
Abstract
Background and Objective
A novel tiotropium bromide monodose capsule dry powder inhaler (DPI) formulation and device have been developed. The formulation was based on a spray-dried matrix that enhances the aerosolizaton properties, allowing a less active tiotropium metered dose (13 µg/capsule) while maintaining the same delivered dose (10 µg/actuation). This study describes the pharmacokinetic bioequivalence to the reference product.
Methods
This randomized, two-stage, crossover, semi-replicate (three-way) study was performed in healthy volunteers. In each study period, subjects received a single dose of two capsules (20 μg delivered dose) of the study medication, separated by a 14-day washout period: tiotropium 10 μg delivered dose (Laboratorios Liconsa, Spain) and Spiriva HandiHaler® (Boehringer Ingelheim Pharma GmbH & Co KG, Germany). Blood samples were obtained up to 48 h post-dose to evaluate the comparative bioavailability. Tiotropium was measured in plasma by means of dual stage liquid–liquid extraction followed by the two-dimensional ultra-high performance liquid chromatography sensitive sub-pg/mL bioanalytical method. The main pharmacokinetic parameters were maximum plasma concentration (C max), area under the concentration–time curve (AUC) from time zero hours to the last observed concentration at time t (AUC t ), and AUC from time zero hours to 30 min (AUC0.5). Bioequivalence was accepted if the 90.20 % confidence interval (CI) for the ratio test/reference of the primary pharmacokinetic parameters lay within the acceptance range of 80–125 %. Safety assessment was a secondary endpoint.
Results
A total of 30 subjects were randomized and bioequivalence was demonstrated for all primary pharmacokinetic parameters: C max (CI 87.26–106.60 %), AUC t (CI 101.33–111.64 %), and AUC0.5 (CI 97.95–113.49 %). Both study treatments were well tolerated (four non-serious adverse events [AEs] were reported in four subjects: one AE before any product administration, two AEs after test product administration; and one AE after reference product administration).
Conclusions
Both products containing tiotropium 10 µg delivered-dose DPI were bioequivalent and showed good tolerability and a similar safety profile.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A novel tiotropium bromide monodose capsule dry powder inhaler formulation with enhanced aerosolization properties has been developed in association with a new device. |
This manuscript describes the bioequivalence to reference product (Boehringer Ingelheim Pharma GmbH & Co KG, Germany) carried out by means of a randomized, two-stage, crossover, semi-replicate (three-way), pharmacokinetic bioequivalence study performed in healthy volunteers. |
The new tiotropium formulation and device represent an alternative to current first-line treatment options for patients with chronic obstructive pulmonary disease. |
1 Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory airway disease characterized by persistent airflow limitation that is usually progressive and associated with an enhanced chronic inflammatory response in the airways and the lung to noxious particles or gases. COPD is a leading cause of morbidity and mortality worldwide and results in an economic and social burden that is projected to increase in the coming decades due to continued exposure to COPD risk factors (such as tobacco smoking, air pollution) and the changing age structure of the world’s population [1].
The preferred therapeutic administration route for COPD patients is via inhalation because it maximizes the concentration of the drug in the target tissue (the lung), providing the greatest potential therapeutic effect while minimizing systemic concentration. As a consequence, the advantages include a more rapid onset of action, lower dosing, avoidance of the first-pass metabolism and fewer systemic side effects [2].
Inhaled medications are administered through appropriate inhalation devices. The choice of device is of major importance because differences in engineering may result in differences in the efficiency of dosing [3, 4]. In addition, properly designed inhalation devices improve patient adherence to the prescribed therapy, resulting in successful disease control with better clinical outcomes and reduced costs [5].
Current international guidelines recommend the use of inhaled long‐acting bronchodilators for first‐line management of COPD [1]. Tiotropium bromide is a quaternary ammonium class of anticholinergic bronchodilators that dilate bronchial smooth muscle through antagonism of muscarinic receptors located in airway smooth muscle. Tiotropium has a similar affinity to subtype M1, M2 and M3 muscarinic receptors, but dissociates much more slowly from M1 and M3 receptors than from M2 receptors, resulting in a long duration of action [6]. Compared with previous anticholinergic agents such as ipratropium, tiotropium has the advantage of its long duration of action that allows once-daily administration, and thus classifies as a long‐acting muscarinic antagonist. Tiotropium is indicated as a maintenance bronchodilator treatment to relieve symptoms of COPD and to reduce the frequency of exacerbations [7, 8].
In general, the inhaled route of administration results in the majority of the delivered tiotropium dose deposited in the gastrointestinal tract and, to a lesser extent, in the intended target organ, the lung [7]. Previous pharmacokinetic investigations have revealed that, following dry powder inhalation of tiotropium by young healthy volunteers, there is an absolute bioavailability of 19.5 %, and the remaining approximately 80 % of inhaled tiotropium dose is swallowed. This suggests that the fraction reaching the lung is highly bioavailable as it is expected, from the chemical structure, that tiotropium bromide is poorly absorbed from the gastrointestinal tract [7]. The terminal elimination half-life of tiotropium bromide is between 5 and 6 days following inhalation [7].
Tiotropium is currently commercially available under two different formulations: an aqueous solution delivered by the Respimat® device (Boehringer Ingelheim Pharma GmbH & Co KG, Germany) and a dry powder for inhalation (DPI) formulation delivered via the HandiHaler® device (Boehringer Ingelheim Pharma GmbH & Co KG, Germany) (Fig. 1). In addition, Laboratorios Liconsa, S.A., Spain, has developed a novel formulation of tiotropium bromide DPI and a new monodose capsule-based device. The developed product was designed to deliver tiotropium 10 µg with a pre-metered dose of tiotropium 13 µg in a hard capsule, in contrast with the reference product DPI delivered via the HandiHaler® device, which also delivers 10 µg but requires a pre-metered dose of 18 µg in the capsule. The test tiotropium 10 µg delivered-dose DPI was based on a spray-dried matrix formulation with enhanced aerosolization properties of the tiotropium active pharmaceutical ingredient (API), allowing less API metered dose per capsule while maintaining the same delivered dose and product performance.

Devices used in the study for the administration of medication. Top HandiHaler®; bottom Zonda®
The new product is delivered through a novel monodose DPI inhaler device (Zonda®) (Fig. 1), which was designed and developed based on the basic principles of HandiHaler®, and hence both have similar airflow resistance in vitro (Table 1) [data on file]. In addition, the inhalation characteristics were comparable between the devices in two different populations—healthy volunteers and COPD patients (Table 1) [data on file].
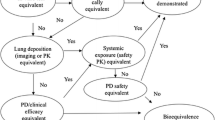
Development of the new tiotropium bromide formulation and device followed the European Medicines Agency (EMA) guidelines on the requirements for therapeutic equivalence for orally inhaled products (OIPs) [9]. This guideline has a stepwise approach, and bioequivalence for abridged applications can be accepted only with comparative in vitro data if some criteria are satisfied, including that the products must contain the same active substance (i.e. same salt, ester, hydrate or solvate, etc.). This first criterion was not satisfied because the experimental product is an anhydrous tiotropium whereas the reference is the monohydrated form. Since a biowaiver is not possible, the second step is to perform an in vivo bioequivalence study. The same guideline recommends that pharmacokinetic trials for the demonstration of bioequivalence are to be carried out in the intended patient population (COPD patients in the case of tiotropium bromide). Conversely, the EMA guidance on the investigation of bioequivalence [10] recommends that bioequivalence studies should normally be performed in healthy volunteers to reduce variability not related to differences between the assayed products (with the exception of drugs with safety concerns). Hence, studies with healthy subjects are considered as adequate in most instances to detect formulation differences and to allow extrapolation of the results to the intended populations for which the reference medicinal product is approved.
Finally, the questions and answers document recently released by the EMA that established the use of the area under the concentration–time curve (AUC) from time zero hours to 30 min (AUC0.5) as an acceptable surrogate for lung deposition for OIPs such as tiotropium with very quick absorption in the lung, was also taken into consideration [11].
The aim of the present publication was to show the results of the comparative bioavailability study carried out in healthy volunteers to demonstrate the bioequivalence between two tiotropium bromide DPI formulations, as recommended by the EMA guidelines for OIPs [9] and bioequivalence [10].
2 Methods
2.1 Ethical Considerations and Study Design
The study protocol was approved by the Independent Ethics Committee of MHAT Tokuda Hospital Sofia AD, Sofia, Bulgaria. The study was conducted in accordance with the Declaration of Helsinki and in full compliance with Good Clinical Practice guidelines. All study participants provided signed informed consent and had the right to withdraw their consent at any time, without giving reason and without detriment. The trial was registered to the European Union Drug Regulating Authorities Clinical Trials database (EudraCT No: 2013-002277-21).
The study was a single-center, single-dose, randomized, open-label, three-way, semi-replicate, crossover study in a two-stage design with one interim analysis and sample size re-assessment conducted in healthy volunteers between 5 January and 29 April 2014. Subjects were assigned to each of the three sequences (Test, Reference, Reference; Reference, Test, Reference; Reference, Reference, Test) following a randomization code (Fig. 2). A washout time of 14 days between study periods was chosen to prevent any carryover effect.

Study design. Test investigational medicinal product: 20 μg delivered dose of tiotropium (test product). Reference investigational product: 20 μg delivered dose of tiotropium (reference product)
2.2 Subjects
Healthy male and female Caucasian subjects aged 18–55 years, weighing within the normal range according to accepted normal values for body mass index (18.5–30.0 kg/m2), were enrolled in the study. All were non-smokers or ex-smokers ≥6 months prior to enrolment in the study, confirmed by urine cotinine test and with forced expiratory volume in 1 s (FEV1) ≥80 % of the predicted value regarding age, height, sex and ethnicity (according to The European Community of Coal and Steel/European Respiratory Society) [12]. A complete medical and surgical history, physical examination, 12-lead electrocardiogram, FEV1 (by means of spirometery), and clinical laboratory examination (hematology, blood chemistry, serology, urinalysis) were performed at screening examination. Physical examination, 12-lead electrocardiogram, vital signs and clinical laboratory (blood and urine) analysis were also repeated 3–9 days after the last dosing day. Urine drug-abuse screens for amphetamines, cannabinoids, benzodiazepines, cocaine, opioids, barbiturates and cotinine, as well as breath alcohol tests and pregnancy tests (females only) were carried out at screening and at hospitalization for each study period.
In the evening prior to each dosing day, subjects were trained in the correct inhalation technique, which was deemed necessary as healthy subjects usually have no experience in the correct inhalation technique. Assessment of the appropriate peak inspiratory flow (between 20 and 90 L/min) was carried out by the use of an inspirometer tool (In Check DIAL®; Clement Clarke International Limited, Harlow, Essex, UK). This training was repeated in the morning of each dosing day. In addition, as the test and reference devices were slightly different by their appearance and use, the training included the use of both devices (Zonda® and Spiriva®) with placebo.
Subjects fasted for at least 10 h before and until at least 4 h after administration of study medication. The dosing time for an individual subject was the same in all three study periods.
The sample size was calculated according to a two-stage design, using the statistical analysis of the first stage data as an interim analysis to calculate the sample size for the second stage [13]. Eighteen subjects were included in the first stage because this number of subjects is regarded as sufficient for a proper estimation of both the residual variance and the mean difference between the test and reference products while preserving a certain chance of proving bioequivalence in the first stage [10]. After analysis of the results obtained in the first stage, the sample size was recalculated considering a remaining α2 of 0.049 and β < 0.20. An additional group of 12 subjects was included, resulting in a total of 30 subjects being included in the study.
2.3 Treatments
The test product was a tiotropium 10 µg delivered-dose DPI, hard hypromellose capsules for inhalation containing tiotropium 13 µg as the metered dose, manufactured by Laboratorios Liconsa, S.A., Spain, administered through the Zonda® device. The reference product was Spiriva® (Boehringer Ingelheim Pharma GmbH & Co KG, Germany), hard gelatin capsules containing tiotropium 18 μg/capsule and delivering 10 μg [7], administered through the HandiHaler® device. Descriptions of the test and reference products, as well as the administration procedure, are summarized in Table 1.
In accordance with the crossover and partial replicate design, subjects received three single doses of two capsules of tiotropium (one test and two references).
On each day of drug administration and during blood sampling, the identity of each subject was compared with the national identification (ID) card. Subjects were then further identified by a wristband containing the study number and randomization number. This wristband was not able to be removed by the subject and was removed by the investigator at clinic discharge. On the following ambulatory visits, the identity of the subjects was checked by using the national ID card only. The study medication was inhaled on the morning of day 1 in each study period, at the same time (between 7:30 and 9:00 a.m.) and under fasting conditions. Subjects fasted from food at least 10 h before and until at least 4 h after administration of study medication on day 1.
All subjects received two capsules (20 μg delivered dose of tiotropium) as a single dose, resulting in four inhalations (two inhalations for each capsule) from the test or reference product. The required number of four inhalations were inhaled by subjects, with a 20-s interval from the start of one inhalation to the next, including a 10-s breath hold after each inhalation. After the second inhalation from the first capsule, the used capsule was replaced by the second capsule of the same drug within 10 s. Two more inhalations were then performed, with a 20-s interval from the start of one inhalation to the next, including a 10-s breath hold after each inhalation.
For dosing, subjects were required to be sitting in an upright position, with a nose clip attached to block breathing through the nose, and were asked to perform the inhalations under supervision of the principal investigator, controlled by a second investigator or study nurse. Correct administration of the study drug was documented in the case report form.
2.4 Bioanalytical Method
Blood samples (8 mL each) were taken pre-dose and 16 times post-dose at 0:01, 0:02, 0:04, 0:06, 0:08, 0:15, 0:30, 0:45, 1:00, 2:00, 4:00, 8:00, 12:00, 24:00, 36:00 and 48:00 (hour:minute) after the end of the last inhalation. Blood samples (17 in each period) were taken by means of a short intravenous catheter on day 1 of each period and by venipuncture thereafter (ambulatory samples). Blood samples (8 mL) were collected in tubes, using EDTA K2 as an anticoagulation agent. The total amount of blood taken from each subject was 428–438 mL per subject (n = 51 blood samples for analysis of tiotropium; 10–20 mL blood for the initial laboratory examination and 10 mL blood for the final safety laboratory examination). Samples were immediately centrifuged, and two plasma aliquots were frozen and stored at less than –18 °C until analysis.
Samples were analyzed after a dual-stage liquid–liquid extraction (LLE), followed by a two-dimensional (2D) ultra-high performance liquid chromatography (UHPLC) with greatly reduced matrix effects and increased assay sensitivity.
A rapid, selective, and highly sensitive bioanalytical method for tiotropium bromide was developed and validated for the analysis of tiotropium in human plasma containing dipotassium ethylenediamine tetraacetic acid (K2EDTA) using a two-stage LLE followed by a 2D UHPLC [14]. The assay was validated within a nominal range of 0.200–100 pg/mL using a 0.500 mL human plasma aliquot. Linearity over the calibration range was demonstrated, with an average correlation coefficient (R 2) of 0.9985. The inter-assay precision and accuracy were 10.8 and 101.8 % (n = 18), respectively, at the lower limit of quantification (LLOQ) of 0.200 pg/mL. The inter-assay precision for five other quality control (QC) levels (0.500, 1.20, 4.50, 15.0 and 75.0 pg/mL) was between 3.1 and 6.4 % (n = 18). The accuracy for the same five levels of QCs was between 96.4 and 98.2 % (n = 18). The bioanalytical method was successfully used to analyze over 3000 clinical samples, with an overall incurred sample reanalysis passing rate of 93.7 %.
2.5 Pharmacokinetic Parameters and Bioequivalence Analysis
The pharmacokinetic and biostatistical evaluation was carried out by means of the validated statistical software package SAS for Windows last available version, v.9.2 (Statistical Analysis System, SAS Institute, Cary NC, USA). The main pharmacokinetic parameters were the observed maximal concentration after administration (C max), obtained directly from data; the AUC, calculated by the trapezoidal rule from time zero hours to the last observed concentration at time t (AUC t ); and the AUC, calculated by the trapezoidal rule from time zero hours to concentration at time 30 min (AUC0.5). Bioavailability can be considered as adequately characterized with the primary parameters since C max reflects the rate of absorption of tiotropium in the lungs and AUCs are used as a measure of the extent of this absorption. As secondary endpoints, and planned to undergo descriptive statistical evaluation, the following parameters were also calculated: time to maximum plasma concentration (t max), AUC from time zero to infinity (AUC∞), AUC residual area (AUCres), terminal half-life (t ½), mean residence time, and terminal rate constant in plasma (λ z) of tiotropium.
For the analysis of bioequivalence, data were log-transformed prior to the analysis of variance according to a general lineal model (GLM-ANOVA). For evaluation of the first stage, sequence, treatment, period and subject sequence were included as effects. For the second stage (final evaluation), treatment, sequence, stage, period (stage) and subject (sequence stage) were included as effects.
According to the two-stage design, the nominal α values used at stage 1 and stage 2 evaluations were chosen according to the Haybittle–Peto method of group sequential designs as α1 = 0.001 and α2 = 0.049, to maintain an overall α = 0.05 of the two-stage test procedure [15, 16].
The 90.20 % confidence interval (CI; second stage) for the ratio test versus reference of the primary endpoints were calculated by the parametric method (ANOVA-log) and then compared with the predefined acceptance ranges (80–125 %).
2.6 Safety
Tolerability assessment was based on the evaluation of adverse events (AEs) for the entire study. An AE was defined as any untoward medical occurrence in a clinical investigation subject administered a pharmaceutical product and which does not necessarily have to have a causal relationship with this treatment. An AE can therefore be any unfavorable and unintended sign (including an abnormal laboratory finding), symptom, or disease temporally associated with the use of a medical product, whether or not considered related to the medical product. One AE was considered serious if it resulted in death, was life-threatening, required inpatient hospitalization or prolongation of existing hospitalization, resulted in persistent or significant disability or incapacity, caused a congenital anomaly/birth defect or, according to medical and scientific judgment, was an important medical event that may not be immediately life-threatening or result in death or hospitalization but may jeopardize the subject or may require intervention to prevent one of the other outcomes listed above. The event was classified as unexpected when it was not consistent with the applicable product information (not listed in the summary of product characteristics of the reference medication). Regardless of the above classification as serious/non-serious and/or unexpected/expected, its severity was assessed as mild, moderate or severe according to medical criteria exclusively.
Clinical safety was assessed through the physical examinations and vital signs recorded throughout the study. Laboratory safety examination and electrocardiograms were performed before, during and after the end of the study.
3 Results
A total of 44 subjects (in stages 1 and 2) were screened, 14 of whom were not randomized (eight subjects did not meet the eligibility criteria, four subjects were included as reserve volunteers, and two subjects did not attend the scheduled hospital appointment). A total of 30 subjects completed the study and were included in the analysis of bioequivalence. Demographic data are summarized in Table 2.
The concentration–time curves of tiotropium after administration of an oral inhalation of a single 20 µg delivered dose of test and reference are shown in Fig. 3. The main pharmacokinetic parameters are described in Table 3.

Concentration–time curves for tiotropium test and reference products. a 0–48 h; and b 0–30 min (mean [standard deviation] value)
The evaluation of comparative bioavailability of C max, AUC t and AUC0.5 of tiotropium was based on a parametric method (ANOVA-log) and is presented in Table 4. In brief, the 90.20 % CIs (second stage) calculated by means of ANOVA-log for the ratio (test:reference [T:R]) of C max, AUC t and AUC0.5 of tiotropium were 87.26–106.60 %, 101.33–111.64 % and 97.95–113.49 %, respectively. The 90.20 % CI for all three primary pharmacokinetic parameters (C max, AUC t and AUC0.5) lay within the 80–125 % acceptance range, and therefore bioequivalence between the assayed products was demonstrated.
Both formulations were well tolerated. No severe or unexpected AEs were recorded. Three non-serious AEs were reported in three subjects (10 %) included in the safety analysis population. One AE (asthenia) and another AE (dizziness) were reported in two subjects randomized to the test formulation. Both AEs were deemed unrelated to the test formulation (as assessed by the study investigator [asthenia was not a listed AE in the summary of product characteristics of the reference tiotropium product]). One AE (dizziness) was reported in one subject randomized to the reference tiotropium product. All AEs resolved without the use of any concomitant medication.
4 Discussion
A novel therapeutic alternative for COPD patients was recently developed—tiotropium 10 µg delivered-dose DPI to be used with the Zonda® inhaler. This pharmacokinetic study demonstrates bioequivalence to the Spiriva HandiHaler®.
Overall development was carried out in accordance with EMA guidance [10], and a single dose and crossover design was then chosen. The crossover design has the advantage of requiring a smaller population. Although the possibility of a within-subjects crossover effect was remote after a single dose of tiotropium, the washout period was long enough as to control the potential impact of the effect. Noteworthy is the two-stage design that is seen less frequently in pharmacokinetic studies, although its use is accepted by the current EMA guideline on bioequivalence [10]. A two-stage design was chosen for accurate estimate of a final sample size for proving bioequivalence and to minimize the risk of any potential AEs.
In the EU, pharmacokinetics bioequivalence studies are considered an acceptable methodology to compare the lung deposition of two inhalation products containing the same active substance. Pharmacokinetic studies are usually considered more discriminative than studies based on pharmacodynamic or clinical endpoints [17]. The crossover design is the most powerful design for a bioequivalence study since it removes the intersubject variability from the comparison of average bioavailability between the formulations. The planned washout period was of at least 10 days’ duration between the subsequent study periods, considered appropriate in view of the long terminal elimination half-life of tiotropium [7]. Finally, and due to practical reasons, the washout time between periods was 14 days.
The use of healthy volunteers instead of patients is supported by the EMA guidance on bioequivalence [10]. In addition, healthy volunteers are considered more discriminative than patients because the presence of bronchoconstriction may result in greater central lung deposition, and hence two inhaled products may appear to be more similar than they actually are [17]. Moreover, in the case of tiotropium, a lung deposition study using radiolabeled tiotropium and assessed by γ scintigraphy showed that tiotropium inhaled from the HandiHaler® DPI has similar delivered doses and lung deposition in healthy subjects than in patients with different stages of COPD disease (mild, moderate and severe) [18]. To our knowledge, at present no pharmacokinetic single-dose study has been published including COPD patients to allow the comparison of the bioavailability between populations.
In this study, the widening of the conventional 90.20 % CI acceptance range for C max was not necessary (the C max values [87.26–106.60 %] were within the preset acceptance range within the 80–125 %). Outcomes from this study have demonstrated that tiotropium is not a drug with high variability (<30 %).
Generally, the OIP guidelines advocate that pharmacokinetic studies should be conducted in patients [10]. However, the available evidence suggests that pulmonary drug deposition is more central in asthmatic patients versus healthy subjects [19], bioavailability is generally greater in healthy volunteers versus asthmatic patients (presumably as the drug is less susceptible to removal by mucociliary clearance prior to absorption in the former) [20–22], and the extent of drug deposition in asthmatic patients is related to the magnitude of airways obstruction [23]. In addition, variability of bronchoconstriction or other underlying pathological changes in asthmatic (and COPD) subjects between study periods may confound the bioequivalence assessments.
A post hoc statistical analysis of the study was performed with the aim of evaluating the bioequivalence of the truncated AUC0.5 as a primary parameter. This is justified for OIPs such as tiotropium with a very quick absorption in the lung (<5 min) where absorption occurs before the contribution of gastrointestinal absorption is significant. Hence, AUC0.5 is an acceptable surrogate for lung deposition and, subsequently, efficacy, whereas AUC t is a surrogate for safety [11]. In the present study, t max for the test formulation and the reference was approximately 3 min, and the subsequent analysis of bioequivalence on AUC0.5 resulted in bioequivalence. Thus, since the results of the study fulfilled the conditions of the EMA [11], a study with charcoal was not necessary and only the hereby presented pharmacokinetic study without active charcoal blockade is sufficient to demonstrate comparable pulmonary deposition and equivalent systemic exposure between the two inhalation products.
A limitation for any pharmacokinetic study of tiotropium is the low plasma concentrations reached after inhaled administration. In consequence, a rapid, selective and highly sensitive bioanalytical method that allows the measurement of tiotropium with an LLOQ of sub-pg/mL was developed and validated for this study. This LLOQ is significantly lower than that used in other similar recent publications [24], and to our knowledge is the lowest LLOQ published for tiotropium. The dual-stage LLE and a 2D UHPLC greatly reduced matrix effects and increased assay sensitivity. Moreover, a supratherapeutic dose of two capsules was administered to better perform the pharmacokinetic profile. The use of this sensitive method was crucial for the determination of bioequivalence because it allows the appropriate characterization of pharmacokinetic parameters.
Also essential for the determination of bioequivalence was the adequate description of absorption curve and C max. It is well known that tiotropium is very rapidly absorbed after inhalation, but the early and frequent sampling times around the expected t max (six samples up to 15 min post-dose) allowed a good estimate of C max. Furthermore, tiotropium also has a long half-life (32–36 h in this study), therefore we were obliged to extend the sample collection up to 48 h. Noteworthy is the fact that as a result of the rapid absorption, all subjects had a quantifiable concentration at the last extraction sample.
5 Conclusion
This study has demonstrated the bioequivalence of a tiotropium 10 µg delivered-dose DPI to be used with the Zonda® inhaler to Spiriva Handihaler®. Both products showed good tolerability with a similar safety profile.
References
Global Initiative for Chronic Obstructive Lung Disease, Inc. Global strategy for the diagnosis, management, and prevention of copd (updated 2016). Available at: http://www.goldcopd.org. Accessed 22 July 2016.
Wright J, Brocklebank D, Ram F. Inhaler devices for the treatment of asthma and chronic obstructive airways disease (COPD). Qual Saf Health Care. 2002;11(4):376–82.
Fink JB. Metered-dose inhalers, dry powder inhalers, and transitions. Respir Care. 2000;45(6):623–35.
Hoppentocht M, Hagedoorn P, Frijlink HW, de Boer AH. Technological and practical challenges of dry powder inhalers and formulations. Adv Drug Deliv Rev. 2014;75:18–31.
Makela MJ, Backer V, Hedegaard M, Larsson K. Adherence to inhaled therapies, health outcomes and costs in patients with asthma and COPD. Respir Med. 2013;107(10):1481–90.
Hvizdos KM, Goa KL. Tiotropium bromide. Drugs. 2002;62(8):1195–203 (discussion 204–5).
Price D, Sharma A, Cerasoli F. Biochemical properties, pharmacokinetics and pharmacological response of tiotropium in chronic obstructive pulmonary disease patients. Expert Opin Drug Metab Toxicol. 2009;5(4):417–24.
Alvarado-Gonzalez A, Arce I. Tiotropium bromide in chronic obstructive pulmonary disease and bronchial asthma. J Clin Med Res. 2015;7(11):831–9.
European Medicines Agency. Committee for medicinal products for human use: guideline on the requirements for clinical documentation for orally inhaled products (OIP) including the requirements for demonstration of therapeutic equivalence between two inhaled products for use in the treatment of asthma and COPD in adults and for the use in the treatment of asthma in children and adolescents.2009. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003504.pdf. Accessed 22 July 2016.
EMA/CHMP. Guideline on the investigation of bioequivalence. 2010. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf. Accessed 22 July 2016.
EMA/CHMP. Questions & answers: positions on specific questions addressed to the Pharmacokinetics Working Party (PKWP). 2015. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002963.pdf. Accessed 22 July 2016.
Quanjer PH, Tammeling GJ, Cotes JE, Pedersen OF, Peslin R, Yernault JC. Lung volumes and forced ventilatory flows. Eur Respir J. 1993;6(Suppl 16):5–40.
Potvin D, DiLiberti CE, Hauck WW, Parr AF, Schuirmann DJ, Smith RA. Sequential design approaches for bioequivalence studies with crossover designs. Pharm Stat. 2008;7(4):245–62.
Chi J, Li F, Jenkins R. Ultrasensitive sub-pg/ml determination of tiotropium bromide in human plasma by 2D-UHPLC-MS/MS: challenges and solutions. Bioanalysis. 2016;8(5):385–95.
Haybittle JL. Repeated assessment of results in clinical trials of cancer treatment. Br J Radiol. 1971;44(526):793–7.
Peto R, Pike MC, Armitage P, Breslow NE, Cox DR, Howard SV, et al. Design and analysis of randomized clinical trials requiring prolonged observation of each patient: I. Introduction and design. Br J Cancer. 1976;34(6):585–612.
García-Arieta A. A European perspective on orally inhaled products: in vitro requirements for a biowaiver. J Aerosol Med Pulm Drug Deliv. 2014;27(6):419–29.
Brand P, Meyer T, Weuthen T, Timmer W, Berkel E, Wallenstein G, et al. Lung deposition of radiolabeled tiotropium in healthy subjects and patients with chronic obstructive pulmonary disease. J Clin Pharmacol. 2007;47:1335–41.
Melchor R, Biddiscombe MF, Mak VH, Short MD, Spiro SG. Lung deposition patterns of directly labelled salbutamol in normal subjects and in patients with reversible airflow obstruction. Thorax. 1993;48(5):506–11.
Brutsche MH, Brutsche IC, Munawar M, Langley SJ, Masterson CM, Daley-Yates PT, et al. Comparison of pharmacokinetics and systemic effects of inhaled fluticasone propionate in patients with asthma and healthy volunteers: a randomised crossover study. Lancet. 2000;356(9229):556–61.
Dalby C, Polanowski T, Larsson T, Borgstrom L, Edsbacker S, Harrison TW. The bioavailability and airway clearance of the steroid component of budesonide/formoterol and salmeterol/fluticasone after inhaled administration in patients with COPD and healthy subjects: a randomized controlled trial. Respir Res. 2009;10:104.
Singh SD, Whale C, Houghton N, Daley-Yates P, Kirby SM, Woodcock AA. Pharmacokinetics and systemic effects of inhaled fluticasone propionate in chronic obstructive pulmonary disease. Br J Clin Pharmacol. 2003;55(4):375–81.
Thorsson L, Geller D. Factors guiding the choice of delivery device for inhaled corticosteroids in the long-term management of stable asthma and COPD: focus on budesonide. Respir Med. 2005;99(7):836–49.
Hohlfeld JM, Sharma A, van Noord JA, Cornelissen PJG, Derom E, Towse L, et al. Pharmacokinetics and pharmacodynamics of tiotropium solution and tiotropium powder in chronic obstructive pulmonary disease. J Clin Pharmacol. 2014;54(4):405–14.
Acknowledgments
Writing support was provided by Nadia Korfali of GeoMed, an Ashfield company, part of UDG Healthcare plc, and was funded by Teva Europe. Teva provided a full review of the article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Laboratorios Liconsa.
Conflict of interest
Jamie Algorta and Laura Andrade are employees of Laboratorios Liconsa. Marta Medina was an employee of Laboratorios Liconsa at the time the study was conducted. Sacha Arsova is an employee of Cooperative Clinical Drug Research and Development AG. Valentin Kirkov, Fumin Li, and Jingduan Chi have no conflicts of interest to declare.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The study protocol was approved by the Independent Ethics Committee of MHAT Tokuda Hospital Sofia AD, Sofia, Bulgaria.
Informed consent
Subjects were informed orally and by means of the Institutional Review Board-approved informed consent sheet. Prior to any activity in the trial, all study participants provided signed informed consent and had the right to withdraw their consent at any time, without giving reason and without detriment.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Algorta, J., Andrade, L., Medina, M. et al. Pharmacokinetic Bioequivalence of Two Inhaled Tiotropium Bromide Formulations in Healthy Volunteers. Clin Drug Investig 36, 753–762 (2016). https://doi.org/10.1007/s40261-016-0441-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-016-0441-8