concentration and, on average, attained a peak at 12 h after the last injection. On the fifth treatment day, systemic FSH attained 96.87 % of steady state and AUCτ was consistent with the data in the literature. Afterwards, the FSH concentration declined and, on average, returned to the baseline levels at 192 h after the last injection.
The FSH pharmacokinetic parameters obtained after multiple subcutaneous doses of hp-FSH (study 6) are consistent with those of the literature [42, 48, 49] for rFSH. In particular, C max is consistent with the data in the literature, though the treatment duration was 7 days in the literature studies of uFSH (150 IU intramuscular) [48] and of subcutaneous rFSH (225 IU) [49] and (150 IU) [42]. AUCτ is also consistent with the same literature data [42, 48, 49]. Results from study 6 are compared with those of the literature in Table 7.
As expected [49], multiple subcutaneous injections of hp-FSH administered to pituitary-suppressed subjects were able to stimulate the increase in endogenous estradiol levels. According to the observations of study 6, the baseline-corrected estradiol concentration attained a peak at 48 h after the fifth injection of hp-FSH. Seven days after the end of the treatment (192 h after the last injection), serum estradiol fell to levels very close to the baseline. Generally, follicular growth and development can also take place with extremely low levels of estrogens in women treated with hp-FSH [27, 52]. Notably, the effect on follicular estrogen production depends on the presence and the amount of endogenous LH when pure exogenous FSH is administered [27]. Previous literature studies demonstrated that minimal LH levels are sufficient to sustain the secretion of estradiol in response to the treatment with hp-FSH even when the pituitary activity is suppressed [48, 53]. In other words, according to the speculations by Mannaerts et al. [48] and Devroey et al. [53], a persisting minimal production of endogenous LH is sufficient to concur with the exogenous FSH to stimulate estrogen biosynthesis and secretion in women with a normal menstrual cycle, even if under pituitary suppression. In conclusion, the results of study 6 show consistency with the data in the literature.
After multiple subcutaneous doses of hp-FSH, the observed increase in the mean follicular size showed a qualitative correlation with the increase in estradiol levels and both correlated with the pharmacokinetics of FSH. The pharmacodynamic effect of FSH was appreciable on the fifth treatment day when the majority of the follicles, which had a size of 1–3 mm at baseline, attained a size of 7–9 mm after five doses. The follicular growth showed a good correlation with the increase in estradiol concentration. In fact, the women who showed the highest estradiol levels at the end of the treatment (206.07 and 614.93 pg/mL) also had the highest number of follicles ≥7 mm. Up to 12 days later, the effect of FSH on the follicles had diminished inasmuch as the majority of the follicles had a size of 4–6 mm. The present results in terms of number and size of follicles are consistent with data published by Voortman et al. [49]. In particular, the number of follicles in the size categories of 7–9 and 10–12 mm observed on the fifth day of treatment with subcutaneous injections of 225 IU of hp-FSH are in agreement with the number of follicles in the size categories 8–9.9 and 10–11.9 mm observed by Voortman et al. [49] on the fifth day of treatment with 225 IU of rFSH.
Meanwhile, the clinical efficacy of both hp-hMG and hp-FSH as tested in the studies reported here has been demonstrated in phase III studies [54, 55].
With respect to the safety of both hp-hMG and hp-FSH, the only untoward effect with a frequency >10 % was headache, the frequency of which showed a correlation with the dose. Indeed, the highest frequency of headache observed across the six studies (24 %) was reported for the hp-FSH dose of 445 IU. Notably, headache had a frequency of 15.4 % in study 6 during the multiple-dose treatment. All other reported untoward effects had a frequency ≤8 %. Nausea and vomiting were reported at a frequency of 8 % with 445 IU of hp-FSH. No adverse events were reported after a single dose of the marketed references in studies 2 and 3.
With respect to local tolerability, the injections of hp-hMG and hp-FSH were mildly painful for about 20–60 % of the subjects. Painful injections of moderate severity occurred at a lower frequency than did mildly painful injections. Painful injections of moderate severity also had a lower frequency with the test treatments than with the reference treatments (studies 2–3). Generally, the pain was limited to the time of injection and did not last longer than 4 h. Other local reactions had a frequency not higher than 15 %, similar to the reference treatments.
5 Conclusions
On the basis of the results of the six clinical trials in healthy fertile women pituitary suppressed by oral hormonal contraceptives, the following conclusions can be drawn: hp-hMG proved to be equivalent when administered by the intramuscular and the subcutaneous routes in terms of extent of absorption; both hp-hMG and hp-FSH were bioequivalent to their respective marketed reference; FSH pharmacokinetic parameters following injection of hp-FSH were dose proportional and independent from the administered concentration; multiple doses of hp-FSH increased estradiol levels and enhanced growth of follicles with a good dose-response correlation; and local tolerability was excellent throughout the six studies.
References
Van Wiessenbruch MM, Schoemaker HC, Drexhage HA, Schoemaker J. Pharmaco-dynamics of human menopausal gonadotrophin (HMG) and follicle-stimulating hormone (FSH). The importance of the FSH concentration initiating follicular growth in polycystic ovary-like disease. Hum Reprod. 1993;8:813–21.
van der Meer M, Hompes PGA, Scheele F, Schoute E, Veersema S, Schoemaker J. Follicle stimulating hormone (FSH) dynamics of low dose set-up ovulation induction with FSH in patients with polycystic ovary syndrome. Hum Reprod. 1994;9:1612–7.
Fauser BCJM, Van Heusden AM. Manipulation of human ovarian function: physiological concepts and clinical consequences. Endocr Rev. 1997;18(1):71–106.
van Santbrink EJP, van Dessel HJHM, Hop WC, De Jong FH, Fauser BCJM. Decremental follicle-stimulating hormone and dominant follicle development during the normal menstrual cycle. Fertil Steril. 1995;64:37–43.
Shin SY, Lee JR, Noh GW, Kim HJ, Kang WJ, Kim SH, et al. Analysis of serum levels of anti-Müllerian hormone, inhibin B, insulin-like growth factor-I, insulin-like growth factor binding protein-3 and follicle-stimulating hormone with respect to age and menopausal status. J Korean Med Sci. 2008;23:104–10.
Henrich JB, Hughes JP, Kaufman SC, Brody DJ, Curtin LR. Limitations of follicle-stimulating hormone in assessing menopause status: findings from the National Health and Nutrition Examination Survey (NHANES 1999–2000). Menopause. 2006;13(2):171–7.
Gemzell CA, Diczfalusy E, Tillinger G. Clinical effect of human pituitary follicle-stimulating hormone (FSH). J Clin Endocrinol Metab. 1958;18(12):1333–48.
Lunenfeld B, Sulimovici S, Rabau E, Eshkol A. L’induction de l’ovulation dans les amenorrhees hypophysaires par un traitement combine de gonadotrophines urinaires menopausiques et de gonadotropins chronique. CR Soc Franc Gynecol. 1962;35:346–56.
Lunenfeld B, Sulimovici S, Rabau E. Urinary gonadotropins in treatment of pituitary amenorrhea. Proc Tel-Hashomer Hosp. 1962;1:25.
Breadbury JT, Brown ES, Brown WE. Adsorption of urinary gonadotrophins on kaolin. Proc Soc Exp Biol Med. 1949;71:228–32.
Giudice E, Crisci C, Eshkol A, Papoian R. Composition of commercial gonadotrophin preparations extracted from human post-menopausal urine: characterization of non-gonadotrophin proteins. Hum Reprod. 1994;9:2291–9.
Van den Weijer BH, Mulders JW, Bos ES, Verhaert PD, Van den Hooven HW. Compositional analyses of a human menopausal gonadotrophin preparation extracted from urine (menotrophin). Identification of some of its major impurities. Reprod Biomed Online. 2003;7:547–57.
Selman HA, De Santo M, Sterzik K, Coccia E, El-Danasouri I. Effect of highly purified urinary follicle-stimulating hormone on oocyte and embryo quality. Fertil Steril. 2002;78(5):1061–7.
Fleming R, Chung CC, Yates RW, Coutts JR. Purified urinary follicle stimulating hormone induces different hormone profiles compared with menotrophins, dependent upon the route of administration and endogenous luteinizing hormone activity. Hum Reprod. 1996;11:1854–8.
Alviggi C, Revelli A, Anserini P, Ranieri A, Fedele L, Strina I, et al. A prospective, randomised, controlled clinical study on the assessment of tolerability and of clinical efficacy of Merional (hMG-IBSA) administered subcutaneously versus Merional administered intramuscularly in women undergoing multifollicular ovarian stimulation in an ART programme (IVF). Reprod Biol Endocrinol. 2007;4(5):45.
Saal W, Happ J, Cordes U, Baum RP, Schmidt M. Subcutaneous gonadotropin therapy in male patients with hypogonadotropic hypogonadism. Fertil Steril. 1991;56(2):319–24.
Schmoutziguer APE, Van Kooij RJ, Te Velde ER, Geurts TBP, de Leeuw R, Rombout F. Retrospective analysis of subcutaneous administration of urinary gonadotrophins in in vitro fertilisation. J Obstet Gynaecol. 1996;16(5):409–13.
The Practice Committee of the American Society for Reproductive Medicine, Birmingham, Alabama. Gonadotropin preparations: past, present, and future perspectives. Fertil Steril. 2008;90(Suppl 5):S13–20.
Al-Inany HG, Abou-Setta AM, Aboulghar MA, Mansour RT, Serour GI. HMG versus rFSH for ovulation induction in developing countries: a cost-effectiveness analysis based on the results of a recent meta-analysis. Hum Reprod. 2003;18:305–13.
Al-Inany H, Aboulghar MA, Mansour RT, Serour GI. Ovulation induction in the new millennium: recombinant follicle-stimulating hormone versus human menopausal gonadotropin. Gynecol Endocrinol. 2005;20:161–9.
Van Wely M, Westergaard LG, Bossuyt PM, Van der Veen F. Human menopausal gonadotropin versus recombinant follicle stimulation hormone for ovarian stimulation in assisted reproductive cycles. Cochrane Database Syst Rev. 2003;1:CD003973.
Al-Inany HG, Abou-Setta AM. Are all human-derived FSH products the same? A systematic review and meta-analysis using direct and adjusted indirect analyses to determine if Fostimon® is more efficient than Metrodin-HP®. Gynecol Endocrinol. 2012;28(2):94–101.
Van Wely M, Kwan I, Burt AL, Thomas J, Vail A, Van der Veen F, et al. Recombinant versus urinary gonadotrophin for ovarian stimulation in assisted reproductive technology cycles. Cochrane Database Syst Rev. 2011;2:CD005354.
National Institute for Health and Care Excellence (NICE). Fertility: assessment and treatment for people with fertility problems. NICE guideline CG156. London: NICE; 2013.
le Cotonnec JY, Porchet HC, Beltrami V, Howles C. Comparative pharmacokinetics of two urinary huma follicle stimulating hormone preparations in healthy female and male volunteers. Hum Reprod. 1993;8(10):1604–11.
Flamigni C, Venturoli S, Dal Parto L, Porcu E. Purified FSH: characteristics and applications. In: Follicori M, Flamigni C, editors. Ovulation induction: basic science and clinical advances. Amsterdam: Elsevier Science; 1994. p. 125–34.
Lunenfeld B. Historical perspectives in gonadotrophin therapy. Hum Reprod Update. 2004;10(6):453–67.
Out HJ, Schnabel PG, Rombout F, Geurts TBP, Bosschaert MAR, Coelingh Bennink HJT. A bioequivalence study of two urinary follicle stimulating hormone preparations: Follegon and Metrodin. Hum Reprod. 1996;11:61–3.
Voortman G, van de Post J, Schoemaker RC, van Gerven JMA. Bioequivalence of subcutaneous injection of recombinant human FSH (Puregon®) by Pen-injector and syringe. Hum Reprod. 1999;14:1698–702.
Huisman JA, Paulussen RJ, Geurts TB, Odink J, Rekers H. Assessment of bioequivalence after subcutaneous and intramuscular administration of urinary gonadotrophins. Hum Reprod. 1997;12:34–8.
Westlake WJ. Use of confidence intervals in analysis of comparative bioavailability trials. J Pharm Sci. 1972;61:1340–1.
Westlake WJ. Symmetrical confidence intervals for bioequivalence trials. Biometrics. 1976;32:741–4.
Schuirmann DJ. A comparison of the two one-sided tests procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm. 1987;15:657–80.
Schuirmann DJ. Design of bioavailability/bioequivalence studies. Drug Inf J. 1990;24:315–23.
Duijkers IJM, Klipping C, Mulders TMT, Out HJ, Ceolingh Bennink HJT, Vemer HM. Pharmacodynamics and pharmacokinetics after repeated subcutaneous administration of three gonadotrophin preparations. Hum Reprod. 1997;12:2379–84.
Dericks-Tan JSE, Krög W, Aktories K, Taubert HD. Dose-dependent inhibition by oral contraceptives of the pituitary to release LH and FSH in response to stimulation with LHRH. Contraception. 1976;14:171–81.
Hemrika DJ, Slaats EH, Kennedy JC, de Vries Robles-Korsen TJ, Schoemaker J. Pulsatile luteinizing hormone patterns in long term oral contraceptive users. J Clin Endocrinol Metab. 1993;77(2):420–6.
Jung-Hoffmann C, Heidt F, Kuhl H. Effect of two oral contraceptives containing 30 µg ethinylestradiol and 75 µg gestodene or 150 µg desogestrel upon various hormonal parameters. Contraception. 1988;38(6):593–603.
Nuttall ID, Elstein M, McCafferty E, Seth J, Cameron ED. The effect of ethinylestradiol 20 µg and levonorgestrel 250 µg on the pituitary-ovarian function during normal tablet-taking and when tablets are missed. Contraception. 1982;26(2):121–35.
Fotherby K. Clinical experience and pharmacological effects of an oral contraceptive containing 20 µg oestrogen. Contraception. 1992;46:477–88.
Dissanayake S. Assessing the bioequivalence of analogues of endogenous substances (‘endogenous drugs’): considerations to optimize study design. Br J Clin Pharmacol. 2010;69(3):238–44.
le Cotonnec JY, Loumaye E, Porchet HC, Beltrami V, Munafo A. Pharmacokinetic and pharmacodynamic interactions between recombinant human luteinizing hormone and recombinant human follicle-stimulating hormone. Fertil Steril. 1998;69(2):201–9.
le Cotonnec JY, Porchet HC, Beltrami V, Khan A, Toon S, Rowland M. Clinical pharmacology of recombinant human follicle-stimulating hormone (FSH). I. Comparative pharmacokinetics with urinary human FSH. Fertil Steril. 1994;61(4):669–78.
Owen JA. Physiology of the menstrual cycle. Am J Clin Nutr. 1975;28:333–8.
Naftolin F, Yen SSC, Perlman D, Tasi CC, Parker DC, Vargo T. Nocturnal patterns of serum gonadotropins during the menstrual cycle. J Clin Endocrinol Metab. 1973;37:6–10.
Klingman KM, Marsh EE, Klerman EB, Anderson EJ, Hall JE. Absence of circadian rhythms of gonadotropin secretion in women. J Clin Endocrinol Metab. 2011;96(5):1456–61.
Lavoie HB, Marsh EE, Hall JE. Absence of apparent circadian rhythms of gonadotropins and free α-subunit in postmenopausal women: evidence for distinct regulation relative to other hormonal rhythms. J Biol Rhythms. 2006;21:58–67.
Mannaerts BMJL, Rombout F, Out HJ, Coelingh Bennink H. Clinical profiling of recombinant follicle stimulating hormone (rFSH; Puregon): relationship between serum FSH and efficacy. Hum Reprod Update. 1996;2(2):153–61.
Voortman G, Mannaerts BM, Huisman JA. A dose proportionality study of subcutaneously and intramuscularly administered recombinant human follicle-stimulating hormone (Follistim*/Puregon) in healthy female volunteers. Fertil Steril. 2000;73:1187–93.
Mannaerts B, Fauser B, Lahlou N, Harlin J, Shoham Shoham Z, Bennink HC, et al. Serum hormone concentrations during treatment with multiple rising doses of recombinant follicle stimulating hormone (Puregon) in men with hypogonadotropic hypogonadism. Fertil Steril. 1996;65(2):406–10.
Schoot DC, Harlin J, Shoham Z, Mannaerts BM, Lahlou N, Bouchard P, et al. Recombinant human follicle-stimulating hormone and ovarian response in gonadotropin deficient women. Hum Reprod. 1994;9(7):1237–42.
Rabinovici J, Blankstein J, Goldman B, Rudak E, Dor Y, Pariente C, et al. In vitro fertilization and primary embryonic cleavage are possible in 17α-hydroxylase deficiency despite extremely low intrafollicular 17-estradiol. J Clin Endocrinol Metab. 1989;68:693–7.
Devroey P, Mannaerts B, Smitz J, Coelingh Bennink H, Van Steirteghem A. Clinical outcome of a pilot efficacy study on recombinant human FSH (Org 32489) combined with various GnRH agonist regimens. Hum Reprod. 1994;9:1064–9.
Alviggi C, Cognigni GE, Morgante G, Cometti B, Ranieri A, Strina I, et al. A prospective, randomised, investigator-blind, controlled, clinical study on the clinical efficacy and tolerability of two highly purified hMG preparations administered subcutaneously in women undergoing IVF. Gynecol Endocrinol. 2013;29(7):695–9.
Baker VL, Fujimoto VY, Kettel LM, Adamson GD, Hoehler F, Jones CE, et al. Clinical efficacy of highly purified urinary FSH versus recombinant FSH in volunteers undergoing controlled ovarian stimulation for in vitro fertilization: a randomized, multicenter, investigator-blind trial. Fertil Steril. 2009;91(4):1005–11.
Acknowledgments
The authors acknowledge the following laboratories for their contribution in performing the bioanalysis of the presented studies: Viollier SA, Switzerland; Eurofins Medinet SAS (formerly Bio-Inova), France; CentraLabS, UK; Nuvisan GmbH (formerly AAI Deutschland GmbH & Co KG), Germany; and ABL, Analytisch Biochemisch Laboratorium BV, the Netherlands.
Authors’ contributions
The sponsor, IBSA, reviewed and approved the study design of each study, was informed about the collection of data, and reviewed and approved the analysis and the interpretation of data. IBSA reviewed and approved the manuscript for publication. A.R. and M.M.R. reviewed and approved the design of the studies, were responsible for the clinical activities and collected the data, L.L. participated in the design of the studies and performed the pharmacodynamics and pharmacokinetics analysis, D.B. performed the statistical analysis, G.C. performed the gynaecological visits including ultrasonography (number and size of follicles) during study 6, B.C. reviewed and approved the design of the studies and the draft manuscript, and A.F.D.D. participated in the clinical trial reporting and drafted the manuscript. All authors read and approved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The sponsor of the six studies, Institut Biochimique S.A. (IBSA), provided financial support for this project. The relationship between IBSA and CROSS Research S.A. was regulated by financial agreements. The relationship between IBSA and Ospedale Regionale di Mendrisio was regulated by a financial agreement.
Conflict of interest
All authors had potential conflicts of interest. In detail, Antonio Rusca, Milko Massimiliano Radicioni, Luca Loprete, Daniela Binelli and Andrea Francesco Daniele Di Stefano are employees of CROSS Research S.A., whose relationship with the sponsor was regulated by financial agreements. Giorgio Caccia received a consultancy fee for his activities. Barbara Cometti is an employee of IBSA, the sponsor of the studies.
Ethical approval
All six studies were conducted in compliance with the Swiss ordinance on clinical trials of therapeutic agents and in accordance with the Declaration of Helsinki and the general principles of the of International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Harmonised Tripartite Guidelines for Good Clinical Practice (GCP) and after approval by the competent Ethics Committee of Canton Ticino, Switzerland.
Informed consent
Study subjects did not undergo any study procedure before signing the written informed consent form.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Di Stefano, A.F.D., Rusca, A., Radicioni, M.M. et al. Pharmacokinetics and Pharmacodynamics of Follicle-Stimulating Hormone in Healthy Women Receiving Single and Multiple Doses of Highly Purified Human Menotrophin and Urofollitrophin. Clin Drug Investig 36, 1031–1044 (2016). https://doi.org/10.1007/s40261-016-0451-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-016-0451-6